

Abstract
Background
For potential future human missions to the Moon or Mars and sustained presence in the International Space Station, a safe enclosed habitat environment for astronauts is required. Potential microbial contamination of closed habitats presents a risk for crewmembers due to reduced human immune response during long-term confinement. To make future habitat designs safer for crewmembers, lessons learned from characterizing analogous habitats is very critical. One of the key issues is that how human presence influences the accumulation of microorganisms in the closed habitat.
Results
Molecular technologies, along with traditional microbiological methods, were utilized to catalog microbial succession during a 30-day human occupation of a simulated inflatable lunar/Mars habitat. Surface samples were collected at different time points to capture the complete spectrum of viable and potential opportunistic pathogenic bacterial population. Traditional cultivation, propidium monoazide (PMA)–quantitative polymerase chain reaction (qPCR), and adenosine triphosphate (ATP) assays were employed to estimate the cultivable, viable, and metabolically active microbial population, respectively. Next-generation sequencing was used to elucidate the microbial dynamics and community profiles at different locations of the habitat during varying time points. Statistical analyses confirm that occupation time has a strong influence on bacterial community profiles. The Day 0 samples (before human occupation) have a very different microbial diversity compared to the later three time points. Members of Proteobacteria (esp. Oxalobacteraceae and Caulobacteraceae) and Firmicutes (esp. Bacillaceae) were most abundant before human occupation (Day 0), while other members of Firmicutes (Clostridiales) and Actinobacteria (esp. Corynebacteriaceae) were abundant during the 30-day occupation. Treatment of samples with PMA (a DNA-intercalating dye for selective detection of viable microbial population) had a significant effect on the microbial diversity compared to non-PMA-treated samples.
Conclusions
Statistical analyses revealed a significant difference in community structure of samples over time, particularly of the bacteriomes existing before human occupation of the habitat (Day 0 sampling) and after occupation (Day 13, Day 20, and Day 30 samplings). Actinobacteria (mainly Corynebacteriaceae) and Firmicutes (mainly Clostridiales Incertae Sedis XI and Staphylococcaceae) were shown to increase over the occupation time period. The results of this study revealed a strong relationship between human presence and succession of microbial diversity in a closed habitat. Consequently, it is necessary to develop methods and tools for effective maintenance of a closed system to enable safe human habitation in enclosed environments on Earth and beyond.
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-016-0167-0) contains supplementary material, which is available to authorized users.
Keywords: Closed habitat, Surface, Bacteriome, Microbial succession
Background
In the industrialized world, humans are spending the majority of their lives indoors—some up to 90 % of their time [1, 2]. Built environments are complex ecosystems known to have specific microbiomes [3]. Bio-contamination of these controlled and enclosed environments can present health risks to inhabitants [4–7]. An airborne microbial biodiversity investigation of Halley Station, an isolated scientific research station in continental Antarctica, was conducted to discern the potential source of microbial population and determined no significant patterns in the aerial biodiversity between the austral summer and austral winter [8]. This investigation, however, did not examine the succession of the microbial population for a defined time period. Subsequently, a 1-year investigation of the environmental airborne bacterial population was conducted in the human occupied Concordia Research Station based on conventional cultivation assays [9]. The airborne cultivable bacterial density of the Concordia Research Station was low (<1.0 × 103 CFU/m3), and bacterial contamination was found to increase over time during confinement but diminish after reopening of the station. The predominant cultivable bacterial genera were related to humans (Staphylococcus sp. and Bacillus sp.), and environmental species, such as Sphingomonas paucimobilis, were also found in the air along with a few opportunistic pathogens.
An air sampling study at Providence Milwaukie Hospital revealed that ventilation (air and airflow rates) influenced the microbial community composition. For this study, outdoor and indoor air samples were collected from mechanically ventilated and “naturally” ventilated rooms of the hospital. The relative abundance of bacteria closely related to human pathogens was higher indoors than outdoors and in rooms with lower airflow rates [10]. This study suggests that reducing the airflow rate with the outdoor environment to a minimum allowed the existence of more pathogenic microorganisms. This is an important factor to consider during the habitat design for space missions. In the closed built environments used for space missions, it is not possible to have a constant air flow from the surrounding environment. Therefore, it is very important to understand the ecology of the microbiome in built and closed environments to maximize the health and performance of crewmembers [11].
Similarly, examination of surfaces in 30 different offices in three different cities (Tucson, New York, and San Francisco) found significant microbial diversity based on geographical location. The main sources of contamination were human and several of the bacterial genera found were pathogens (e.g., Neisseria, Shigella, Streptococcus, and Staphylococcus) [12]. These results suggest that potential pathogens may only be problematic for severely immune-compromised individuals in indoor offices. Furthermore, such studies shed significant scientific knowledge about the microbial diversity and possible health threats to humans living in sealed and enclosed environments for prolonged periods of time.
Humans have a reduced immune response when exposed to unfavorable environmental conditions like long-term confinement and spaceflight [13, 14]. In addition, several microorganisms have the capability to degrade habitat materials [9, 15, 16] and exhibited increased pathogenic characteristics during spaceflight [17]. Previous environmental microbiological investigations, like the one of the International Space Station (ISS), suggested that microbial species may undergo permanent changes, such as mutations in the genome over time, and consequently shift microbial population dynamics [5, 11, 18–20]. This finding emphasizes the importance of monitoring and preventing bio-contamination of enclosed environments inhabited by humans and measuring microbial succession to develop strategies in mitigating harmful microbial contamination [18]. The MARS 500 study’s microbiological estimation is the first full-duration simulation of a manned flight to Mars that measured microbiological changes of the air and surfaces using cultivation and molecular methods [21]. However, the MARS 500 study did not account for viable microorganisms utilizing iTag Illumina sequencing technology as reported here but rather adopted Sanger sequencing for evaluating both dead and live microorganisms. Moreover, the study did not include sampling of the facility prior to human occupation to determine a baseline and the change of the microbiome due to human presence.
In early 2009, a team led by the Department of Space Studies at the University of North Dakota (UND), USA, developed advanced inflatable habitat architecture concepts that could be adapted for use on surfaces of the Moon and Mars. An inflatable lunar/Mars analogous habitat (ILMAH) was built to occupy four student crews for mission durations up to 6 months. After completion of the habitat, three test subjects (graduate students of the UND; hereafter called student crews) occupied the ILMAH for two different periods of time (10 and 30 days) [22]. Among others, one purpose of the study was to identify psychological and behavioral problems related to a potential human mission to Moon or Mars. However, the study also provided the opportunity to measure microbiological changes during the student crew’s occupation of the ILMAH.
Here, we present the results of the microbial succession study in the ILMAH, which was conducted by monitoring the bacteriome of several surfaces before human occupation and during the 30-day occupation by the three student crew. To address the microbial divergence in closed indoor environments, surface samples from defined locations were sampled before and after the occupation, as well as three consecutive time periods during the habitation. In addition to the total bacteriome (iTag Illumina sequencing), cultivable and viable microbial populations of the ILMAH surfaces were elucidated. Viable but not-yet-cultivable microbial populations were determined by using well-established propidium monoazide (PMA) and adenosine triphosphate (ATP) assays [23, 24]. Microbiological characterization has been conducted for human habitats, such as indoor buildings [25, 26], hospitals [27], and the MARS 500 habitat [21]; however, this is the first study to characterize the viable microbial community of a closed habitat utilizing state-of-the-art technologies.
Results
Microbial burden
The microbial populations of various surfaces of the ILMAH, as estimated by culture-dependent and independent analyses, are summarized in Table 1. The cultivable bacterial counts ranged from 103 to 107 CFU/m2. After an initial decline of ~1 to 3 logs of colony counts from Day 0 (before occupation) to Day 13, the counts were exhibited an increase in order from Day 13, to Day 20, to Day 30 for locations in front of the sleeping area (bedroom) and bathroom, whereas the colony counts in the kitchen area decreased over these time points. However, the cultivable counts did not change during the 30-day occupation period in the lab locations. Changes in Day 0 cultivable bacterial population were significant (Additional file 1: Table S1), where Day 0 possessed a significantly higher cultivable population than the samples collected in subsequent time points (Table 1).
Table 1.
