

Abstract
The importance of TANK binding kinase-1 (TBK1), a multimeric kinase that modulates inflammation and autophagy, in human health has been highlighted for the first time by the recent discoveries of mutations in TBK1 that underlie amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), normal tension glaucoma (NTG) or childhood herpes encephalitis (HSE). Gain-of-function mutations in TBK1 are associated with NTG, whereas loss-of-function mutations result in ALS/FTD or in HSE. In light of these new findings, we review the role of TBK1 in these seemingly unrelated, yet allelic diseases, and discuss the role of TBK1 in neurological diseases. This discovery has the potential to significantly increase our understanding of the molecular basis of these poorly understood neurological disorders.
Keywords: TBK1, autophagy, interferon, herpes simplex encephalitis, amyotrophic lateral sclerosis, dementia, glaucoma
TBK1 At Multiple Crossroads
TBK1 (tumor necrosis factor (TNF) receptor associated factor NF-κB activator (TANK)-binding kinase 1) (see Glossary), also known as NAK or T2K, has recently attracted the attention of human geneticists, immunologists and neurologists alike for its critical role in central nervous system (CNS) pathology. It is an ubiquitously expressed serine-threonine kinase, belonging to the ‘non-canonical IκB kinases (IKKs)’, recognized for its critical role in regulating type I interferon (IFN) production [1]. TBK1 is involved in the activation of various cellular pathways leading to IFN and pro-inflammatory cytokine production following infection [1], autophagic degradation of protein aggregates or pathogens [2–4], and homeostatic cellular functions such as cell growth and proliferation [5]. Research in genetics has experienced an increased pace of discovery owing to the advances in sequencing technologies, which have begun to reveal a number of new genetic etiologies underlying various diseases. The recent discoveries of TBK1 heterozygous mutations in multiple human diseases has demonstrated the non-redundant role of this multifaceted protein in the CNS in particular [6–11] (Figure 1). Here, we review the pleiotropic role of TBK1 in light of new discoveries of human germline TBK1 mutations underlying neuroinflammatory diseases, including herpes simplex encephalitis (HSE), amyotrophic lateral sclerosis (ALS), frontal temporal lobe dementia (FTD) and normal tension glaucoma (NTG). These novel findings have emerged following the first identification of a series of genetic etiologies defining disease (HSE), or after a period of stagnant gene discovery (ALS, NTG). The data implicate new molecular pathways in disease pathogenesis. Consequently, discerning these pathways may lead to a better understanding of the causal mechanisms underlying such neurological disorders. Moreover, knowledge gained from further mechanistic insight could be used to develop new and more effective therapies for these neurological diseases which currently bear limited treatment options.
Figure 1. Disease-causing Mutations in Human TBK1.
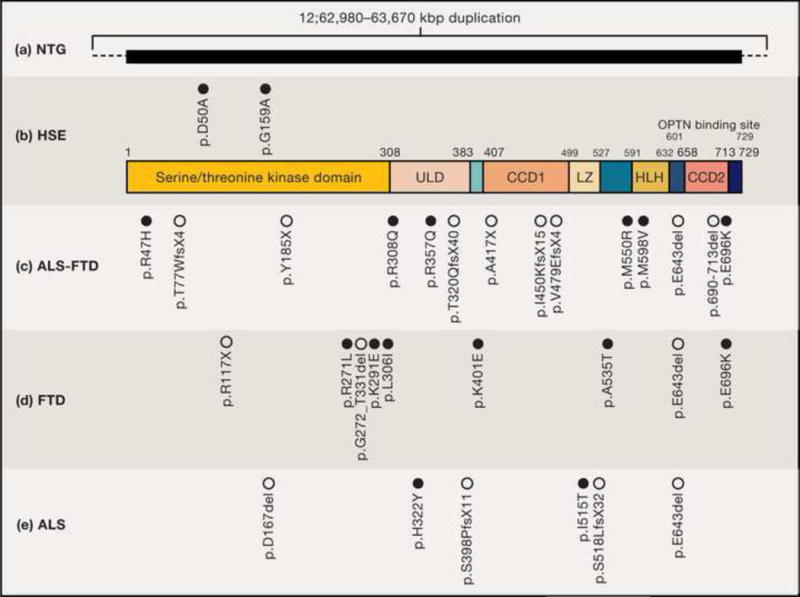
