

Abstract
Rationale
Sirtuin (SIRT3), a major nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase in mitochondria, declines with aging and its ablation is associated with accelerated development of cardiovascular diseases. However, the role of SIRT3 in coronary microvascular function and post-MI recovery has not been incompletely understood.
Objective
The goal was to investigate whether ablation of SIRT3 causes coronary microvascular dysfunction, exacerbates post-myocardial ischemia (MI) cardiac dysfunction and impairs cardiac recovery.
Methods and results
Using endothelial cells (ECs) isolated from SIRT3 knockout (KO) mice, we revealed that the angiogenic capabilities were significantly reduced in SIRT3 deficient ECs. SIRT3 KO mice presented a pre-existing coronary microvascular dysfunction and microvascular rarefaction, as evidenced by a reduction in hyperemic peak diastolic blood flow velocity and coronary flow reserve (CFR), accompanied by loss of capillary-pericytes in the heart. Furthermore, SIRT3 KO mice subjected to myocardial ischemia by the ligation of left anterior descending coronary artery (LAD) exhibited more severe cardiac dysfunction together with decreased pericyte/EC coverage than that of WT mice. In contrast, overexpression of SIRT3 preserved cardiac function in post-MI mice. Immunoblot analysis further showed that the expression of angiopoietin-1 (Ang-1), vascular endothelial growth factor (VEGF) and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) were significantly decreased in the SIRT3-deficient ischemic hearts than those of WT ischemic hearts. This was accompanied by higher levels of cleaved caspase-3 and apoptosis.
Conclusion
Our results reveal a potential mechanism by which SIRT3 deletion exacerbates post-MI cardiac dysfunction and impairment of cardiac recovery involving microvascular rarefaction and pre-existing coronary microvascular dysfunction.
Keywords: SIRT3, pericyte, CFR, myocardial ischemia
1. Introduction
The incidence and prevalence of coronary heart disease including myocardial ischemia (MI) increases with advanced age [1]. MI results in left ventricle dysfunction and subsequent heart failure, which is the leading cause of death in the USA [1;2]. Mitochondrial dysfunction, oxidative stress, disrupted calcium homeostasis and cellular metabolism contribute to MI injury [3]. MI activates a series of endogenous protective mechanisms including myocardial angiogenesis in the ischemic area of MI that depends on endothelial function and promotes collateral vessel formation, to maintain basic cardiac function, homeostasis and recovery [4]. It is a critical adaptive mechanism to restore blood perfusion in post-MI. The percutaneous coronary intervention (PCI) that reduces infarct size by restoration of blood flow to the ischemic area is the most common therapy for patients with acute MI [5]. Although the door-to-balloon time declined significantly in patients receiving PCI, in-hospital mortality remained unchanged [6]. Coronary flow reserve (CFR) is widely used as a marker of coronary microvascular dysfunction which has been shown to increase the risk of cardiovascular events and is associated with increased MI size, reduced left ventricle (LV) ejection fraction, adverse LV remodeling, and reduced long-term survival [3]. Advanced age, hypertension and diabetes usually have pre-existing coronary microvascular dysfunction [5;7]. Moreover, the benefits from PCI therapies have been shown compromised in patients with pre-existing microvascular dysfunction. Therefore, preserving and restoring a normal microvascular function before or after acute MI have become a more attractive target in the setting of PCI [5].
Sirtuins are a family of Class III histone deacetylases (HDACs), which require nicotinamide adenine dinucleotide (NAD+) for their lysine residue deacetylase activity [8;9]. Sirtuins have been shown to regulate almost every aspect of cellular function, including energy metabolism, reactive oxygen species (ROS) scavenging and cell survival [10;11]. Of these, SIRT3 is predominately localized to the mitochondria in the heart where it regulates mitochondrial function and cellular metabolism [11–14], which has been shown to correlate with longevity in humans and protects cardiomyocytes from aging, hypertrophy and oxidative stress-mediated cell death [15;16]. In addition, SIRT3 levels have been shown decreased in old sedentary adults compared to younger individuals [17]. Our previous study has shown that the level of SIRT3 was decreased in the heart of db/db diabetic mice, and was associated with diabetes-induced impairment of angiogenesis, whereas over-expression of apelin increased the myocardial expression of SIRT3 and myocardial vascular density in db/db mice [18]. In addition, apelin gene therapy in streptozotocin (STZ)-induced diabetic mice improved myocardial vascular density and ameliorated MI injury in diabetic STZ-control mice, but not in STZ-SIRT3 knockout (KO) mice [4]. These studies imply an important role of SIRT3 in the process of angiogenesis and endothelial function. In the present study, we hypothesized that pre-existing coronary microvascular dysfunction and microvascular rarefaction exacerbates MI-induced cardiac dysfunction and impairs cardiac repair and recovery. Using SIRT3 KO mice subjected to MI, our data revealed a potential novel mechanism of SIRT3 ablation-mediated exacerbation of cardiac dysfunction and cardiac recovery that is through impairment of myocardial angiogenesis and coronary microvascular dysfunction.
2. Methods
2.1. Mice
Male global SIRT3 KO (129-SIRT3tm1.1Fwa/J) mice and their respective wild-type (WT) control (129S1/SvImJ) mice, and C57BL6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). All animals were fed with laboratory standard chow and water, and housed in individually ventilated cages in the Laboratory Animal Facilities at the University of Mississippi Medical Center. All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Mississippi Medical Center (Protocol ID: 1280A) and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85–23, Revised 1996).