Total, viable, and cultivable microbiological characteristics of ILMAH surface samples
| Sample location | Cultivable bacterial population (CFU/m2) | QPCR-based bacterial population (16S rRNA copies/m2) | ATP-based microbial population (relative luminescence unit/m2) | ||||
|---|---|---|---|---|---|---|---|
| (A) Total bacterial burden (PMA-untreated) | (B) Viable bacterial burden (PMA-treated) | Percentage of viable bacterial population (B/A × 100) | (C) Total microbial burden (total ATP content) | (D) Viable microbial burden (intracellular ATP content) | Percentage of viable microbial population (D/C × 100) | ||
| North Dakota ILMAH 30-day mission (before crew occupation) | |||||||
| Bedroom 1 | 1.26 × 106 | 1.44 × 106 | 3.69 × 105 | 25.68 | 3.93 × 106 | 7.65 × 106 | >100 |
| Bedroom 2 | 1.86 × 106 | 1.10 × 106 | 8.42 × 105 | 76.62 | 1.27 × 106 | 1.13 × 106 | 88.89 |
| Kitchen 3 | 4.63 × 106 | 5.83 × 105 | 6.23 × 105 | >100 | 9.80 × 105 | 4.43 × 105 | 45.16 |
| Kitchen 4 | 4.25 × 106 | 2.44 × 106 | 1.50 × 106 | 61.46 | 1.56 × 106 | 2.28 × 106 | >100 |
| Bathroom 5 | 1.41 × 106 | 9.41 × 106 | 1.70 × 106 | 18.02 | 9.29 × 105 | 3.36 × 105 | 36.19 |
| Laboratory 6 | 1.51 × 107 | 1.33 × 106 | 4.28 × 105 | 32.23 | 2.15 × 106 | 5.47 × 106 | >100 |
| Laboratory 7 | 2.08 × 107 | 6.43 × 103 | BDL | ND | 2.03 × 106 | 1.04 × 107 | >100 |
| Laboratory 8 | 8.10 × 105 | BDL | BDL | ND | 1.89 × 106 | 1.02 × 106 | 53.63 |
| North Dakota ILMAH 30-day mission (Day 13) | |||||||
| Bedroom 1 | 3.80 × 103 | 1.23 × 106 | 6.49 × 104 | 5.28 | 7.82 × 105 | 3.36 × 105 | 43.04 |
| Bedroom 2 | 1.85 × 104 | 5.32 × 105 | 7.80 × 104 | 14.66 | 4.82 × 106 | 6.63 × 105 | 13.76 |
| Kitchen 3 | 2.60 × 104 | 8.88 × 105 | 1.36 × 105 | 15.28 | 6.96 × 106 | 8.75 × 105 | 12.58 |
| Kitchen 4 | 7.35 × 104 | 1.43 × 107 | 4.66 × 105 | 3.26 | 2.48 × 106 | 1.20 × 106 | 48.43 |
| Bathroom 5 | 1.05 × 104 | 7.95 × 105 | 3.44 × 104 | 4.33 | 3.50 × 106 | 7.02 × 105 | 20.07 |
| Laboratory 6 | 8.93 × 104 | 2.06 × 106 | 6.46 × 105 | 31.47 | 4.75 × 106 | 1.89 × 106 | 39.81 |
| Laboratory 7 | 1.19 × 105 | 4.16 × 106 | 3.68 × 105 | 8.85 | 4.60 × 106 | 9.76 × 105 | 21.22 |
| Laboratory 8 | 3.33 × 104 | 1.11 × 106 | 3.25 × 105 | 29.30 | 3.87 × 106 | 1.03 × 106 | 26.54 |
| North Dakota ILMAH 30-day mission (Day 20) | |||||||
| Bedroom 1 | 2.05 × 104 | 6.93 × 105 | 8.52 × 104 | 12.31 | 6.04 × 106 | 1.31 × 106 | 21.65 |
| Bedroom 2 | 1.62 × 105 | 2.89 × 105 | 4.45 × 104 | 15.40 | 6.04 × 106 | 1.78 × 106 | 29.43 |
| Kitchen 3 | 4.63 × 104 | 4.38 × 106 | 3.79 × 105 | 8.66 | 5.05 × 106 | 1.94 × 106 | 38.48 |
| Kitchen 4 | 2.10 × 104 | 2.00 × 106 | 1.01 × 105 | 5.04 | 2.47 × 106 | 5.97 × 105 | 24.19 |
| Bathroom 5 | 1.90 × 104 | 2.84 × 105 | 1.75 × 105 | 61.86 | 4.05 × 106 | 1.75 × 106 | 43.15 |
| Laboratory 6 | 5.58 × 104 | 1.09 × 106 | 2.16 × 105 | 19.76 | 4.40 × 106 | 2.10 × 106 | 47.72 |
| Laboratory 7 | 1.47 × 104 | 3.71 × 105 | 1.74 × 104 | 4.70 | 2.65 × 106 | 1.01 × 106 | 38.05 |
| Laboratory 8 | 5.55 × 104 | 8.90 × 105 | 1.86 × 105 | 20.86 | 3.88 × 106 | 1.47 × 106 | 37.91 |
| North Dakota ILMAH 30-day mission (Day 30) | |||||||
| Bedroom 1 | 1.12 × 105 | 3.81 × 106 | 1.60 × 105 | 4.19 | 1.61 × 106 | 4.14 × 105 | 25.77 |
| Bedroom 2 | 2.33 × 105 | 3.28 × 106 | 3.38 × 105 | 10.30 | 4.10 × 106 | 1.83 × 106 | 44.75 |
| Kitchen 3 | 3.24 × 104 | 3.98 × 106 | 1.94 × 105 | 4.88 | 2.42 × 106 | 9.63 × 105 | 39.85 |
| Kitchen 4 | 2.15 × 103 | 2.41 × 106 | 8.69 × 104 | 3.61 | 3.66 × 106 | 3.61 × 105 | 9.87 |
| Bathroom 5 | 1.03 × 105 | 3.29 × 106 | 1.44 × 105 | 4.38 | 2.17 × 106 | 6.15 × 105 | 28.37 |
| Laboratory 6 | 1.27 × 105 | 5.92 × 106 | 1.09 × 106 | 18.36 | 1.48 × 106 | 1.07 × 106 | 72.35 |
| Laboratory 7 | 6.30 × 104 | 6.89 × 106 | 7.78 × 105 | 11.30 | 2.96 × 106 | 1.25 × 106 | 42.19 |
| Laboratory 8 | 5.85 × 104 | 4.86 × 106 | 3.41 × 105 | 7.02 | 4.12 × 106 | 7.29 × 105 | 17.68 |
BDL below detection limit, ND since one of the values is BDL, the ratio cannot be determined
The qPCR assay that measured 16S ribosomal RNA (rRNA) gene copies from both dead and live bacterial cells showed an increase in bacterial density over the 30-day occupation period. In general, samples treated with PMA (viable bacterial burden) revealed that ~60 % of the bacterial population was dead at Day 0, whereas the reduction in bacterial population was ~90 % for the subsequent days of occupation. The viable bacterial burden as measured by the PMA-qPCR assay ranged from below detection limit to 106 16S rRNA gene copies/m2. Furthermore, during the 30-day occupation period, the ratio of cultivable bacteria to viable bacterial burden (ATP assay results) was at least twice more on the final day (Day 30; 33.4 %) than before occupation (Day 0; 18.9 %). The percent cultivable bacteria among viable bacterial burden were greater at the surfaces in front of the bathroom (~47 %) or in bedroom area (~55 %) when compared to other locations (~12 to 14 %). These differences were highly significant when considering qPCR results of PMA vs. non-PMA samples in a paired t test (p = 0.000106).
The total (ATP content from both dead and live microbes) and viable microorganisms (intracellular ATP content) were in the range of 105–106 relative luminescence unit per m2. In general, when samples from all sampling locations were pooled together, the total microbial burden did not show any changes in microbial accumulation over the time period. However, ~55 % of the microbes were viable at Day 1, and the viable microorganisms were reduced to ~28 % on Day 13 and ~35 % on the last day of occupation. The percentage of viable bacterial burden (PMA-qPCR assay) among viable microbial burden (intracellular ATP content) was highest in the lab area (~40 %) and lowest in the bathroom area (~10 %). A closer look at the microbial burden as measured by ATP content of the different locations revealed that the succession of bacterial burden was not linear throughout the 30-day occupation. The bacterial density initially decreased in all the locations (from Day 13 to Day 20) and then drastically increased (Day 30) in the bedroom area, the bathroom area, and the lab area, whereas samples from the kitchen area showed no fluctuation. From a statistical standpoint, the microbial density fluctuations in various locations were not significant (Additional file 1: Table S2).
Cultivable bacterial diversity
Phylogenetic characterization of 150 strains isolated during this study and identified via 16S rRNA gene analysis revealed a total of 62 known bacterial species and eight lineages yet to be described (based on 97 % similarity of 16S rRNA genes to publicly available sequences; Fig. 1, Additional file 1: Table S3). More than half of the identified isolates belonged to Firmicutes (76 strains), 26 % to Proteobacteria (39 strains), and 20 % to Actinobacteria (30 strains). Bacillus species represented the highest number of isolates (43 strains), followed by Staphylococcus (24 strains) and Pseudomonas (17) species.
Fig. 1.
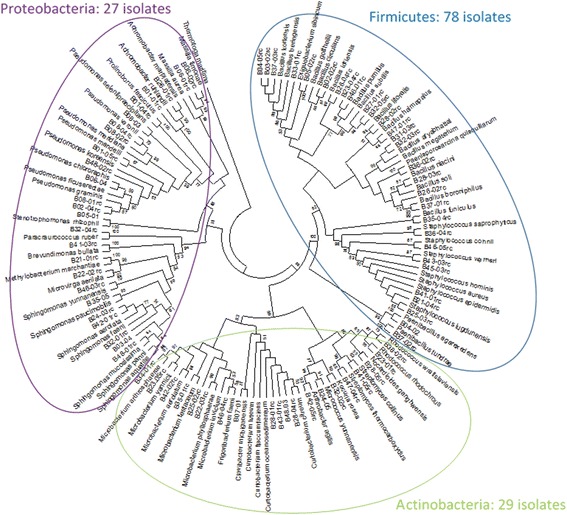
Cultivable bacterial diversity detected through the 30-day habitation period at all the locations based on 16S rRNA gene sequences. The strain designation or type strain of the closest neighbor followed by the GenBank EZtaxon-e is given. The phylogenetic tree was constructed using neighbor-joining method (bootstrap 1000). In total, 150 isolates were collected, 147 of which were successfully sequenced (3 strains did not respond to the sequencing methods attempted and hence excluded from the tree). The numbering of the isolates is explained as follows. B = bacteria, first number (0–4) will be the sample collection day (0 = Day 0, 2 = Day 13, 3 = Day 20, 4 = Day 30), second number (1–8) will be sampling location and the third number (01–05) is the replicate number of the isolate. For example, B-38-02 will be a bacterial strain, isolated from Day 30, at location number 8, and a second isolate. Frequency of isolates is given in parenthesis after the name of the bacterium
Throughout the occupation period, changes in the abundance of the cultivable bacterial species occurred. Before the student crew moved in (Day 0 sampling), almost two thirds of the isolates were Proteobacteria (14 strains), 28 % Firmicutes (8 strains), 8 % Actinobacteria (2 strains), and 3 % Bacteroidetes (1 strain). The majority of the proteobacterial isolates (13 out of 14 strains) belonged to Gammaproteobacteria and most Firmicute isolates (6 out of 8 strains) were Bacillus species.
At Day 13, the relative abundance pattern of cultivable bacteria changed drastically. The frequency of Proteobacteria declined to 10 %, whereas Firmicutes and Actinobacteria increased to 60 and 29 %, respectively. None of the Gammaproteobacteria recovered at Day 0 were retrieved at Day 13 (or Day 20 or Day 30), instead Alphaproteobacteria was present. A few of the Bacillus species found at Day 0 recurred at Day 13 (Bacillus idriensis, Bacillus litoralis, and Bacillus niacini) and additional Bacillus species were isolated (e.g., Bacillus soli, Bacillus megaterium). Actinobacteria that were recovered for the first time were represented by the genera Microbacterium, Curtobacterium, Micrococcus, and Rhodococcus.
Day 20 samples showed a similar relative abundance pattern of the phyla as the Day 13 samples. No species of Proteobacteria and almost no species of Actinobacteria (one exception: Curtobacterium faccumfaciens) from Day 13 were isolated again at Day 20. However, bacterial species from all of the same genera at Day 13 also recurred at Day 20. Some of the Firmicutes from Day 13 reappeared, whereas some Bacillus species emerged for the first time.
Day 30 samples revealed a slight change in the relative abundance pattern compared to Day 20: less Firmicutes (51 %) were retrieved in favor of Proteobacteria (21 %). The recovery rate of Actinobacteria did not change (24 %), and one Bacteroidetes was isolated (3 %). Consistently, almost no Proteobacteria and Actinobacteria from the earlier time points were detected again. However, other species of the previously recovered genera were found and some of the Firmicutes from Day 13 and/or Day 20 were isolated again and some undetected Firmicutes species emerged (Fig. 1).
The analysis of the cultivable bacteria indicates a strong decline in the number of Proteobacteria from Day 0 to Day 13 and an increase in Firmicutes and Actinobacteria over this time. Changes within the later three time points (Day 13, Day 20, and Day 30) were only detectable at species level, not at phyla level. Almost all representative species from Actinobacteria and Proteobacteria were isolated only once throughout the whole occupation period, whereas representatives of the Firmicutes were recovered multiple times.