TBK1 is an 82 kDa, 729 amino-acid protein composed of a kinase domain, an ubiquitin-like domain (ULD), and coiled-coiled domains 1 and 2 (CCD1; CCD2). The kinase domain is critical for the phosphorylation of various substrates such as IRF3 [15], whereas the ULD domain regulates kinase activation and interactions with other proteins in the TBK1 pathway [112]. The CCD1 domain harbors leucine zipper (LZ) and helix-loop-helix (HLH) domains which specifically control dimerization. The C-terminus CCD2 harbors an adaptor-binding motif facilitating the interaction of TBK1 with its adaptors TANK, NAK–associated protein (NAP1), and Similar to NAP1 TBK1 Adaptor (SINTBAD) [89]. Germline human TBK1 mutations have been reported to be disease-causing in (a) normal tension glaucoma (NTG), (b) herpes simplex encephalitis (HSE), (c) amyotrophic lateral sclerosis-frontotemporal dementia ALS-FTD, (d) FTD and (e) ALS : these mutations are shown with respect to their amino acid positions within the TBK1 protein. The black horizontal box in (a) indicates duplications in kb reported to include TBK1. Open circles represent LoF variants; filled circles represent missense variants. (See Table 1).
TBK1 in Inflammatory Pathways
TBK1 was first identified as a TANK interacting protein in mice[12] with a role in controlling NF-κB-mediated responses as demonstrated in luciferase reporter assays of HEK293T cells co-transfected with TBK1 and the NF-kB promoter [13]. However, in contrast to canonical IKKs (IKKα and IKKβ) that control NF-κB activation, the non-canonical IKKs (TBK1 and IKKε) have since been found to play a more important role in the activation of transcription factors of the IFN-inducing interferon regulatory factor (IRF) family [14]. Indeed, TBK1 has been shown to play a key role in multiple cellular pathways, particularly inflammation and autophagy. Consequently, TBK1 sits at the crossroad of multiple inflammatory pathways, including NF-κB, and multiple IFN-inducing pathways.
Pattern recognition receptors (PRRs) such as toll-like (TLRs), retinoic acid-inducible gene I (RIG-I)-like (RLRs), and cytosolic DNA receptors all play important roles in the recognition of invading pathogens leading to IFN production (Key Figure, Figure 2). The engagement of such innate immune sensors by their cognate ligands, such as LPS, double stranded RNA (dsRNA) or DNA, results in the production of cytokines which alert neighboring cells (including immune cells) of danger and foreign invasion, subsequently promoting the early events of defense against infection. Engagement of TLR3 by dsRNA recruits its adaptor TRIF (TIR-domain-containing adaptor-inducing interferon-β), eventually activating TBK1, found in complex with NAK-associated protein 1 (NAP1) and IKKɛ (see Figure 1). Activated TBK1 phosphorylates IRF3 leading to its homodimerization and translocation to the nucleus, where they drive the expression of antiviral type-I and type-III IFNs (IFNα/β/λ) [1,15,16]. Apart from membrane-bound TLRs, cytosolic RLRs (RIG-I, melanoma differentiation-associated 5 (MDA5) activated by viral RNA, [17–19] and cytosolic DNA receptors (cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS), stimulator of IFN genes (STING)) activated by dsDNA [20], all activate downstream TBK1 and induce IRF3, and in some cases, IRF7 [21–23]. Finally, another DEAD (Asp-Glu-Ala-Asp)-box helicase 3, X-linked protein (DDX3X) has also been shown to directly interact with TBK1 in RAW264.7 murine macrophages following DNA and viral RNA recognition, thus leading to IFNβ production [24] (summarized in Key Figure, Figure 2).
Figure 2. TBK1 Molecular Pathways.