2.2. Mouse Myocardial Ischemia Model
WT, SIRT3 KO mice and C57BL6 mice were subjected to myocardial ischemia by the ligation of left anterior descending coronary artery (LAD) for 48h or 14 days as indicated in the text [19;20]. Briefly, the mice were initially anesthetized with the mixture of ketamine (100 mg/kg) and xylazine (15 mg/kg) and maintained via supplemental doses of ketamine/xylazine mixture as needed. The mice were then intubated to allow artificial ventilation with room air. A left thoracotomy was performed to expose the heart. The left anterior descending coronary artery was ligated using an 8-0 nylon suture. At day 2 or day 14 after myocardial infarction, the mice were sacrificed and the heart tissue was collected for immunoblot analysis or immunohistochemistry.
2.3. Overexpression of SIRT3 in C57BL6 mice
C57BL6 mice subjected to myocardial ischemia were intramyocardially injected with recombinant adenovirus overexpressing SIRT3 (Ad-SIRT3) or adenovirus overexpressing β-galactosidase (Ad-β-gal) at ischemic area with a dose of 1 × 109 PFU per heart at four sites at the time of ligation. Experimental mice were divided into three groups: (i) C57 sham; (ii) C57 + Ad-β-gal; and (iii) C57 + Ad-SIRT3. After 2 weeks of Ad-SIRT3 or Ad-β-gal gene therapy, cardiac function was assessed using transthoracic echocardiography and the mice were then euthanized by cervical dislocation under anesthesia with isoflurane [4].
2.4. Echocardiography
Transthoracic echocardiograms were performed on mice using a Vevo770 High-Resolution In Vivo Micro-Imaging System equipped with a RMV 710B scanhead (VisualSonics Inc, Canada), as described previously [4;21]. The studied mouse was anesthetized by inhalation of 1.5–2% isoflurane mixed with 100% medical oxygen in an isolated chamber for induction. Anesthesia was maintained with 1–1.5% isoflurane to control the heart rate between 400 and 450 beats per minute. M-mode cine loops were recorded and analyzed by High-Frequency Ultrasound Imaging software (VisualSonics Inc, Canada) to assess myocardial parameters and cardiac functions of left ventricle, including LV end-systolic diameter (LVESD), LV end-diastolic diameter (LVEDD), LV end-systolic volume (LVESV), LV end-diastolic volume (LVEDV), thickness of the anterior wall (LVAW) and posterior wall (LVPW) at end systole and end diastole, and stroke volume (SV), as well as ejection fraction (EF%) and fractional shortening (FS%).
For measurement of coronary flow velocity, a cine loop of left proximal coronary artery (LCA) is recorded in pulsed-wave Doppler-mode at baseline, and under hyperemic conditions induced by inhalation of 1% and 2.5% isoflurane, respectively. The coronary flow reserve (CFR) is expressed as the ratio of peak blood flow velocity during hyperemia to peak blood flow velocity at baseline [22;23].
2.5. Primary Cell Culture
Microvascular endothelial cells (MECs) were isolated from WT or SIRT3 KO mice (n=3) as described previously [24]. Briefly, The WT and SIRT3 KO mice were anesthetized with ketamine (100 mg/kg) and xylazine (15 mg/kg). The lung was perfused with 10 ml of 2.5 mM ethylenediaminetetraacetic acid (EDTA) in phosphate-buffered saline (PBS), followed by perfusion of 5 ml of 0.25% trypsin/2 mM EDTA. The whole lung were then removed en bloc and incubated at 37 °C for 20 min. Small cuts were made and the EC suspension can be collected by pipetting up and down with 1.5 ml culture medium. The cell suspension was filtered through a 100 μm filter and seeded into a 60 mm cell culture dish. After 3 days of incubation in EGM-2 medium (Lonza, MD) supplemented with growth factors and 10% fetal bovine serum (FBS), non-ECs were removed by vacuum and ECs formed small colonies. Staining with rabbit anti-von Willebrand factor (vWF) polyclonal antibody (Santa Cruz Biotechnology, TX, USA) confirmed the purity of cultured ECs. Cells between passage 4 and 10 were used for all studies.
2.6. Tube Formation Assay
Tube formation assay was performed as previously described [25]. Briefly, ECs (2 × 104 cells/well) were seeded into a 96-well plate pre-coated with 40 μL reduced-growth factor ECM gel (Life Technologies, NY, USA) and incubated in a regular incubator or in an air-tight chamber (MIC-101, Billups-Rothenberg, Inc., CA, USA) flushed with 95% N2/5% CO2, at 37 °C for 6 hours. Photos of tube-like structure were captured with an AMG inverted phase-contrast microscope (AMG, Life Technologies, NY, USA). The numbers of branching points and segments were quantified by NIH Image J software with angiogenesis analyzer plug-in (developed by Gilles Carpentier, http://image.bio.methods.free.fr/ImageJ/?Angiogenesis-Analyzer-for-ImageJ).
2.7. Histology and Immunofluorescence
Cryostat ventricular sections were stained with fluorescerin-labeled Griffonia Bandeiraea Simplicifolia Isolectin B4 (1:200; IB4, Invitrogen, OR, USA) to visualize capillaries. Pericytes were labeled with mouse monoclonal antibody specific to neural/glial antigen-2 (NG2 proteoglycan) (1:200; Abcam, MA, USA). The pericyte/capillary coverage was expressed as the ratio of the number of NG2-positive cells to the number of IB4-positive cells.