Controls
Sampling device control, environmental controls, DNA extraction reagent controls, and no-template PCR controls included in this study yielded no sequence reads. The colony counts, qPCR, and ATP-based analyses to estimate microbial burden also exhibited values below detection limit.
Significant differences in viable and total bacteriome
The bacterial richness between PMA and non-PMA samples declined significantly (paired t test based on number of operational taxonomic units (OTUs) revealed a p value <0.0001). Moreover, 208 genera were detected in the non-treated samples, 37 of which were not identified in the PMA-treated samples. The two sample types also differed significantly in community relationships (NMDS analysis in Fig. 2a, c, Adonis p = 0.034 and MRPP, significance of delta = 0.023, A = 0.01888) and their Shannon diversity index indicated a significant reduction (paired t test p < 0.001). Due to these differences in the total and viable bacteriome, it could be concluded that the total bacteriome (including dead cells) does not give a true picture of the causative bacterial agent(s) that trigger illness. Consequently, all following results are based on data generated from the viable bacteriome only.
Fig. 2.
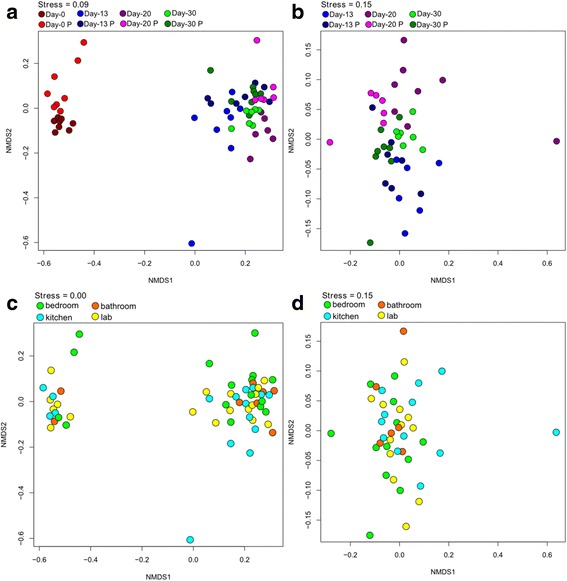
NMDS ordinations based on Bray-Curtis distances between all samples; a NMDS ordination displaying the distance between samples taken at the different time points; b NMDS ordination displaying the distance between samples taken at the different time points excluding D0 samples; c NMDS ordination displaying the distance between samples taken at the different locations; Adonis p-value 0.452, MRPP, significance of delta 0.957, A: –0.02321. d NMDS ordination displaying the distance between samples taken at the different locations excluding D0 samples; Adonis p-value 0.486, MRPP, significance of delta 0.254, A: 0.005762. A suffix “P” after the respective time points indicates that these samples are treated with PMA. a, b Various time points: Adonis p = 0.001 and MRPP significance of delta = 0.001 and A = 0.02813. c, d Various locations: Adonis p value of 0.452, MRPP, significance of delta = 0.957, A = −0.02321. The permanova and MRPP when looking at two different test (being treated with PMA or being not treated with PMA) are Adonis p = 0.034 and MRPP, significance of delta = 0.023, A = 0.01888. The Day 30; Kitchen-4 (see Table 1) sample is a potential outlier but removing this sample from the NMDS permutations did not change the significance values of the different variables but this sample was not included in this figure
Substantial changes in the viable bacterial community composition after occupation
The diversity of the overall viable bacteriome was dominated by Actinobacteria, Firmicutes, and Proteobacteria (97 % of all characterized OTUs) and, in combination with the Bacteroidetes, these four phyla accounted for 99 % of the characterized OTUs. A closer examination of the OTUs on genus level indicated a predominance of nine taxa, which were primarily comprised of by Corynebacteria (20 % of total OTUs), Bacilli (15 %), Staphylococci (18 %), Anaerococci (11 %), and Fusobacterii (14 %). In sum, the sequences from the aforementioned five genera constituted 78 % of all OTUs found in the PMA-treated samples (Table 2).
Table 2.
Bacterial taxa retrieved from ILMAH surfaces sampled at various time points of human occupation
| Bacterial taxon | Number of sequences from sampling time at | |||||||
|---|---|---|---|---|---|---|---|---|
| Day 0 | Day 13 | Day 20 | Day 30 | |||||
| Total | Viable | Total | Viable | Total | Viable | Total | Viable | |
| Acidobacteria | 15 | 11 | 89 | 47 | 15 | 49 | 41 | |
| Actinobacteria | 2803 | 1156 | 17631 | 10697 | 14865 | 4806 | 23314 | 17663 |
| Bacteroidetes | 2733 | 538 | 2418 | 169 | 1719 | 1517 | 538 | |
| Candidatus Saccharibacteria | 91 | 52 | 106 | 39 | 56 | 99 | 7 | |
| Chlamydiae | 22 | 23 | 31 | 8 | ||||
| Chloroflexi | 94 | 42 | 49 | 59 | 19 | 28 | 43 | 11 |
| Cyanobacteria | 129 | 53 | 567 | 29 | 96 | 1704 | 541 | |
| Deinococcus-Thermus | 50 | 9 | 7 | 1 | 3 | 2 | 4 | |
| Firmicutes | 6303 | 4719 | 17282 | 9604 | 13910 | 2212 | 23312 | 15422 |
| Fusobacteria | 11 | 54 | 1 | 13 | 111 | 5 | ||
| α-proteobacteria | 15956 | 4082 | 2959 | 1196 | 396 | 29 | 1893 | 1881 |
| β-proteobacteria | 10664 | 3051 | 1835 | 980 | 261 | 39 | 1242 | 1065 |
| δ-proteobacteria | 21 | 5 | 113 | 88 | 34 | 8 | 13 | 16 |
| ε-proteobacteria | 3 | 6 | 1 | 2 | 2 | |||
| γ-proteobacteria | 3088 | 1279 | 3561 | 1777 | 3997 | 27 | 2340 | 412 |
| Parcubacteria | 19 | 54 | 3 | 5 | 13 | |||
| Others (4 phyla) | 1 | 3 | 21 | 20 | 5 | 19 | 3 | |
| Unclassified bacteria | 131 | 63 | 247 | 620 | 71 | 10 | 215 | 150 |
To investigate differences in the bacteriome between samples, multivariate statistics were applied using ordination analyses and Monte Carlo-based permutation tests. Viable bacterial communities formed significantly different groups in NMDS plots based on the factor time, i.e., the different sampling days showed distinct microbiome profiles (Fig. 2a, various time points: Adonis p = 0.001 and MRPP, significance of delta = 0.001 and A = 0.02813; Fig. 2c, various locations: Adonis p value of 0.452, MRPP, significance of delta = 0.957, A = −0.02321). Notably, the bacteriome of Day 0 samples was substantially different from the bacteriome of the later time points. For that reason, Day 0 samples were excluded in another analysis (Fig. 2b, d), which also revealed that the community profiles of the later three sampling events (Day 13, Day 20, and Day 30) were significantly different from each other, even though the differences were smaller compared to Day 0 (Fig. 2b; various time points: Adonis p = 0.001 and MRPP, significance of delta = 0.001 and A = 0.06871). The NMDS ordinations based on Bray-Curtis distances between all samples that were not treated with PMA (Additional file 2: Figure S2) and PMA (Additional file 2: Figure S3) are shown. The UniFrac distance of various ILMAH bacteriome datasets are shown in Additional file 2: Figure S4. The bacteriome distribution patterns were similar whether Bray-Curtis distance or UniFrac distance was used (UniFrac for various time points: Adonis p value of 0.001, MRPP significance of delta = 0.001, A = 0.2988; UniFrac for various locations: Adonis p value of 0.801, MRPP significance of delta = 0.955, A = −0.03882; different treatments (PMA and no PMA): Adonis p value of 0.012, MRPP significance of delta = 0.004, A = 0.03886).
Due to the differences in multivariate statistics, the bacteriome change on a single OTU level was investigated. Prior to the occupation (Day 0 sampling), the majority of the microbial community consisted of Firmicutes (59 %), followed by Proteobacteria (27 %), and Actinobacteria (9 %). By the end of the occupation (Day 30 sampling), the relative abundance of Firmicutes in the habitat had not changed significantly (61 %), whereas Proteobacteria decreased (16 %) and Actinobacteria increased (21 %) (Table 2). The bacteriome profiles of various locations of ILMAH surfaces are shown in Table 3.
Table 3.
Bacterial taxa retrieved from ILMAH surfaces sampled at various locations during the human occupation
| Bacterial taxon | Number of sequences from | |||||||
|---|---|---|---|---|---|---|---|---|
| Bedroom | Kitchen | Bathroom | Lab | |||||
| Total | Viable | Total | Viable | Total | Viable | Total | Viable | |
| Acidobacteria | 83 | 2 | 43 | 44 | 16 | 47 | 26 | 6 |
| Actinobacteria | 22177 | 5326 | 20602 | 14580 | 6168 | 3764 | 9666 | 10652 |
| Bacteroidetes | 2511 | 73 | 3733 | 396 | 613 | 55 | 1530 | 721 |
| Candidatus Saccharibacteria | 147 | 78 | 50 | 81 | 10 | 46 | 38 | |
| Chlamydiae | 35 | 9 | 10 | 14 | 7 | 9 | ||
| Chloroflexi | 57 | 6 | 86 | 91 | 16 | 8 | 46 | 35 |
| Cyanobacteria | 396 | 4 | 1845 | 37 | 107 | 13 | 148 | 569 |
| Deinococcus-Thermus | 5 | 39 | 7 | 2 | 1 | 18 | 4 | |
| Firmicutes | 22694 | 5259 | 21324 | 13813 | 6557 | 3323 | 10232 | 9562 |
| Fusobacteria | 43 | 1 | 124 | 11 | 11 | 5 | ||
| α-proteobacteria | 6737 | 825 | 8090 | 2586 | 1384 | 712 | 4993 | 3065 |
| β-proteobacteria | 4321 | 1062 | 5303 | 2236 | 760 | 363 | 3618 | 1474 |
| δ-proteobacteria | 71 | 9 | 46 | 100 | 51 | 13 | 8 | |
| ε-proteobacteria | 5 | 5 | 1 | 1 | 2 | |||
| γ-proteobacteria | 1661 | 292 | 8988 | 2121 | 560 | 200 | 1777 | 882 |
| Parcubacteria | 19 | 5 | 54 | 13 | 3 | |||
| Others (4 phyla) | 4 | 24 | 23 | 18 | 3 | |||
| Unclassified bacteria | 180 | 58 | 301 | 669 | 42 | 33 | 141 | 83 |
To identify viable taxa that significantly correlated with the various occupation time points, a Spearman rank correlation was applied individually to each OTU’s abundance pattern and sampling time. The results are displayed as a heat map (Fig. 3, PMA-treated samples) presenting only those OTUs that showed a significant correlation (76 in number) with a p value of 0.01. Most of the OTUs belonged to the phylum Firmicutes (23 OTUs), followed by Proteobacteria (20 OTUs) and Actinobacteria (19 OTUs); the rest of the taxa were classified as Bacteroidetes (9 OTUs) and Candidatus Sachcharibacteria (formerly known as TM7), Cyanobacteria, Deinococcus-Thermus, Fusobacteria and Acidobacteria (1 OTU, respectively). Actinobacteria and Firmicutes that increased throughout the occupation period were dominated by two families: Corynebacteriaceae (7 OTUs), Clostridiales Incertae Sedis XI (8 OTUs). Other OTUs that increased throughout the occupation period were mostly Proteobacteria (7 OTUs) belonging to different families. Members of Proteobacteria that decreased throughout time (13 OTUs) were dominated by Oxalobacteraceae (4 OTUs), Comamonadaceae (2 OTUs), and Pseudomonadaceae (2 OTUs). Among Firmicutes (9 OTUs), members of the family Bacillaceae (3 OTUs) increased and all but one Bacteroidetes (8 OTUs) decreased within the course of time.