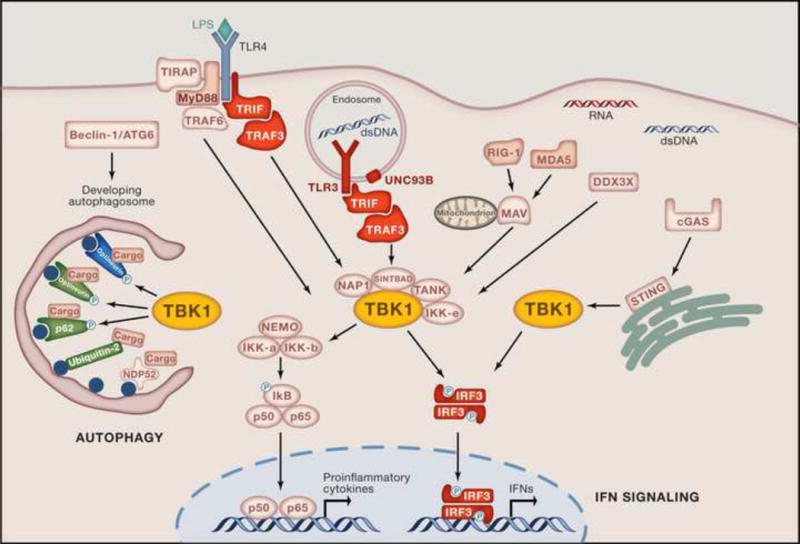
TBK1 and IKKɛ function as the non-canonical IkB kinases downstream of TLRs, RLRs, DDX3X, and DNA receptors leading to the activation of the transcription factors NF-kB (p65/p50) and IRFs (IRF3), resulting in the production of proinflammatory cytokines and antiviral IFNs. TLR3 recognizes dsRNA initiating the recruitment of adaptors such as TRIF and TRAF3 (TNF receptor-associated factor 3), which then activate TBK1 found in complex with its interacting proteins NAP1 (NF-kB-activating kinase-associated protein-1), SINTBAD (similar to NAP1 TBK1 adaptor) and TANK. LPS recognition by TLR4 can also recruit TRIF and subsequently TRAF3, mediating TBK1 activation. Activated TBK1 can then phosphorylate IRF3, leading to its homodimerization and subsequent translocation into the nucleus where it induces the production of IFNs. Cytosolic RLRs and DDX3X, as well as DNA sensor cGAS signal via TBK1 following recognition of their ligands viral 5′-ppp RNA and DNA, respectively. RLRs typically signal via the adaptor MAVS (mitochondrial antiviral-signaling protein; also known as IPS-1, CARDIF or VISA), which activates TBK1. cGAS detects dsDNA and stimulates STING (stimulator of interferon genes) to bind and activate TBK1 directly. TBK1 is also involved in autophagy where it directly phosphorylates the autophagy receptors optineurin and p62, which target cargo to the autophagosome. Ubiquilin-2 can also target ubiquitinated cargo to autophagosomes [113]. Target cargo may be comprising pathogen or ubiquitinated protein aggregates. Proteins whose genes have been reported to predispose to diseases are indicated in red, HSE; green, ALS or ALS-FTD; blue, NTG. Yellow denotes TBK1, where all pathways converge.
TBK1 in Autophagy
Recent studies have described TBK1 as an important player in yet another critical cellular function, autophagy. Autophagy is an evolutionarily conserved homeostatic process of self-degradation that contributes to the maintenance of cell function at critical times by balancing resources through the turnover of long-lived proteins and organelles, and also, in the clearance of intracellular pathogens [25]. Autophagy is achieved by directing bulk cargo, such as protein aggregates, for degradation and/or recycling in lysosomes. It is a highly regulated process that is orchestrated by a variety of autophagy-related proteins (ATGs) such as beclin-1 (ATG6) that functions as a promoter of autophagy upstream of the TBK1 pathway (reviewed in [26,27]). Autophagy, traditionally thought to be a non-selective process, there is accumulating evidence that autophagy proteins recognize specific cargo. This specificity is mediated by recruitment of autophagy receptors such as optineurin, p62, nuclear dot protein 52 kDa (NDP52) and neighbour of BRCA1 gene 1 (NBR1) [3,27–30] (Key Figure, Figure 2). These proteins bind simultaneously to ubiquitin residues on target cargo via their ubiquitin-binding domain, and to phosphatidyletholamine-conjugated microtubule-associated protein light chain 3 (LC3-II) proteins, which are found on the inner leaflet of a forming autophagosomal membrane [27]. For post-mitotic cells such as neuronal cells, autophagy is an essential survival mechanism by which toxic proteins are eliminated, as they are not able to dilute these proteins through mitosis [31,32]. A direct role of TBK1 in recycling protein aggregates has been shown via its role in phosphorylating the autophagy receptor optineurin [33]. TBK1 has been reported to co-localize with optineurin and cell aggregates in HeLa cells in vitro, as well as in vivo in an SOD1 transgenic mouse model of ALS [33]. TBK1 has also been found to play a role in the autophagic elimination of invading intracellular pathogens such as Salmonella, Mycobacteria, and herpes simplex virus-1 (HSV1) in human and murine cell lines [2–4].