2.8. Terminal Deoxynucleotidyl Transferase dUTP Nick end Labelling (TUNEL) Assay
In situ DeadEnd™ Colorimetric Apoptosis Detection System (Promega, Madison, WI, USA) was used to detect apoptotic cells in frozen heart sections according to the manufacturer’s instructions. The sections were counterstained with DAPI to visualize nuclei.
2.9. Western Blot Analysis
Heart ventricle samples were homogenized in lysis buffer with protease inhibitor cocktail. The PVDF membranes were probed with antibodies specific to cleaved caspase-3 (Cell Signaling Technology, MA, USA), vascular endothelial growth factor (VEGF), VEGF receptor 2 (VEGFR2), angiopoietin-1 (ANG1) (Santa Cruz Biotechnology, TX, USA), or 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) (Abcam, MA, USA). The membranes were then washed and incubated with an anti-rabbit or anti-mouse secondary antibody conjugated with horseradish peroxidase. Densitometries were analyzed using TINA 2.0 image analysis software.
2.10. Statistical Analysis
The data are expressed as mean ± S.E.M. Statistical analysis was performed using Student’s unpaired two-tailed t-test for comparisons between two groups, one-way followed by post hoc test for multiple comparisons. For the statistical analysis of the coronary flow velocity and echocardiographic parameters, we performed a two-way ANOVA test to examine the differences between the genotypes. p values < 0.05 were considered as statistically significant. Analysis was conducted using SPSS v23 (IBM, NY, USA).
3. Results
3.1. SIRT3 ablation impairs angiogenic properties of endothelial cells
In our previous study, we have revealed reduced expression of angiogenic growth factors and capillary rarefaction in the heart of SIRT3 KO mice [18], suggesting impaired endothelial angiogenic properties in SIRT3 deficient mice. Therefore, we performed tube formation assay to further investigate the role of SIRT3 ablation in EC on angiogenic capabilities. As shown in Fig. 1A, there was a significant decrease in network formation in SIRT3 KO-ECs, as evidenced by the decreased number of branching points and segments. Furthermore, hypoxia-induced network formation in SIRT3 KO ECs was also significantly reduced when compared to WT-ECs (Fig. 1B).
Fig. 1.

SIRT3 deletion reduces EC angiogenic capabilities. A. EC angiogenic capabilities was assessed using tube formation assay on ECM gel. Images represent WT and SIRT3 KO-ECs network formation after 6 hours of incubation. Quantifications of EC network formation revealed a significant decrease in the number of branching points and segments in SIRT3 KO-ECs when compared to WT-ECs. The measurements were done in triplicates. B. Representative images of WT and SIRT3 KO-ECs network formation after 6 hours of incubation under hypoxia. Quantifications of EC network formation revealed significant decrease in the number of branching points and segments in SIRT3 KO-ECs compared to WT-ECs. The measurements were done in triplicates. n=3 per group. *p<0.05. Data are shown as the means ± s.e.m.
3.2. Deletion of SIRT3 causes coronary microvascular dysfunction
Given the observation that SIRT3 KO mice exhibited decreased capillary density [18] and impaired endothelial angiogenic capability, we hypothesized that this may lead to a decline in coronary microvascular function. Echocardiography examination showed that SIRT3 deletion did not result in significant difference in cardiac function and parameters between WT and SIRT3 KO mice at early age (5 months old) (Table 1). However, pulsed-wave Doppler showed that the hyperemic peak diastolic blood flow velocity of LCA was significantly decreased in SIRT3 KO mice, which led to a significant reduction in CFR (Fig. 2A). The effect of SIRT3 gene deletion depends on the concentration of isoflurane. In addition, pericyte/EC coverage was significantly reduced in the heart of SIRT3 KO mice (Fig. 2B). These results suggest impaired coronary microvascular function and a possible reduction of myocardial perfusion under stress.
Table 1.
Morphological parameters and cardiac function
| Parameters | WT baseline (n=7) | WT + IS 2d (n=6) | WT + IS 2W (n=8) | SIRT3 KO baseline (n=7) | SIRT3 KO + IS 2d (n=5) | SIRT3 KO + IS 2W (n=5) |
|---|---|---|---|---|---|---|
| LVESD (mm) | 2.15 ± 0.08 | 3.04 ± 0.15* | 2.65 ± 0.12* # | 2.19 ± 0.05 | 3.14 ± 0.14* | 3.25 ± 0.12* ‡ |
| LVEDD (mm) | 3.54 ± 0.12 | 4.04 ± 0.13* | 3.84 ± 0.10 | 3.42 ± 0.09 | 3.76 ± 0.10 | 3.88 ± 0.18* |
| LVESV (μl) | 15.60 ± 1.49 | 36.98 ± 4.65* | 26.46 ± 2.70* # | 16.11 ± 1.01 | 39.51 ± 4.35* | 42.84 ± 3.66* ‡ |
| LVEDV (μl) | 52.58 ± 4.19 | 72.22 ± 5.82* | 64.22 ± 4.02 | 48.58 ± 3.33 | 60.75 ± 3.99 | 65.77 ± 7.13* |
| SV (μl) | 37.12 ± 2.72 | 35.27 ± 1.72 | 37.77 ± 2.13 | 32.44 ± 2.55 | 21.24 ± 3.52* † | 22.93 ± 3.92* ‡ |
| LVAW, s (mm) | 1.17 ± 0.06 | 1.06 ± 0.04 | 0.89 ± 0.04 | 1.16 ± 0.06 | 0.64 ± 0.09 | 0.71 ± 0.04 |
| LVAW, d (mm) | 0.71 ± 0.05 | 0.73 ± 0.03 | 0.64 ± 0.03 | 0.71 ± 0.05 | 0.47 ± 0.08* † | 0.51 ± 0.03* |
| LVPW, s (mm) | 0.99 ± 0.05 | 0.84 ± 0.03* | 0.86 ± 0.04* | 0.88 ± 0.04 | 0.72 ± 0.06* | 0.89 ± 0.02# |
| LVPW, d (mm) | 0.58 ± 0.04 | 0.60 ± 0.04 | 0.52 ± 0.02 | 0.61 ± 0.01 | 0.62 ± 0.05 | 0.64 ± 0.03‡ |
Data are means ± s.e.m.