Fig. 3.
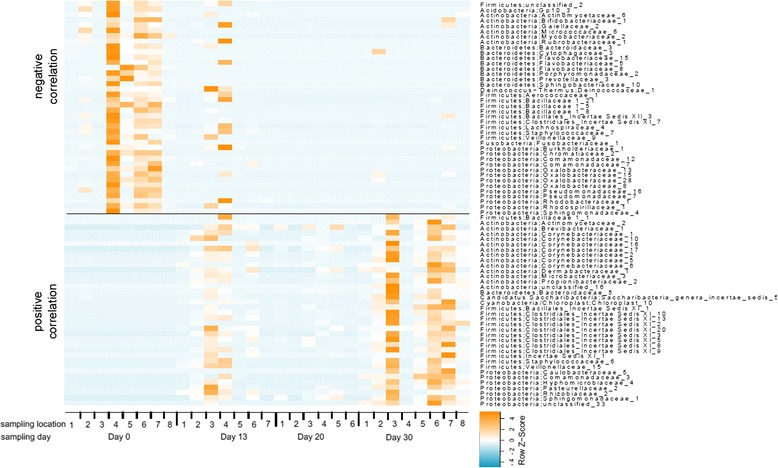
Heat map of the taxa that showed a significant correlation (p value of 0.01) with the factor time in the PMA-treated sample set. The color blue indicates a low abundance of the single OTU in the respective sample and orange indicates a high abundance of the single OTU in the respective sample
Since the Day 0 samples exhibited a fundamentally different bacteriome composition, two additional analyses were employed. First, Spearman rank correlation was applied on the PMA-treated dataset excluding Day 0 samples. At a p value of 0.05, 26 OTUs showed a significant correlation with the factor time, six of which decreased throughout the occupation period, whereas 20 OTUs increased. Most of the OTUs that exhibited a positive correlation belonged to the phyla Proteobacteria (8 OTUs; dominated by Caulobacteraceae (2 OTUs)), followed by Firmicutes (7 OTUs; dominated by Clostridiales Incertae Sedis XI (4 OTUs)), followed by Actinobacteria (3 OTUs), and Actinobacteria (2 OTUs). Of those 26 OTUs, three families were identical with the correlation analyses above, confirming that OTUs belonging to Clostridiales Incertae Sedis XI, Comamonadaceae, and Corynebacteriaceae truly increased.
Second, an ANOVA was applied on OTU abundances across all the PMA-treated samples comparing data at Day 0 with those at the later three time points (p = 0.01). Thirty-four OTUs that were more abundant at Day 0 were identified as belonging to similar genera as OTUs identified in the correlation analysis. They belonged mainly to Oxalobacteraceae (8 genera) and different genera belonging to Proteobacteria, Actinobacteria, and Firmicutes. The predominant and differential bacterial taxa (>0.75 % of overall microbial abundance) of various time points of the ILMAH surfaces are depicted in Fig. 4.
Fig. 4.
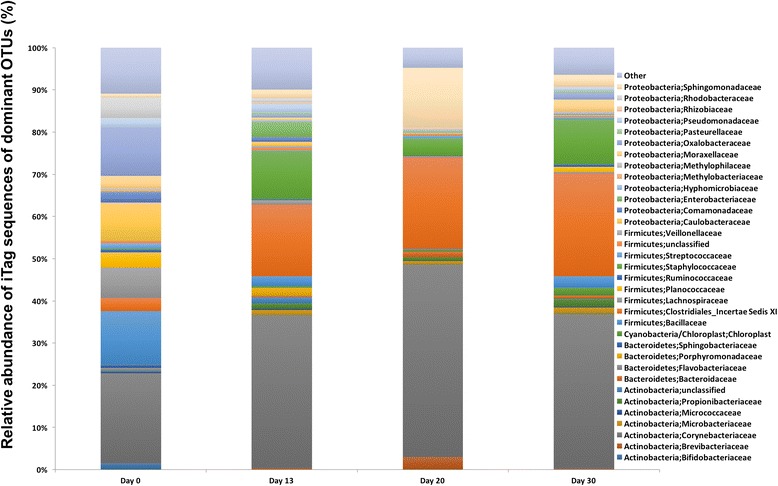
Dominant microbial population and microbial succession patterns observed in 30-day occupation period of the ILMAH system. The OTU abundance that are >0.75 % of the total microbial population are computed in this bar graph. Day 0 surfaces show completely different microbial profile when compared to the subsequent sampling days of the crew occupation
In sum, statistical analyses revealed a significant difference in community structure of samples over time, particularly of the bacteriomes existing before human occupation of the habitat (Day 0 sampling) and after occupation (Day 13, Day 20, and Day 30 samplings). Actinobacteria (mainly Corynebacteriaceae) and Firmicutes (mainly Clostridiales Incertae Sedis XI a) were shown to increase over the occupation time period.
Archaea
In general, archaea were of low abundance in the samples collected from the ILMAH surfaces. The archaeal microbial burden detected via specific qPCR was below detection level in most of the samples tested (28 out of 32 samples) and in positive samples, archaea were between one and two logs lower than the bacterial microbial burden. The overall low abundance of archaea in the community was confirmed by the iTag data, since 92 % of all the OTUs found with archaea-specific primers were identified as bacteria indicating low abundance of archaea resulting in unspecific priming. Due to the low number of sequences detected it was not possible to depict any change in the archaeal community structure throughout the occupation period (Additional file 1: Table S4). It is important to note that Nitrososphaera represented the majority of all OTUs found in the non-PMA-treated samples (94 %). Along with Nitrososphaera, three other archaeal genera were detected: Methanocaldococcus, Methanosarcina, and Nitrosopumilus. However, in the PMA-treated sample set, Nitrososphaera was the only archaeal genera observed, indicating that the latter three genera might be dead highlighting the importance of using viability assays for archaea in indoor microbiomes.
Discussion
Human missions to other celestial bodies like the Moon or Mars are currently being planned for the future by space-faring nations [28]. When building a self-sufficient settlement, microbial accumulation of viable cells that may potentially cause disease will be a concern for crewmembers’ health. Application of validated microbial reduction technologies for a closed habitat may substantially reduce microbial populations [29] but leave behind genetic materials that could be falsely diagnosed for the presence of potentially dangerous biological contamination. Hence, it is necessary to develop microbial detection technologies to target viable cells that cause disease (human and plants) and deteriorate the human habitat. Findings from microbiological characterization of the controlled Mars analogous habitat (ILMAH, MARS 500, etc.) and other closed systems (ISS) will help space agencies in developing appropriate countermeasures to eradicate viable microorganisms that could be problematic to human health.
Previous studies have shown that meaningful data can often only be retrieved if the dead fraction of the cells is excluded [23, 30]. Otherwise, the significance of viable, but low in abundance microbial communities might be underestimated, because their molecular signals are masked by other taxa that are dominant but dead. As shown in this ILMAH study and, other reports, more than 50 % of the cells were dead and the bacterial diversity was reduced significantly in samples treated with PMA [11, 23]. In addition, low-abundant bacterial taxa were more present in the PMA-treated samples when compared with the non-treated samples, which is statistically significant (p = 0.04813) and had been reported before for a variety of microorganisms including viruses [31]. This approach confirmed that a PMA treatment or other validated life/dead-detection method is essential when characterizing molecular microbial communities, thus eliminating the nucleic acids from dead cells. In this study, only the viable microbial community structure was discussed in terms of predicting changes in microorganisms over the successive sampling time points.
Various studies report that human presence is the most common source of contamination in strictly controlled rooms [21, 32, 33]. Dispersal of microorganisms by humans is dependent on their activities and time spent in the closed habitats. Typically, human natural skin renewal and shedding are 106 to 107 particles per day [34]; perspiration, coughing, or speaking expels 103 to 104 droplets per (re)action; [35, 36]. In addition, shoes and clothing introduced into a controlled room by the inhabitants are the other sources of microorganisms [37]. The ILMAH study results also indicate that the presence of humans influenced the microbial diversity and composition in the closed habitat. Most of the observed variations in the ILMAH microbial community were due to the move-in of the student crew. Both the cultivable bacteria and the analysis of the bacterial community via iTaq sequencing showed that samples taken at Day-0 (before occupation) contained an ecologically distinctive set of microbial taxa that were not abundant in the samples taken at Day-13, -20, and -30 (Fig. 4). Microbial community changes within the later three time points (Day-13, -20, and -30) were seen only at species level, not at phyla level. It is becoming increasingly evident that quorum sensing enhances the ability of bacteria to access more favorable environmental niches and increase bacterial defenses against eukaryotic hosts, competing bacteria, and environmental stresses [38]. The physiological and clinical aspects of quorum sensing have received considerable attention and are starting to be studied at the molecular level. However, little is known about whether quorum sensing plays an important role in indoor environments. Additional research is needed to understand the mechanism(s) of biofilm formation by the predominant environmental microbial species of the closed systems and the influence of cell-to-cell signaling. With a greater understanding, it may be possible to maintain an environment that interferes with quorum sensing, thus inhibiting growth of potential corrosive microorganisms, virulence, and biofilm formation, which would greatly benefit the health and safety of humans in closed systems [39].