The role of TBK1 in selective autophagy has been extensively studied in Salmonella where it associates with optineurin and NDP52, targeting ubiquitinated Salmonella for autophagic clearance (Figure 2) [2,34]. NDP52 is thought to act upstream of optineurin by directing TBK1 into the vicinity; TBK1 is then able to phosphorylate optineurin. TBK1 is also involved in autophagic clearance of Mycobacterium tuberculosis in RAW264.7 murine macrophages where it has been shown to phosphorylate the autophagy receptor p62, enhancing its binding to polyubiquitinated bacteria [4]. Moreover, TBK1 is particularly crucial for the maturation of the autophagosome into the hydrolytic autophagolysosome leading to degradation of p62 and its affiliated cargo [4]. Autophagy is also critical in HSV1 infections, demonstrated by the virus’ ability to inhibit host autophagy through two virally-encoded products US11 and ICP34.5 [35–37]. And, although TBK1 has not been directly implicated in HSV1-mediated autophagy, the virally-encoded autophagy antagonist ICP34.5 has been shown to bind and inhibit TBK1 in a mouse model of HSV1 infection [38]. This interaction has been been suggested to play a role in limiting the propagation and dissemination of HSV1 to the CNS [35]. Hence, TBK1 has been implicated in pathogen clearance via autophagy, contributing to cell-autonomous immunity. The two TBK1-regulated processes, autophagy and IFN signaling are not mutually exclusive, as crosstalk between them has been reported. For instance, upon HSV1 infection, cGAS has been shown to bind Beclin-1, leading to the suppression of IFN production, and simultaneously, to an increase in the autophagosomal clearance of cytosolic viral DNA in mouse bone marrow-derived macrophages (BMDMs) [39]. Similarly, following mycobacterial infection, mouse BMDMs were shown to induce type-I IFN and to trigger autophagic clearance of the pathogen in a TBK1-dependent manner, via cGAS [40]. Although mouse models of TBK1 deficiency have contributed to our fundamental understanding of TBK1 function, particularly in immune signaling (see Box 1), they have not been predictive of the phenotypes associated with human TBK1 mutations, since neurological phenotypes have not been assessed.
Box 1. Mouse Models of TBK1 Deficiency.
TBK1 is highly conserved in mammals, with human TBK1 protein sharing 99% homology with its mouse ortholog [13]. However, characterization of TBK1 function in vivo remains a major challenge, as homozygous deletion of TBK1 in mice results in embryonic lethality at embryonic day 14.5 due to severe hepatic tissue loss and apoptosis [97].
However, mice homozygous for a truncated allele (TBK1Δ/Δ) are viable, with minimal expression of truncated TBK1, which lacks kinase activity [98]. Macrophages from these mice have shown reduced IRF3 DNA-binding activity and IFNβ induction upon LPS induction. Heterozygous mice with one truncated allele (TBK1Δ/+) are also viable although their immunological response to infection has not been studied [98].
Much of our understanding of TBK1 function in viral infections and upon stimulation with the synthetic analog of dsRNA, polyinosinic:polycytidylic acid (poly I:C) in vitro, has mainly come from observations in TBK1-deficient (TBK1−/−) mouse embryonic fibroblasts (MEFs) or from macrophages exhibiting either impaired IFN responses (IFNβ/α) or IFN-induced responses such as IP-10 (IFN-gamma-inducible protein 10) and Mx1 [38,99–101]. It should be noted that the functional role of autophagy in TBK1-deficient mouse models has yet to be characterized.