p < 0.05 vs. corresponding baseline;
p < 0.05 vs. corresponding IS 2d;
p<0.05 vs. WT+IS 2d;
p<0.05 vs. WT+IS 2W.
p values were determined using two-way ANOVA to test the interaction between gene deletion and MI time. LVESD, left ventricle end-systolic diameter; LVEDD, left ventricle end-diastolic diameter; LVESV, left ventricle end-systolic diameter volume; LVEDV, left ventricle end-diastolic volume; SV, stroke volume; EF, ejection fraction; FS, fractional shortening; LVAW, s, left ventricle systolic anterior wall thickness; LVAW, d, left ventricle diastolic anterior wall thickness; LVPW, s, left ventricle systolic posterior wall thickness; LVPW, d, left ventricle diastolic posterior wall thickness.
Fig. 2.

SIRT3 deletion reduces coronary microvascular function and pericyte coverage. A. Representative Pulsed-wave Doppler images indicated that the peak hyperemic diastolic blood flow velocity and CFR were significantly decreased in SIRT3 KO mice (n=8) vs. WT mice (n=5). B. Pericyte (red)/ECs (green) coverage were assessed by labeling with anti-NG2 antibody for pericyte and IB4 for ECs and presented in the bar graph as the ratio of the number of NG2-positive cells to the number of IB4-positive signals. Note that the pericyte coverage in the LV myocardium of the SIRT3 KO mice (n=3) was decreased when compared to WT mice (n=3). *p<0.05. Data are shown as the means ± s.e.m.
3.3. Ablation of SIRT3 exacerbates cardiac dysfunction and impairs cardiac recovery
Since we observed decreased CFR and coronary microvascular dysfunction in SIRT3 KO mice, we further hypothesized that reduced myocardial perfusion (CFR) may lead to more severe cardiac dysfunction and impairment of cardiac recovery. The experimental mice were then subjected to myocardial infarction by permanent LAD ligation. Two days after MI, cardiac function was measured by echocardiography, as shown in the Table 1. The stroke volume (SV), EF% and FS% was significantly reduced in both WT and SIRT3 KO mice 2 days post-MI (Table 1 and Fig. 3A–C). Intriguingly, SIRT3 KO mice exhibited significant lower EF% and FS% when compared to WT mice with MI (Fig. 3A–C). Immunostaining with pericyte marker (NG2) and capillary marker (IB4) showed that pericyte/EC coverage in the WT mice was decreased two days after MI, but pericyte/EC coverage in SIRT3 KO mice did not further decrease after MI (Fig. 3D–E). Moreover, SIRT3 KO mice exhibited increased apoptotic cardiomyocytes in the border zone (Fig. 3F). This was accompanied by higher level of cleaved caspase-3 after MI (Fig. 3G), which supported the findings that there were increased apoptotic cardiomyocytes in the border zone. Since pre-existing microvascular dysfunction impacts the long-term outcomes, we further examine the cardiac functional recovery at two weeks post MI. As shown in Fig. 3A–C, there was significant recovery of cardiac function in WT mice two weeks post-MI as compared to that at 2 days post MI. However, there was no improvement of cardiac function in SIRT3 KO mice at 2 weeks post MI (Fig. 3A–C).
Fig. 3.

SIRT3 deletion exacerbates MI-induced cardiac dysfunction and cell death. A. Representative M-mode images of WT and SIRT3 KO mice at baseline, 2 days post-MI, and 2 weeks post-MI. n=5–8 per group. B. Quantification of ejection fraction. C. Quantification of fraction shortening. *p < 0.05 vs. corresponding baseline; #p < 0.05 vs. corresponding IS 2d; †p<0.05 vs. WT+IS 2d; ‡p<0.05 vs. WT+IS 2W. p values were determined using two-way ANOVA to examine the differences between the genotypes. D and E. IB4 (green) and NG2 (red) staining in the LV of WT (n=3) and SIRT3 KO mice (n=3) and quantification indicated that the pericyte coverage of SIRT3 KO mice was decreased when compared with WT mice. F. The number of apoptotic cells was higher in the border zone of SIRT3 deficient heart than that of WT heart. G. SIRT3 KO mice exhibited higher level of cleaved caspase-3 post-MI. n=5–6 per group. Data are shown as the means ± s.e.m.