The viable bacterial communities (PMA-treated samples) retrieved from samples at Day 0 (Flavobacteriaceae, Caulobacteraceae, and Oxalobacteraceae) were reported to be common in aquatic and soil habitats [40, 41]. The 10 % bleach cleaning solution used on the ILMAH surfaces was likely detrimental to these bacteria and eradicated most of the soil microorganisms documented in Day 0, since their presence in subsequent sampling periods was not in great abundance (Fig. 4). The bacterial communities noticed at Day-13, -20, and -30 were dominated by taxa mostly associated with humans as commensals or pathogens, such as Corynebacteriaceae, Clostridiales Incertae Sedis, and Staphylococcaceae [42–45], and were reported to be associated with the human skin or gut microbiome [46]. Actinobacteria (Corynebacterium, Propionibacterium, etc.) were more abundant on skin, with Firmicutes (Clostridium) and Bacteroidetes more abundant in the gastrointestinal tract. Sequences of notable pathogens, such as Brevibacteria (osteomyelitis and otitis; [47]), Actinomycetae (actinomycosis; [48]), Propionibacteria (acne; [49]), Corynebacteraceae (nosocomical; [50–52]), Staphylococcaceae (skin rashes; [53]), and Clostridium (tetanus and botulism; [54]) were retrieved from the samples that were treated with PMA and thus may be viable. Even though sequences of potential pathogens were retrieved from the ILMAH surfaces and phylogenetically identified, their pathogenicity could not be confirmed with the available data. Future metagenomic study might shed light on the presence of the virulence genes in addition to the phylogenetic signatures.
Several studies have already been conducted on the microbial population in confined habitats, including several office buildings [12, 55, 56], hospitals [57], the Concordia Research Station [9], spacecraft surfaces and associated cleanrooms [7, 58–60], the MARS 500 habitat [21], and the ISS [5, 11, 19]. These studies also confirm that the sources of microbial contamination are human and opportunistic pathogens able to thrive in enclosed environments. All monitored confined habitats reported having restricted waste disposal and limited fresh air supply, which may possibly lead to bad air quality, water condensation, and accumulation of biological residues [9, 21]. Unlike human-inhabited enclosed environments, cleanrooms (pharmaceutical, medical, and spacecraft assembly) restrict human access (e.g., generally, ~8 h per shift). Various researchers have investigated these highly maintained cleanroom surfaces using iTag sequencing and metagenomic analyses [31, 59, 60] and found that the majority of the microbial contaminations were of human origin despite restricted human access. However, when specific cultivation-based assays were performed, domination of spore-forming bacteria was reported in these cleanrooms [4, 61–64]. The cultivable bacterial burden was significantly higher in ILMAH surfaces before occupation (mean 4.9 × 106/m2) compared to the occupation period (Day 13, mean 3.8 × 104/m2). The bacterial burden in cleanrooms was comparatively low (1- to 3-logs less) than the ILMAH and MARS 500 habitats [21] and may be attributed to the relatively low human activity, enforced dress regulations, and controlled entrances.
Of all the microbial characterizations studies in confined habitats, the MARS 500 project (June 2010 to November 2011) had the most similar study objectives to the ILMAH study. However, the MARS 500 study did not measure the viable portion of the community. Hence, the measured bioburden and diversity characterization of the MARS 500 report is an overestimation. This has been confirmed since the MARS 500 study revealed a pattern: members of the class Proteobacteria, followed by Bacteroidetes and Firmicutes, decreased over the occupation period; this was also noticed in the ILMAH samples that were not treated with PMA (including both dead and viable bacteria). But such trend was not found for the viable community of the ILMAH. Furthermore, the MARS 500 microbial analysis did not contain control, i.e., preoccupation bacterial diversity measurements, to compare the microbial accumulation as measured in this study. We conclude that it is critical to compare the microbiome of habitat without human occupation to habitat with human occupation since humans appeared to be the primary source of contamination in such habitats. Such a comparison may enable development of cleaning and maintenance protocols. Nonetheless, the MARS 500 project [21] provided important insight about the succession of a microbial community over time and, when compared to this study, the need for live/dead differentiation protocol(s) to elucidate the presence of viable pathogens in closed systems for prolonged periods of time become essential. These studies would help space-faring nations mitigate microbiological problems by developing countermeasures to eradicate unwanted microbial pathogens.
However, the microbial risk assessment in an Earth setting might underestimate the importance of humans living in stressed situations, such as microgravity and long-term travel in confined spaces [65]. Under microgravity conditions, the human immune system is compromised [13, 14] and bacteria exhibit enhanced virulence, antibiotic resistance [17, 66], and increased biofilm formation [67, 68]. The microbial monitoring of the ISS by various space-faring agencies revealed that Staphylococcus and Aspergillus were the dominant cultivable species [5, 69]. However, state-of-the-art molecular techniques to elucidate viable microbial communities of various ISS surfaces and microbial accumulation and succession (biofilm formation) analyses are warranted. The recent ISS air filters and vacuum debris analyses showed that cultivable Staphylococcus species are dominant but also reported retrieval of a large percent (>90 %) of molecular signatures of viable Corynebacterium/Propionibacterium [11].
Studies on archaea have concentrated on natural biotopes where they have been found in overwhelming numbers and with versatile properties [70]. Moreover, extremophilic archaea are considered the terrestrial life most likely capable of surviving on Mars [71], thus it is important to characterize the archaeal community in closed habitats such as the ILMAH. The role of archaea in artificial, human-controlled environments is still unclear, since there are few studies monitoring archaea in households, offices, airplanes, clinical environments, or other restricted environments like pharmaceutical and industrial cleanrooms and spacecraft assembly cleanrooms [72–74]. Previous archaeal surveys suggest that spacecraft assembly cleanroom facilities inhabit a restricted diversity of archaea. Examination of cleanrooms in Europe, South America, and the USA found archaeal signatures belonging to Thaumarchaeota and Euryarchaeota in one third of the collected surface samples (and none from the air samples). In this ILMAH study, only 4 out of the 80 samples (5 %) tested positive for archaea. The majority of the ILMAH archaeal community comprised OTUs of Thaumarchaeota (over 94 %) and the rest were Euryarchaeota (6 %), which supports the results from the aforementioned studies. Particularly, Thaumarchaeota have been recognized to inhabit human skin, another indication that the ILMAH microbiome was influenced by human activity [74].
Conclusions
Accumulation of cultivable and viable (as per molecular methods) bacteria was evident during the 30-day occupation period of the enclosed habitat. No observed change in the total microbial burden (including dead cells) stressed the importance of differentiating dead organic matter from live cells. Thorough maintenance procedures adapted to keep the ILMAH system clean might have eradicated the microorganisms but such processes would not have removed the biomolecules from the surfaces, hence implementation of genetic methods estimating total microbial community structures including dead cells would result in overestimation. However, when viability assays were used, only ~55 % of the microbes were viable at Day 0, and they were reduced to ~28 % on Day 13 and ~35 % on the last day and this might be due to the prolonged stay by the human and their microbial shedding. Among the cultivable bacterial diversity members of the Firmicutes, Proteobacteria, and Actinobacteria phyla were dominant and such results also supported by the Illumina-based sequencing studies. Changes in the abundance of the cultivable bacteria and molecular signatures of viable bacterial species were noticed throughout the occupation period and are statistically significant. Actinobacteria (mainly Corynebacteriaceae) and Firmicutes (mainly Clostridiales Incertae Sedis XI and Staphylococcaceae) were shown to increase over the occupation time period. As seen in other studies where confined habitats were investigated, this study also concluded that humans are the primary source of contamination. A combination of cultivation-based analysis and viability assays is warranted to elucidate the significance of bioaccumulation that might be problematic to the inhabiting human health.
Methods
Sample locations and sampling
Habitat
The ILMAH is approximately 12 m long, 10 m wide, and 2.5 m high. The ILMAH interior consists of four sleeping compartments where the student crews are able to rest as well as stow their personal belongings, a small galley/dining room, a bathroom, and plenty of laboratory space (Additional file 2, Figure S1). Three student crews inhabited the ILMAH for 30 days collecting several surface samples at four different time points for further microbiological analyses.
Habitat preparation and cleaning procedures
Twenty-four hours prior to inhabitation, the interior surfaces of the habitat were cleaned with a 10 % bleach solution. The cleaning reagent used was a prediluted, stabilized sodium hypochlorite solution to disinfect hard surfaces in labs and production areas. The product is made up of 0.525 % sodium hypochlorite (1:10 bleach solution), the strength recommended by the Center for Disease Control for inactivating viruses and other pathogens. Immediately after cleaning, the ILMAH habitat was closed and undisturbed until 10 min prior to inhabitation. The student crews cleaned all the rooms and surfaces of the habitat once a week after they had taken the microbial samples. Cleaning included wiping down the surfaces (kitchen, tables, and bathroom) with antibacterial wipes (Catalog #:TX3214; Texwipes, Kernersville, NC), dusting, sweeping, and wet mopping the floor. Finally, the student crews performed additional cleaning duties, as required to keep the ILMAH system tidy. Cleaning procedures utilizing bleach, as carried out prior to occupation (Day 0), were not allowed during human occupation.
Sample location, collection, and processing
The architectural scheme of the ILMAH system is depicted in Additional file 2: Figure S1. All samples were collected from the ILMAH floor surfaces. Among the eight prescribed locations (1 m2 each), two sample locations were in the sleeping compartment area, two in the dining room area, one in front of the bathroom, and three in the lab area. Samples from the closed habitat were collected using premoistened biological sampling kits (BiSKits; QuickSilver Analytics, Abingdon, MD, USA) from the eight prescribed locations at four time points (Day 0, Day 13, Day 20, and Day 30). Sampling point Day 0 was taken prior to the occupation of the ILMAH system by the student crew, and subsequent samplings were conducted on Day 13, Day 20, and Day 30, the last day of the occupancy. When particulate materials were collected for microbiological examination from cleanroom surfaces, we have shown that at least 1 m2 surface area should be sampled so that sufficient biological matter was available to conduct several traditional microbiological and molecular microbial community analyses.
One BiSKit was used to collect samples from one location. The selected area was wiped in three different directions (unidirectional horizontal, vertical, and diagonal) while rotating the sampling device. After the sampling, the dropper attachment handle of the BiSKit was turned to expel the sample, soaked in the macrofoam, into the attached collection bottle. The liquid from the collection bottle was transferred into a sterile 50-ml Falcon tube. To increase sample extraction from each macrofoam, the collection bottle was filled with 15 mL of sterile phosphate-buffered saline (PBS) and reattached to the BiSKit and the macrofoam soaked again in PBS. Then, the dropper attachment was turned one more time to release the sample in the collection tube. The liquid was then transferred into the respective sample collection tube. This step was repeated twice. The sampling liquids (45 mL for each BiSKit) were stored at 4 °C and transported to JPL via overnight courier for further processing. For each sampling time point, a field control (BiSKit open and kept in the air for the time equal to collecting samples) and BiSKit control (unused BiSKit prepared the same way as BiSKits used for sampling) were collected. The protocol for BiSKit sampling and processing was followed as published elsewhere [60]. The time taken to process samples from collection to analyses was within 36 h.