3.4. SIRT3 deficiency decreases expression of angiogenic growth factors and PFKFB3 after MI
Myocardial angiogenesis restores blood supply and promotes cardiac repair and functional recovery in post-MI [4], mediated by various angiogenic growth factors released from ischemic tissue. Here, we examined the levels of angiogenic factors after MI. In SIRT3 KO mice the expression of ANG1 and VEGF were significantly lower than those in WT mice (Fig. 4A, B), as well as the expression of VEGFR2 (Fig. 4C). A metabolic shifting from oxygen-dependent fatty acid oxidation to oxygen-independent glucose glycolysis plays an important role for the heart to maintain essential functions and in the ischemic conditions. PFKFB3, a glycolytic enzyme activator, plays an important role in such process. We found that the expression of PFKFB3 was dramatically reduced in SIRT3 KO mice (Fig. 4D).
Fig. 4.
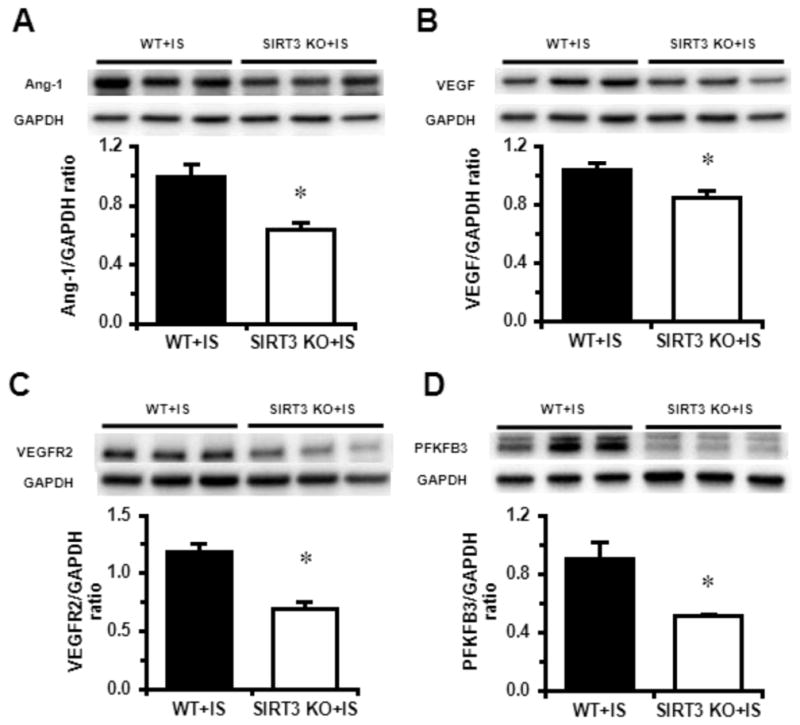
SIRT3 deletion reduces expression of angiogenic growth factors and PFKFB3 post MI. A–D. The expressions of Ang-1, VEGF, VEGFR2 and PFKFB3 were significantly lower in the heart of SIRT3 KO mice (n=5) than that of WT mice (n=6). n=5–6 per group. *p < 0.05. Data are shown as the means ± s.e.m.
3.5. Overexpression of SIRT3 protects heart from MI
Since reduction in SIRT3 resulted in more severe MI-induced cardiac dysfunction, we investigated the effect of overexpression of SIRT3 on preserving cardiac function using Ad-SIRT3. Five-months-old C57BL6 mice were subjected to MI. At the time of LAD ligation, intramyocardial injection of either Ad-β-gal or Ad-SIRT3 was performed at the ischemic area. Cardiac function was evaluated by echocardiography two weeks after MI. As shown in Fig. 5A–C and Table 2, MI induced significant cardiac dysfunction and LV dilation, as evidenced by decreased EF% and FS%, and increased LV dimension and volume. Overexpression of SIRT3, however, significantly improved cardiac function after MI when compared to C57+Ad-β-gal mice (Table 2 and Fig. 5A–C). These data suggest that reduction of SIRT3 is a causative factor regarding to MI-induced cardiac dysfunction.
Fig. 5.

Overexpression of SIRT3 improves post-MI cardiac function. A. Representative M-mode images of C57BL6 mice at baseline, 2 weeks post-MI overexpressing β-gal, and 2 weeks post-MI overexpressing SIRT3. n=6–8 per group. B. Quantification of ejection fraction. D. Quantification of fraction shortening. *p < 0.05. Data are shown as the means ± s.e.m.
Table 2.
Morphological parameters and cardiac function
| Parameters | C57 sham (n=8) | C57+IS+Ad-β-gal (n=6) | C57+Ad-SIRT3+IS (n=7) |
|---|---|---|---|
| LVESD (mm) | 2.09 ± 0.09 | 2.80 ± 0.06* | 2.54 ± 0.06* # |
| LVEDD (mm) | 3.43 ± 0.15 | 3.64 ± 0.07 | 3.67 ± 0.08 |
| LVESV (μl) | 14.66 ± 1.60 | 29.78 ± 1.68* | 23.36 ± 1.36* # |
| LVEDV (μl) | 49.34 ± 4.86 | 56.07 ± 2.59 | 57.24 ± 3.10 |
| SV (μl) | 34.81 ± 3.30 | 26.34 ± 0.96* | 33.87 ± 2.77 |
| LVAW, s (mm) | 1.18 ± 0.05 | 0.98 ± 0.04* | 1.07 ± 0.03 |
| LVAW, d (mm) | 0.71 ± 0.04 | 0.74 ± 0.02 | 0.74 ± 0.01 |
| LVPW, s (mm) | 0.99 ± 0.05 | 0.75 ± 0.02* | 1.00 ± 0.09# |
| LVPW, d (mm) | 0.57 ± 0.04 | 0.58 ± 0.02 | 0.70 ± 0.06* |
Data are means ± s.e.m.
p < 0.05 vs. C57;
p<0.05 vs. C57+IS.
LVESD, left ventricle end-systolic diameter; LVEDD, left ventricle end-diastolic diameter; LVESV, left ventricle end-systolic diameter volume; LVEDV, left ventricle end-diastolic volume; SV, stroke volume; EF, ejection fraction; FS, fractional shortening; LVAW, s, left ventricle systolic anterior wall thickness; LVAW, d, left ventricle diastolic anterior wall thickness; LVPW, s, left ventricle systolic posterior wall thickness; LVPW, d, left ventricle diastolic posterior wall thickness.
4. Discussion
Coronary microvascular dysfunction has been shown to be highly prevalent in post-MI patients, and is associated with poor prognosis regardless of successful reopening of epicardial vessel [3]. Coronary microvascular dysfunction has been described in many cardiac disease associated with aging, diabetes and hypertension. Therefore, novel therapeutic approaches for the treatment of acute MI should consider to target or improve microvascular dysfunction. In this study, we identify a potential mechanism by which SIRT3 deletion exacerbates post-MI cardiac dysfunction and impairment of cardiac recovery. Our data demonstrated that loss of SIRT3 impaired endothelial angiogenic capabilities and coronary microvascular dysfunction, which may contribute to exacerbation of cardiac dysfunction post-MI (Fig. 6). Our study suggests that SIRT3 can be a candidate of selective therapeutic target, especially in the aging population with reduced SIRT3.
Fig. 6.

Potential mechanism of action for impaired cardiac repair and recovery in SIRT3 KO mice post-MI. SIRT3 deletion results in, on the one hand decreased expression of angiogenic growth factors and thus neovascularization, on the other hand endothelial dysfunction and coronary microvascular dysfunction (decreased CFR). Taken together, decreased neovascularization and pre-existing coronary microvascular dysfunction limit cardiac repair and recovery post-MI.
SIRT3 deficient mice have been shown to accelerate aging process and development of cancer, metabolic diseases, as well as cardiovascular disease at younger age than WT mice [26–29]. As humans aging, the level of SIRT3 declines and alterations in the coronary vascular homeostasis and function makes the heart more susceptible to pathological conditions, such as MI. In addition, impairment of myocardial microcirculation results in insufficient perfusion to meet myocardial demand, which may eventually lead to myocardial ischemia [30]. To assess coronary microvascular function, we used pulsed-wave Doppler to measure peak blood flow velocity and CFR in the current study, since combination of these two parameters provides a comprehensive assessment and eliminate many limitations of using CFR alone [30]. We showed that the hyperemic peak blood flow velocity and CFR were significantly decreased in SIRT3 KO mice compared to their control mice, suggesting a reduction in coronary microvascular function and possible impairment of myocardial perfusion under stressed condition. Our data strongly suggest that pre-existing coronary microvascular dysfunction and established microvascular rarefaction may contribute to exacerbated post-MI cardiac dysfunction and impaired post-MI repair and recovery in SIRT3 KO mice.
Angiogenesis is initiated by endothelial cells (ECs), which form vessel network by sprouting, branching, migration, and proliferation. The interactions between EC and cardiomyocyte are critical for maintaining cardiac function [31]. The process of angiogenesis is regulated by the interactional effects of VEGF/VEGFR2 and Angiopoietins/Tie-2 systems [19;32]. Our previous studies have shown that ANG1 and VEGFR2 were dramatically reduced in the heart of SIRT3 KO mice relative to WT mice, indicating impaired angiogenic capabilities [18]. Moreover, SIRT3 KO mice exhibited decreased myocardial capillary, supporting the potential mechanism of coronary microvascular dysfunction mediated by impaired myocardial angiogenesis [18]. In addition, endothelial metabolism also plays a critical role in the regulation of angiogenesis since ECs depend on the energy produced predominantly from glycolysis for migration and proliferation [33;34]. Among the many enzymes in the metabolism of glucose, PFKFB3 is a key regulator of glycolysis in endothelial cells, which has been shown to promote angiogenic sprouting [34]. MI activates a series of endogenous protective mechanisms, including activation of glycolytic enzymes that decrease oxygen demand and MI-induced angiogenesis that promotes collateral vessel formation, to maintain essential function and homeostasis of the heart. Heart is a highly oxygen and energy demanding organ whose function depends on the ATP production from mainly oxidative phosphorylation in mitochondria [35]. During ischemia oxygen supply is limited and depleted quickly, thus mitochondrial oxidative phosphorylation is no longer a reliable source of ATP. In order to maintain the energy homeostasis, up-regulation of glycolysis to enhance the production of ATP is achieved by activation of multiple signaling pathways. Several studies have demonstrated that the expression of PFKFB3 is enhanced during ischemia possibly via VEGFR2 and the accumulation of hypoxia-inducible factors (HIFs) during hypoxia/ischemia [34;36–38]. Here, we showed that the expression of Ang-1, VEGF, and PFKFB3 were decreased in the heart of SIRT3 KO mice. Ang-1 is critical for the vascular integrity, stability, and EC survival, as well as pericyte recruitment. Our recent study demonstrated a critical role of SIRT3 in LPS-mediated pericyte loss, vascular leakage and cardiac dysfunction [39]. Microvascular dysfunction could be related to the changes in both function and morphology of the microcirculation [5;40]. In present study, we also found that the pericyte coverage was decreased in the heart of SIRT3 KO mice at both normal and ischemic conditions, indicating a possibly active role of pericyte during ischemia. A “no-reflow” phenomenon that can be described as inability to reperfusion an ischemic area has been confirmed by many studies [3;7;41]. Most recently clinical studies have demonstrated that dying pericytes that irreversibly constrict capillaries might be the major cause of this phenomenon [42]. Therefore, therapeutic myocardial angiogenesis, improvement of coronary microvascular and cardiac pericyte function become novel therapeutic targets for the treatment of MI.