At JPL, the 45-mL liquid samples from each BiSKit were concentrated to ~4-mL samples using the Innovaprep system (INNOVAPEP, Drexel, MO, USA). The concentrated samples were utilized for cultivation and molecular analyses to measure microbial burden and community structure analyses. Briefly, 200 μL of the concentrated sample was mixed with 1.8 mL sterile PBS and thoroughly mixed before performing appropriate dilution(s) and subsequent plating onto suitable agar media and ATP assay. Remaining solutions were used for DNA-based analysis and ~800-μL sampling fluids were stored at 4 °C and used for any contingency.
Microbiological examination
Cultivable microbial examination
For the analysis of cultivable bacterial population, 100 μL of appropriately diluted sample was spread onto two plates of R2A media (Difco). Plates were incubated at room temperature for 7 days. After the cultivation conditions, colony-forming units (CFUs) were counted and reported as CFU/m2. The identification and phylogenetic affiliations were carried out via Sanger sequencing targeting the 1–5-kb 16S rRNA gene sequencing. Initially, colony PCR was performed to generate appropriate PCR fragments. However, when the colony PCR step did not yield PCR amplicon, those isolates were subjected to a freeze (−80 °C)–thaw (+80 °C) cycle (suspend one colony in 1-ml PBS, freeze suspension for 15 min, thaw suspension for 15 min, repeat 3 times), automated DNA extraction system [60], and finally, the traditional phenol-chloroform steps [75] to break open cells and enabling to extract DNA. Amplification of the bacterial small-subunit rRNA genes was carried out using primers 8F and 1525R [76]. PCR conditions were as follows: 94 °C for 10 min for denaturation, followed by 35 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min 30 s. At the end of the 35 cycles, elongation was carried out at 72 °C for 10 min. The phylogenetic affiliation of the strains was determined by sequencing results with the published type strains sequence database [77] and aligned using ClustalW. Phylogenetic trees were reconstructed using the software MEGA by applying the neighbor-joining method [78].
Quantitation of total and viable microorganisms
ATP assay
Total ATP and intracellular ATP contents representing total and viable microbial population, respectively, were determined using the CheckLite HS kit (Kikkoman, Japan) as previously described [24, 63]. The ATP content was directly correlated with the size of the cells and hence, the ATP values of Gram-positive bacteria were at least 5 times more than Gram-negative cells. Likewise, ~200 times more ATP concentrations were reported in one fungal/yeast-yielding colony compared to 1 CFU of Gram-negative bacteria and spores were devoid of ATP [63]. Similarly, metabolically inactive cells will yield less ATP when compared to the cells that were able to proliferate in favorable conditions [79]. Given these constraints, ATP results were not used to correlate with other bioburden measurements.
Sample preparation for molecular assays
The concentrated samples were divided into two aliquots and one of the aliquots was treated with 12.5 μL of PMA (2 mM; Biotium, Inc., Hayward, CA) to a final concentration of 25 μM [80], followed by thorough mixing and incubation in the dark for 5 min at room temperature. Samples were then exposed to light with the PhAST blue-photoactivation system for tubes (GenIUL, S.L., Terrassa, Spain) for 15 min [81]. Information deduced from PMA-treated samples was documented for viable microbial population and data derived from the PMA-untreated aliquot was reported as total (dead and live) microbial population. Both, the PMA-treated and non-treated samples were further split in half, with one half subjected to bead beating with the Fastprep-24 bead-beating instrument (MP Biomedicals, Santa Ana, CA). The samples were run at 5 m/s for 60 s. After bead beating, the samples were combined with their respective analog, which were not subjected to bead beating, and the DNA from the combined sample was extracted by the Maxwell-16 MDx automated system according to the manufacturer’s instructions (Promega, Madison, WI). The purified DNA was eluted into a final volume of 50 μL.
qPCR assay
For the analysis of the bacterial and archaeal burden in the samples, real-time quantitative polymerase chain reaction (qPCR) assay, which targets the 16S rRNA gene, was performed in triplicate with a qPCR instrument (BioRad, CFX-96 thermal cycling; Hercules, CA). Universal bacterial primers targeting the 16S gene, 1369F (5′-CGG TGA ATACGT TCY CGG-3′) and modified 1492R (5′-GGW TAC CTTGTT ACG ACT T-3′), and universal archaeal primers targeting the 16S gene, 344af (5′-ACG GGG YGC AGC AGG CGC GA-3′) and 517r (5′-GCC AGC AGC CGC GGT AA-3′), were used [76, 82] to measure bacterial and archaeal burden, respectively. Each 25 μL reaction consisted of 12.5 μL of 2X iQ SYBR Green Supermix (BioRad, Hercules, CA), 1 μL each of forward and reverse oligonucleotide primers, 9.5 μL molecular grade water, and 1 μL of template DNA. The qPCR conditions to determine bacterial burden were: 95 °C; 3 min for denaturation followed by 40 cycles, with each cycle consisting of the following conditions: 10 s hold at 95 °C (denature), 55 °C for primer annealing, and 95 °C for 35 s for extension. Parameters to determine archaeal burden were the following: 95 °C; 15 min for denaturation followed by 40 cycles, with each cycle consisting of the following conditions: 15 s hold at 94 °C, 30 s at 60 °C for annealing, and 30 s at 72 °C for 30 s.
Molecular microbial diversity analysis
Illumina sequencing
The DNA samples were quantified using a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA). Bacterial primers 519wF and 1017R were used to amplify a ~500-bp fragment spanning the V4 hypervariable regions of the bacterial 16S rRNA gene [83]. Sequencing was carried out at the Research and Testing Laboratory (Lubbock, TX).
Bioinformatic analysis of Illumina sequences
The bacterial and archaeal Illumina-generated MiSeq quality reads were processed and analyzed using the LotuS software [84]. Sequences are screened for quality and discarded if (a) the quality score falls below a threshold of 25, (b) there is one ambiguous base call or more, (c) there is a homonucleotide run in sequence longer than 8, or (d) the final length after trimming and removal of primer sequences and barcodes is lower than 170 bp. Sequences were demultiplexed and clustered into OTUs based on their sequence similarity (97 %) with UPARSE [85]. Taxonomic classification was assigned using the Ribosomal Database Project (RDP) classifier with a confidence of 0.8, and taxonomic abundance was calculated and tabulated.
Statistical analyses
An in-house R-script employing the libraries vegan, ape, gplots, mgcv, and GUniFrac was used to compare the bacterial and archaeal Illumina data [31, 86]. Each dataset consisting of the OTU abundances per sample was rarefied 1,000 times to the lowest number of reads and an average Bray-Curtis distance was calculated. This distance was then utilized to calculate non-metric multidimensional scaling (NMDS) or principal coordinate analysis (PCoA), PERMANOVA (Adonis test) and multi-response permutation procedure (MRPP). In addition, the OTU abundances per sample of each dataset were sum-normalized and used to employ either an analysis of variance (ANOVA) or a Spearman rank correlation on the statistical significant changing parameters, and to generate a heat map (p value of 0.05). The change of diversity was measured via the Shannon-Wiener diversity index. OTUs that were unclassified at phylum level were removed. When warranted, the p value was decreased to only represent high-abundant OTUs and remove false positive results. Heat maps were presented at family level.
Availability of supporting data
The data set supporting the results of this article is available in the NCBI SRA repository, under accession # SRP069729.
Acknowledgements
The authors are grateful to the three student crews that participated in this program. Special thanks to Mr. Tim Buli who physically collected the samples during occupation. Part of the research described in this publication was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with NASA. This research was funded by a 2012 Space Biology NNH12ZTT001N Grant No. 19-12829-26 under Task Order NNN13D111T award to KV. We would also like to thank the Department of Space Studies, University of North Dakota for allowing sampling in the ILMAH, and the members of the Planetary Protection group at JPL for their technical assistance. We appreciate M. Jones and S. Ozyildirim of JPL for critically reading the manuscript. © 2016. All rights reserved.
Abbreviations
- 16S rRNA
small subunit of ribosomal ribose nucleic acid
- ANOVA
analysis of variance
- ATP
adenosine triphosphate
- BiSKit
Biological Sampling Kit
- CFU
colony-forming units
- DNA
deoxyribose nucleic acid
- ILMAH
inflatable lunar/Mars analogous habitat
- ISS
International Space Station
- MRPP
multi-response permutation procedure
- NCBI
National Center for Biotechnology Information
- NMDS
non-metric multidimensional scaling
- OTU
operational taxonomic unit
- PBS
phosphate-buffered saline
- PCoA
principal coordinate analysis
- PMA
Propidium monoazide
- qPCR
quantitative polymerase chain reaction
- RDP
Ribosomal Database Project
- SRA
Sequence Read Archive
- UND
University of North Dakota
- US
United States
Additional files
Statistical analysis. Tables S1. Statistical analysis (paired t test) to compare the microbial populations of the different time points. Table S2. Statistical analysis (paired t test) to compare the microbial populations of different locations. Table S3. Taxonomic affiliation of cultivable bacterial isolates. Table S4. Number of archaeal OTUs associated with ILMAH surfaces collected at various time points. (DOCX 23 kb)
ILMAH architecture, NMDS ordinations based on Bray-Curtis distances, and UniFrac distance of various ILMAH bacteriome datasets. Figure S1. Schematic representation of the ILMAH architecture with dimensions. The sampling locations are indicated with stars and numbers (1, 2: bedroom; 3, 4: kitchen; 5: bathroom; and 6, 7, 8: laboratory). Figure S2. NMDS ordinations based on Bray-Curtis distances between all samples that were not treated with PMA. (A) All time points, Adonis p-value 0.001, MRPP significance of delta 0.001. (B) all time points except Day 0, Adonis p-value 0.001, MRPP significance of delta 0.001, A: 0.1257. (C) All locations, Adonis p-value 0.493, MRPP significance of delta 0.977, A: –0.09709. (D) all locations except Day 0, Adonis p-value 0.597, MRPP significance of delta 0.413, A: 0.00659. Figure S3. NMDS ordinations based on Bray-Curtis distances between all samples that were treated with PMA. (A) All time points, Adonis p-value 0.001, MRPP significance of delta 0.001, A: 0.2813. (B) All time points except Day 0, Adonis p-value 0.001, MRPP significance of delta 0.001, A: 0.06871. (C) All locations, Adonis p-value 0.515, MRPP significance of delta 1, A: –0.09423. (D) all locations except Day 0, Adonis p-value 0.127, MRPP significance of delta 0.905, A: –0.03109. Figure S4. UniFrac distance of various ILMAH bacteriome datasets. The bacteriome distribution patterns were similar whether Bray-Curtis distance (A) or UniFrac distance (B) was used. Likewise, significance levels were same. UniFrac for various time points: Adonis p value of 0.001, MRPP significance of delta 0.001, A = 0.2988; UniFrac for various locations: Adonis p value of 0.801, MRPP significance of delta 0.955, A = −0.03882; variable test: Adonis p value of 0.012, MRPP significance of delta 0.004, A = 0.03886). (PDF 527 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TM drafted the manuscript, contributed to the sample processing and to the data analysis and interpretation, and carried out the identification of the bacterial isolates in this study. AB coordinated the sample collection, performed the sample processing, quantified the microbial burden of the samples, and finished the identification of the bacterial isolates. AJP contributed to the statistical analysis and data interpretation. TS and PdL collected the samples and sent them to JPL for analysis, PV coordinated the sample collection and contributed to the statistical data analysis, and AC performed the sample processing. KV designed the study, interpreted the data, and drafted the manuscript. All authors read and approved the final manuscript.
References
- 1.Prussin AJ, Marr LC. Sources of airborne microorganisms in the built environment. Microbiome. 2015;3:78. doi: 10.1186/s40168-015-0144-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klepeis NE, Nelson WC, Ott WR, Robinson JP, Tsang AM, Switzer P, Behar JV, Hern SC, Engelmann WH. The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants. J Expo Anal Environ Epidemiol. 2001;11(3):231–52. doi: 10.1038/sj.jea.7500165. [DOI] [PubMed] [Google Scholar]
- 3.Kelley ST, Gilbert JA. Studying the microbiology of the indoor environment. Genome Biol. 2013;14(2):202. doi: 10.1186/gb-2013-14-2-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.La Duc MT, Kern RG, Venkateswaran K. Microbial monitoring of spacecraft and associated environments. Microb Ecol. 2004;47:150–8. doi: 10.1007/s00248-003-1012-0. [DOI] [PubMed] [Google Scholar]
- 5.Pierson D, Botkin D, Bruce R, Castro V, Smith M, Oubre C, Ott C. Microbial monitoring of the International Space Station. In: Moldenhauer J, editor. Environmental Monitoring: A Comprehensive Handbook. River Grove: DHI Publishing, LLC; 2012. [Google Scholar]
- 6.Pierson DL. Microbial contamination of spacecraft. Gravit Space Biol Bull. 2001;14(2):1–6. [PubMed] [Google Scholar]
- 7.Venkateswaran K, La Duc MT, Horneck G. Microbial existence in controlled habitats and their resistance to space conditions. Microbes Environ. 2014;29(3):243–9. doi: 10.1264/jsme2.ME14032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearce DA, Hughes KA, Lachlan-Cope T, Harangozo SA, Jones AE. Biodiversity of air-borne microorganisms at Halley Station, Antarctica. Extremophiles. 2010;14(2):145–59. doi: 10.1007/s00792-009-0293-8. [DOI] [PubMed] [Google Scholar]
- 9.Van Houdt R, De Boever P, Coninx I, Le Calvez C, Dicasillati R, Mahillon J, Mergeay M, Leys N. Evaluation of the airborne bacterial population in the periodically confined Antarctic base Concordia. Microb Ecol. 2009;57(4):640–8. doi: 10.1007/s00248-008-9462-z. [DOI] [PubMed] [Google Scholar]
- 10.Kembel SW, Jones E, Kline J, Northcutt D, Stenson J, Womack AM, Bohannan BJ, Brown GZ, Green JL. Architectural design influences the diversity and structure of the built environment microbiome. ISME J. 2012;6(8):1469–79. doi: 10.1038/ismej.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Checinska A, Probst AJ, Vaishampayan P, White JR, Kumar D, Stepanov VG, Fox GE, Nilsson HR, Pierson DL, Perry J, et al. Microbiomes of the dust particles collected from the International Space Station and spacecraft assembly facilities. Microbiome. 2015;3(1):50. doi: 10.1186/s40168-015-0116-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hewitt KM, Gerba CP, Maxwell SL, Kelley ST. Office space bacterial abundance and diversity in three metropolitan areas. PLoS One. 2012;7(5) doi: 10.1371/journal.pone.0037849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehta SK, Cohrs RJ, Forghani B, Zerbe G, Gilden DH, Pierson DL. Stress-induced subclinical reactivation of varicella zoster virus in astronauts. J Med Virol. 2004;72(1):174–9. doi: 10.1002/jmv.10555. [DOI] [PubMed] [Google Scholar]
- 14.Mehta SK, Stowe RP, Feiveson AH, Tyring SK, Pierson DL. Reactivation and shedding of cytomegalovirus in astronauts during spaceflight. J Infect Discov. 2000;182(6):1761–4. doi: 10.1086/317624. [DOI] [PubMed] [Google Scholar]
- 15.Mijnendonckx K, Provoost A, Ott CM, Venkateswaran K, Mahillon J, Leys N, Van Houdt R. Characterization of the survival ability of Cupriavidus metallidurans and Ralstonia pickettii from space-related environments. Microb Ecol. 2013;65(2):347–60. doi: 10.1007/s00248-012-0139-2. [DOI] [PubMed] [Google Scholar]
- 16.Ilyin VK. Microbiological status of cosmonauts during orbital spaceflights on Salyut and Mir orbital stations. Acta Astronaut. 2005;56(9–12):839–50. doi: 10.1016/j.actaastro.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Wilson JW, Ott CM, Quick L, Davis R, Honer zu Bentrup K, Crabbe A, Richter E, Sarker S, Barrila J, Porwollik S, et al. Media ion composition controls regulatory and virulence response of Salmonella in spaceflight. PLoS One. 2008;3(12):e3923. doi: 10.1371/journal.pone.0003923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.NRC. Committee for the decadal survey on biological physical sciences in space: recapturing a future for space exploration: life and physical sciences research for a new era. Washington D.C., USA: The National Academies Press; 2011.
- 19.Venkateswaran K, Vaishampayan P, Cisneros J, Pierson DL, Rogers SO, Perry J. International Space Station environmental microbiome—microbial inventories of ISS filter debris. Appl Microbiol Biotechnol. 2014;98(14):6453–66. doi: 10.1007/s00253-014-5650-6. [DOI] [PubMed] [Google Scholar]
- 20.Castro VA, Thrasher AN, Healy M, Ott CM, Pierson DL. Microbial characterization during the early habitation of the International Space Station. Microb Ecol. 2004;47(2):119–26. doi: 10.1007/s00248-003-1030-y. [DOI] [PubMed] [Google Scholar]
- 21.Schwendner P. Microbial ecology of the MARS 500 habitat. Regensburg: Universität Regensburg; 2014. [Google Scholar]
- 22.Swarmer TM, Anderson L, de Leon P. International Conference on Environmental Systems. Bellevue: ICES; 2014. Performance review of a pressurized inflatable lunar habitat integrated with an electric rover and pressurized analog planetary suits during an initial ten day simulation. [Google Scholar]
- 23.Vaishampayan P, Probst AJ, La Duc MT, Bargoma E, Benardini JN, Andersen GL, Venkateswaran K. New perspectives on viable microbial communities in low-biomass cleanroom environments. ISME J. 2013;7(2):312–24. doi: 10.1038/ismej.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venkateswaran K, Hattori N, La Duc MT, Kern R. ATP as a biomarker of viable microorganisms in clean-room facilities. J Microbiol Methods. 2003;52(3):367–77. doi: 10.1016/S0167-7012(02)00192-6. [DOI] [PubMed] [Google Scholar]
- 25.Adams RI, Bateman AC, Bik HM, Meadow JF. Microbiota of the indoor environment: a meta-analysis. Microbiome. 2015;3(1):1–18. doi: 10.1186/s40168-014-0066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams RI, Tian Y, Taylor JW, Bruns TD, Hyvärinen A, Täubel M. Passive dust collectors for assessing airborne microbial material. Microbiome. 2015;3(1):1–11. doi: 10.1186/s40168-014-0066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin H, Pei Z, Martinez KA, Rivera-Vinas JI, Mendez K, Cavallin H, Dominguez-Bello MG. The first microbial environment of infants born by C-section: the operating room microbes. Microbiome. 2015;3(1):1–6. doi: 10.1186/s40168-015-0126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.NRC . Pathways to exploration: rationales and approaches for a U.S. program of human space exploration. Washington DC: NRC; 2014. [Google Scholar]
- 29.Fawley WN, Underwood S, Freeman J, Baines SD, Saxton K, Stephenson K, Owens RC, Jr, Wilcox MH. Efficacy of hospital cleaning agents and germicides against epidemic Clostridium difficile strains. Infect Control Hosp Epidemiol. 2007;28(8):920–5. doi: 10.1086/519201. [DOI] [PubMed] [Google Scholar]
- 30.Nocker A, Sossa-Fernandez P, Burr MD, Camper AK. Use of propidium monoazide for live/dead distinction in microbial ecology. Appl Environ Microbiol. 2007;73(16):5111–7. doi: 10.1128/AEM.02987-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinmaier T, Probst AJ, La Duc MT, Ciobanu D, Cheng JF, Ivanova N, Rattei T, Vaishampayan P. A viability-linked metagenomic analysis of cleanroom environments: eukarya, prokaryotes, and viruses. Microbiome. 2015;3(1):62. doi: 10.1186/s40168-015-0129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.La Duc MT, Vaishampayan P, Nilsson HR, Torok T, Venkateswaran K. Pyrosequencing-derived bacterial, archaeal, and fungal diversity of spacecraft hardware destined for Mars. Appl Environ Microbiol. 2012;78(16):5912–22. doi: 10.1128/AEM.01435-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stieglmeier M, Rettberg P, Barczyk S, Bohmeier M, Pukall R, Wirth R, Moissl-Eichinger C. Abundance and diversity of microbial inhabitants in European spacecraft-associated clean rooms. Astrobiology. 2012;12(6):572–85. doi: 10.1089/ast.2011.0735. [DOI] [PubMed] [Google Scholar]
- 34.Reinmuller B, Ljungqvist B. Modern cleanroom clothing systems: people as a contamination source. PDA J Pharm Sci Technol. 2003;57(2):114–25. [PubMed] [Google Scholar]
- 35.Noble WC. Dispersal of skin microorganisms*. Br J Dermatol. 1975;93(4):477–85. doi: 10.1111/j.1365-2133.1975.tb06527.x. [DOI] [PubMed] [Google Scholar]
- 36.Wilson M. Microbial inhabitants of humans: their ecology and role in health and disease. Cambridge, UK: Cambridge University Press; 2005.