In conclusion, our study provides a potential mechanism by which SIRT3 deletion exacerbates post-MI cardiac dysfunction and limits cardiac repair and recovery that is attributable to the impaired angiogenic capabilities of EC, pre-existing coronary microvascular dysfunction and loss of pericytes. Our studies will provide potential therapeutic targets for acute ischemia injury.
Highlights.
SIRT3 deficiency leads to decreased CFR and microvascular dysfunction.
SIRT3 deficiency exacerbates post-MI cardiac dysfunction and impairment of cardiac recovery.
Overexpression of SIRT3 attenuates myocardial ischemia induced cardiac dysfunction.
Acknowledgments
This work was supported by the National Institutes of Health RO1 HL102042 (Chen).
Footnotes
Disclosures
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015 Jan 27;131(4):e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012 Jan 5;366(1):54–63. doi: 10.1056/NEJMra1112570. [DOI] [PubMed] [Google Scholar]
- 3.Fordyce CB, Gersh BJ, Stone GW, Granger CB. Novel therapeutics in myocardial infarction: targeting microvascular dysfunction and reperfusion injury. Trends Pharmacol Sci. 2015 Sep;36(9):605–16. doi: 10.1016/j.tips.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Hou X, Zeng H, He X, Chen JX. Sirt3 is essential for apelin-induced angiogenesis in post-myocardial infarction of diabetes. J Cell Mol Med. 2015 Jan;19(1):53–61. doi: 10.1111/jcmm.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niccoli G, Scalone G, Lerman A, Crea F. Coronary microvascular obstruction in acute myocardial infarction. Eur Heart J. 2015 Sep 12; doi: 10.1093/eurheartj/ehv484. [DOI] [PubMed] [Google Scholar]
- 6.Menees DS, Peterson ED, Wang Y, Curtis JP, Messenger JC, Rumsfeld JS, et al. Door-to-balloon time and mortality among patients undergoing primary PCI. N Engl J Med. 2013 Sep 5;369(10):901–9. doi: 10.1056/NEJMoa1208200. [DOI] [PubMed] [Google Scholar]
- 7.Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007 Feb 22;356(8):830–40. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 8.Pillai VB, Sundaresan NR, Jeevanandam V, Gupta MP. Mitochondrial SIRT3 and heart disease. Cardiovasc Res. 2010 Nov 1;88(2):250–6. doi: 10.1093/cvr/cvq250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J. 2015 Dec 21;36(48):3404–12. doi: 10.1093/eurheartj/ehv290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brouwers FP, de Boer RA, van der Harst P, Voors AA, Gansevoort RT, Bakker SJ, et al. Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J. 2013 May;34(19):1424–31. doi: 10.1093/eurheartj/eht066. [DOI] [PubMed] [Google Scholar]
- 11.Tanno M, Kuno A, Horio Y, Miura T. Emerging beneficial roles of sirtuins in heart failure. Basic Res Cardiol. 2012 Jul;107(4):273. doi: 10.1007/s00395-012-0273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guarani V, Potente M. SIRT1 - a metabolic sensor that controls blood vessel growth. Curr Opin Pharmacol. 2010 Apr;10(2):139–45. doi: 10.1016/j.coph.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev. 2012 Jul;92(3):1479–514. doi: 10.1152/physrev.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tseng AH, Wu LH, Shieh SS, Wang DL. SIRT3 interactions with FOXO3 acetylation, phosphorylation and ubiquitinylation mediate endothelial cell responses to hypoxia. Biochem J. 2014 Nov 15;464(1):157–68. doi: 10.1042/BJ20140213. [DOI] [PubMed] [Google Scholar]
- 15.Matsushima S, Sadoshima J. The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol. 2015 Nov;309(9):H1375–H1389. doi: 10.1152/ajpheart.00053.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008 Oct;28(20):6384–401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, et al. Endurance exercise as a countermeasure for aging. Diabetes. 2008 Nov;57(11):2933–42. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng H, He X, Hou X, Li L, Chen JX. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am J Physiol Heart Circ Physiol. 2014 Feb 15;306(4):H585–H597. doi: 10.1152/ajpheart.00821.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen JX, Stinnett A. Ang-1 gene therapy inhibits hypoxia-inducible factor-1alpha (HIF-1alpha)-prolyl-4-hydroxylase-2, stabilizes HIF-1alpha expression, and normalizes immature vasculature in db/db mice. Diabetes. 2008 Dec;57(12):3335–43. doi: 10.2337/db08-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, Zeng H, Chen JX. Apelin-13 increases myocardial progenitor cells and improves repair postmyocardial infarction. Am J Physiol Heart Circ Physiol. 2012 Sep 1;303(5):H605–H618. doi: 10.1152/ajpheart.00366.