- 37.Moldenhauer J. Environmental monitoring: a comprehensive handbook. Bethesda: Davis Healthcare International Publishing, LLC.; 2005. [Google Scholar]
- 38.Annous AA, Fratamico PM, Smith JL. Quorum sensing in biofilms: why bacteria behave the way they do. J Food Sci. 2009;74:R24–37. doi: 10.1111/j.1750-3841.2008.01022.x. [DOI] [PubMed] [Google Scholar]
- 39.Rutherford ST, Bassler BL. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med. 2012;2(11).pii: a012427. doi:10.1101/cshperspect.a012427. [DOI] [PMC free article] [PubMed]
- 40.Bernardet J-F, Nakagawa Y. An introduction to the family Flavobacteriaceae. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, editors. The Prokaryotes. New York: Springer; 2006. pp. 455–80. [Google Scholar]
- 41.Ofek M, Hadar Y, Minz D. Ecology of root colonizing Massilia (Oxalobacteraceae) PLoS One. 2012;7(7) doi: 10.1371/journal.pone.0040117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernard K. The genus Corynebacterium and other medically relevant Coryneform-like bacteria. J Clin Microbiol. 2012;50(10):3152–8. doi: 10.1128/JCM.00796-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9(4):244–53. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes. 2013;4(2):125–35. doi: 10.4161/gmic.23571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vincent C, Stephens DA, Loo VG, Edens TJ, Behr MA, Dewar K, Manges AR. Reductions in intestinal Clostridiales precede the development of nosocomial Clostridium difficile infection. Microbiome. 2013;1(1):18. doi: 10.1186/2049-2618-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, White O, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neumeister B, Mandel T, Gruner E, Pfyffer GE. Brevibacterium species as a cause of osteomyelitis in a neonate. Infection. 1993;21(3):177–8. doi: 10.1007/BF01710543. [DOI] [PubMed] [Google Scholar]
- 48.Eastridge CE, Prather JR, Hughes FA, Jr, Young JM, McCaughan JJ., Jr Actinomycosis: a 24 year experience. South Med J. 1972;65(7):839–43. doi: 10.1097/00007611-197207000-00014. [DOI] [PubMed] [Google Scholar]
- 49.Perry A, Lambert P. Propionibacterium acnes: infection beyond the skin. Expert Rev Anti Infect Ther. 2011;9(12):1149–56. doi: 10.1586/eri.11.137. [DOI] [PubMed] [Google Scholar]
- 50.Berner R, Pelz K, Wilhelm C, Funke A, Leititis JU, Brandis M. Fatal sepsis caused by Corynebacterium amycolatum in a premature infant. J Clin Microbiol. 1997;35(4):1011–2. doi: 10.1128/jcm.35.4.1011-1012.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hinić V, Lang C, Weisser M, Straub C, Frei R, Goldenberger D. Corynebacterium tuberculostearicum: a potentially misidentified and multiresistant Corynebacterium species isolated from clinical specimens. J Clin Microbiol. 2012;50(8):2561–7. doi: 10.1128/JCM.00386-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Knox KL, Holmes AH. Nosocomial endocarditis caused by Corynebacterium amycolatum and other Nondiphtheriae Corynebacteria. Emerg Infect Dis. 2002;8(1):97–9. doi: 10.3201/eid0801.010151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Archer GL. Staphylococcus aureus: a well-armed pathogen. Clin Infect Dis. 1998;26(5):1179–81. doi: 10.1086/520289. [DOI] [PubMed] [Google Scholar]
- 54.Chen S, Karalewitz AP, Barbieri JT. Insights into the different catalytic activities of Clostridium neurotoxins. Biochemistry. 2012;51(18):3941–7. doi: 10.1021/bi3000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonetta S, Bonetta S, Mosso S, Sampò S, Carraro E. Assessment of microbiological indoor air quality in an Italian office building equipped with an HVAC system. Environ Monit Assess. 2010;161(1-4):473–83. doi: 10.1007/s10661-009-0761-8. [DOI] [PubMed] [Google Scholar]
- 56.Stenberg B, Eriksson N, Hansson MK, Hobog J, Sandström M, Sundell J, Wall, S. The Office Illness Project in northern Sweden. An interdisciplinary study of the ‘‘sick building-syndrome’’ (SBS). In Proceedings of the Sixth International Conference on Indoor Air Quality and Climate. Helsinki, Finland. 1993.
- 57.Leung M, Chan AH. Control and management of hospital indoor air quality. Med Sci Monit. 2006;12(3):SR17–23. [PubMed] [Google Scholar]
- 58.Moissl-Eichinger C, Auerbach AK, Probst AJ, Mahnert A, Tom L, Piceno Y, et al. Quo vadis? Microbial profiling revealed strong effects of cleanroom maintenance and routes of contamination in indoor environments. Sci Rep. 2015;5. [DOI] [PMC free article] [PubMed]
- 59.Mahnert A, Vaishampayan P, Probst AJ, Auerbach A, Moissl-Eichinger C, Venkateswaran K, Berg G. Cleanroom maintenance significantly reduces abundance but not diversity of indoor microbiomes. PLoS One. 2015;10(8) doi: 10.1371/journal.pone.0134848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Venkateswaran K, La Duc MT, Vaishampayan P. Genetic Inventory Task: Final Report, JPL Publication 12-12. Pasadena: Jet Propulsion Laboratory, California Institute of Technology; 2012. pp. 1–117. [Google Scholar]
- 61.Puleo JR, Fields ND, Bergstrom SL, Oxborrow GS, Stabekis PD, Koukol R. Microbiological profiles of the Viking spacecraft. Appl Environ Microbiol. 1977;33(2):379–84. doi: 10.1128/aem.33.2.379-384.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puleo JR, Fields ND, Moore B, Graves RC. Microbial contamination associated with the Apollo 6 spacecraft during final assembly and testing. Space Life Sci. 1970;2(1):48–56. doi: 10.1007/BF00928955. [DOI] [PubMed] [Google Scholar]
- 63.La Duc MT, Dekas A, Osman S, Moissl C, Newcombe D, Venkateswaran K. Isolation and characterization of bacteria capable of tolerating the extreme conditions of clean room environments. Appl Environ Microbiol. 2007;73(8):2600–11. doi: 10.1128/AEM.03007-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.La Duc MT, Nicholson W, Kern R, Venkateswaran K. Microbial characterization of the Mars Odyssey spacecraft and its encapsulation facility. Environ Microbiol. 2003;5(10):977–85. doi: 10.1046/j.1462-2920.2003.00496.x. [DOI] [PubMed] [Google Scholar]
- 65.Horneck G, Klaus DM, Mancinelli RL. Space microbiology. Microbiol Mol Biol Rev. 2010;74(1):121–56. doi: 10.1128/MMBR.00016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilson JW, Ott CM, Honer zu Bentrup K, Ramamurthy R, Quick L, Porwollik S, Cheng P, McClelland M, Tsaprailis G, Radabaugh T, et al. Space flight alters bacterial gene expression and virulence and reveals a role for global regulator Hfq. Proc Natl Acad Sci U S A. 2007;104(41):16299–304. doi: 10.1073/pnas.0707155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lynch SV, Mukundakrishnan K, Benoit MR, Ayyaswamy PS, Matin A. Escherichia coli biofilms formed under low-shear modeled microgravity in a ground-based system. Appl Environ Microbiol. 2006;72(12):7701–10. doi: 10.1128/AEM.01294-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim W, Tengra FK, Young Z, Shong J, Marchand N, Chan HK, Pangule RC, Parra M, Dordick JS, Plawsky JL, et al. Spaceflight promotes biofilm formation by Pseudomonas aeruginosa. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0062437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamaguchi N, Roberts M, Castro S, Oubre C, Makimura K, Leys N, Grohmann E, Sugita T, Ichijo T, Nasu M. Microbial monitoring of crewed habitats in space-current status and future perspectives. Microbes Environ. 2014;29(3):250–60. doi: 10.1264/jsme2.ME14031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castelle Cindy J, Wrighton Kelly C, Thomas Brian C, Hug Laura A, Brown Christopher T, Wilkins Michael J, Frischkorn Kyle R, Tringe Susannah G, Singh A, Markillie Lye M, et al. Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol. 2015;25(6):690–701. [DOI] [PubMed]
- 71.Stevenson A, Burkhardt J, Cockell CS, Cray JA, Dijksterhuis J, Fox-Powell M, Kee TP, Kminek G, McGenity TJ, Timmis KN, et al. Multiplication of microbes below 0.690 water activity: implications for terrestrial and extraterrestrial life. Environ Microbiol. 2015;17(2):257–77. doi: 10.1111/1462-2920.12598. [DOI] [PubMed] [Google Scholar]
- 72.Moissl C, Bruckner JC, Venkateswaran K. Archaeal diversity analysis of spacecraft assembly clean rooms. ISME J. 2008;2(1):115–9. doi: 10.1038/ismej.2007.98. [DOI] [PubMed] [Google Scholar]
- 73.Moissl-Eichinger C. Archaea in artificial environments: their presence in global spacecraft clean rooms and impact on planetary protection. ISME J. 2010;5(2):209–19. doi: 10.1038/ismej.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Probst AJ, Auerbach AK, Moissl-Eichinger C. Archaea on human skin. PLoS One. 2013;8(6) doi: 10.1371/journal.pone.0065388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 76.Suzuki MT, Taylor LT, DeLong EF. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl Environ Microbiol. 2000;66(11):4605–14. doi: 10.1128/AEM.66.11.4605-4614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H, Park SC, Jeon YS, Lee JH, Yi H, et al. Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol. 2012;62(Pt 3):716–21. doi: 10.1099/ijs.0.038075-0. [DOI] [PubMed] [Google Scholar]
- 78.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karl D. Cellular nucleotide measurements and applications in microbial ecology. Microbiol Rev. 1980;44:739–96. doi: 10.1128/mr.44.4.739-796.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brescia CC, Griffin SM, Ware MW, Varughese EA, Egorov AI, Villegas EN. Cryptosporidium propidium monoazide-PCR, a molecular biology-based technique for genotyping of viable Cryptosporidium oocysts. Appl Environ Microbiol. 2009;75(21):6856–63. doi: 10.1128/AEM.00540-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Agusti G, Codony F, Fittipaldi M, Adrados B, Morato J. Viability determination of Helicobacter pylori using propidium monoazide quantitative PCR. Helicobacter. 2010;15(5):473–6. doi: 10.1111/j.1523-5378.2010.00794.x. [DOI] [PubMed] [Google Scholar]
- 82.Gantner S, Andersson AF, Alonso-Saez L, Bertilsson S. Novel primers for 16S rRNA-based archaeal community analyses in environmental samples. J Microbiol Methods. 2011;84(1):12–8. doi: 10.1016/j.mimet.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 83.Moreau MM, Eades SC, Reinemeyer CR, Fugaro MN, Onishi JC. Illumina sequencing of the V4 hypervariable region 16S rRNA gene reveals extensive changes in bacterial communities in the cecum following carbohydrate oral infusion and development of early-stage acute laminitis in the horse. Vet Microbiol. 2014;168(2–4):436–41. doi: 10.1016/j.vetmic.2013.11.017. [DOI] [PubMed] [Google Scholar]
- 84.Hildebrand F, Tadeo R, Voigt AY, Bork P, Raes J. LotuS: an efficient and user-friendly OTU processing pipeline. Microbiome. 2014;2:30. doi: 10.1186/2049-2618-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–8. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 86.R Development Core Team R. A Language and environment for statistical computing (http://www.r-project.org/) [http://www.r-project.org/]. Accessed 2011.