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao S, Ho D, Vatner DE, Vatner SF. Echocardiography in Mice. Curr Protoc Mouse Biol. 2011 Mar 1;1:71–83. doi: 10.1002/9780470942390.mo100130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang WT, Fisch S, Chen M, Qiu Y, Cheng S, Liao R. Ultrasound based assessment of coronary artery flow and coronary flow reserve using the pressure overload model in mice. J Vis Exp. 2015;(98):e52598. doi: 10.3791/52598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.You J, Wu J, Ge J, Zou Y. Comparison between adenosine and isoflurane for assessing the coronary flow reserve in mouse models of left ventricular pressure and volume overload. Am J Physiol Heart Circ Physiol. 2012 Nov 15;303(10):H1199–H1207. doi: 10.1152/ajpheart.00612.2012. [DOI] [PubMed] [Google Scholar]
- 24.Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A. 2000 Feb 29;97(5):2202–7. doi: 10.1073/pnas.040378497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen JX, Lawrence ML, Cunningham G, Christman BW, Meyrick B. HSP90 and Akt modulate Ang-1-induced angiogenesis via NO in coronary artery endothelium. J Appl Physiol (1985) 2004 Feb;96(2):612–20. doi: 10.1152/japplphysiol.00728.2003. [DOI] [PubMed] [Google Scholar]
- 26.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008 Sep 23;105(38):14447–52. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010 Mar 4;464(7285):121–5. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonnell E, Peterson BS, Bomze HM, Hirschey MD. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol Metab. 2015 Sep;26(9):486–92. doi: 10.1016/j.tem.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010 Jan 19;17(1):41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van de Hoef TP, Echavarria-Pinto M, van Lavieren MA, Meuwissen M, Serruys PW, Tijssen JG, et al. Diagnostic and Prognostic Implications of Coronary Flow Capacity: A Comprehensive Cross-Modality Physiological Concept in Ischemic Heart Disease. JACC Cardiovasc Interv. 2015 Nov;8(13):1670–80. doi: 10.1016/j.jcin.2015.05.032. [DOI] [PubMed] [Google Scholar]
- 31.Leucker TM, Bienengraeber M, Muravyeva M, Baotic I, Weihrauch D, Brzezinska AK, et al. Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection. J Mol Cell Cardiol. 2011 Nov;51(5):803–11. doi: 10.1016/j.yjmcc.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tuo QH, Zeng H, Stinnett A, Yu H, Aschner JL, Liao DF, et al. Critical role of angiopoietins/Tie-2 in hyperglycemic exacerbation of myocardial infarction and impaired angiogenesis. Am J Physiol Heart Circ Physiol. 2008 Jun;294(6):H2547–H2557. doi: 10.1152/ajpheart.01250.2007. [DOI] [PubMed] [Google Scholar]
- 33.Goveia J, Stapor P, Carmeliet P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol Med. 2014 Sep;6(9):1105–20. doi: 10.15252/emmm.201404156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De BK, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013 Aug 1;154(3):651–63. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 35.Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM. SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol. 2014 Jun 15;306(12):H1602–H1609. doi: 10.1152/ajpheart.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Y, An X, Guo X, Habtetsion TG, Wang Y, Xu X, et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler Thromb Vasc Biol. 2014 Jun;34(6):1231–9. doi: 10.1161/ATVBAHA.113.303041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Obach M, Navarro-Sabate A, Caro J, Kong X, Duran J, Gomez M, et al. 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J Biol Chem. 2004 Dec 17;279(51):53562–70. doi: 10.1074/jbc.M406096200. [DOI] [PubMed] [Google Scholar]
- 38.Minchenko O, Opentanova I, Caro J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 2003 Nov 20;554(3):264–70. doi: 10.1016/s0014-5793(03)01179-7. [DOI] [PubMed] [Google Scholar]
- 39.Zeng H, He X, Tuo QH, Liao DF, Zhang GQ, Chen JX. LPS causes pericyte loss and microvascular dysfunction via disruption of SIRT3/angiopoietins/Tie-2 and HIF-2a/Notch3 pathways. Sci Rep. 2016 doi: 10.1038/srep20931. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niccoli G, Burzotta F, Galiuto L, Crea F. Myocardial no-reflow in humans. J Am Coll Cardiol. 2009 Jul 21;54(4):281–92. doi: 10.1016/j.jacc.2009.03.054. [DOI] [PubMed] [Google Scholar]
- 41.Ito H. No-reflow phenomenon and prognosis in patients with acute myocardial infarction. Nat Clin Pract Cardiovasc Med. 2006 Sep;3(9):499–506. doi: 10.1038/ncpcardio0632. [DOI] [PubMed] [Google Scholar]
- 42.O’Farrell FM, Attwell D. A role for pericytes in coronary no-reflow. Nat Rev Cardiol. 2014 Jul;11(7):427–32. doi: 10.1038/nrcardio.2014.58. [DOI] [PubMed] [Google Scholar]