TBK1 Variants in Human Diseases
Mutations in Human TBK1 Predispose to HSE: Impairment in IFN Production
Herpes simplex encephalitis (HSE) is a devastating neurological disease caused by HSV1 infection of the CNS. HSV1 is a neurotropic dsDNA alpha-herpesvirus usually causing asymptomatic or benign disease in the general population. With an incidence of 1–2 individuals per million annually, HSE is a sporadic and rare manifestation of HSV1 infection [41]. Peak incidence of HSE follows a bimodal curve, affecting children between three months-six years of age, coincident with the time of primary HSV-1 infection, and adults over 50 years of age, probably due to reactivation of latent HSV1 infection [42]. It is thought to reach the CNS through the nasal or oral epithelium via the olfactory or trigeminal nerves [43]. It exerts a wide spectrum of clinical features ranging from necrosis of brain tissue, fever, altered behavior and disturbed consciousness usually in the absence of viremia. Standard current treatment of acyclovir has greatly improved survival rates among HSE patients, although survivors tend to suffer from lifelong neurological sequelae characterized by global developmental delay, intellectual deficiencies, seizures and motor skill disturbances [42,44,45]. HSE has never been associated with any particularly neurovirulent strain of HSV1, and hence it had been considered a rare idiopathic complication of HSV1 infection until the identification of defects in a single gene within the TLR3-IFN pathway, TBK1, leading to diseases such as autosomal dominant TBK1 deficiency [46].
Isolated childhood HSE can be caused by at least seven different genetic etiologies of the TLR3-IFN pathway. These include autosomal recessive (AR) UNC93B1, autosomal dominant (AD) and AR TLR3, AD and AR TRIF, AD TRAF3, AD TBK1, and AD IRF3 deficiencies, reflecting the importance of IFN production in defense against HSV1 infection (Key Figure, Figure 2) [6,47–52]. For both AD and AR defects however, the clinical penetrance of HSE is incomplete, as healthy family members have also found to carry HSE-causing mutations [6,47–50,52]. This is consistent with HSE being almost invariably sporadic, with only four multiplex families reported since 1941 [6,48,50]. Nevertheless, there is complete penetrance of the mutations at the cellular level. For instance, functional studies of fibroblasts or induced pluripotent stem cell (iPSC)-derived neuronal cells derived from these patients have revealed a common defect in antiviral type-I and type-III IFN production. However, IFN responses have shown to be intact in these patient cells, underscoring the importance of IFN production in clearing HSV1 infection [53,54].
TLR3 signaling has also been studied in cells from patients with HSE. Endosomal TLR3 recognizes dsRNAs [55], produced during the HSV1 life cycle [56,57], triggering the production of anti-viral type-I and type-III IFNs (IFNα/β, IFNλ) (Key Figure, Figure 2). These IFNs are essential in controlling viral infection and establishing an anti-viral state by activating various host mechanisms that inhibit viral propagation and spread, such as translational arrest, and the induction of apoptosis [35,58].
Surprisingly, despite having impaired TLR3-mediated IFN production by their fibroblasts, these patients are otherwise healthy and are not susceptible to other viral infections, presumably because of the presence of intact and protective TLR3-independent IFN signaling mediated by cytosolic receptors such as RLRs [17].
Two different heterozygous missense TBK1 mutations have also been found in two unrelated European children with HSE (p.G159A and p.D50A respectively) (Figure 1, Table 1). Both heterozygous mutations occur in the kinase domain of the protein; however, one produces its effect in a dominant negative fashion (p.G159A) whilst the other is dominant by haploinsufficiency (p.D50A) [6]. The patient carrying the G159A mutation developed HSE at 7 years of age and subsequently developed epilepsy and cognitive disabilities [6]. The patient carrying the D50A mutation developed HSE at 11 months of age and suffered from obesity as well as cognitive and motor dysfunctions thereafter [6]. Despite normal protein and mRNA expression, the G159A mutant allele produced a kinase-dead TBK1. And, in terms of IFN signaling, the G159A mutation led to impairment of IRF3 phosphorylation, resulting in lack of IFNβ and IFNλ production, but normal IL-6 production upon TLR3 stimulation of patient dermal fibroblasts in vitro [6]. Because overexpression of this mutant allele in control human fibroblasts (with endogenous wild type TBK1) led to blocked IFN production, this suggested that the impaired signaling occurred due to the dominant negative effect of the mutant allele over the wild type allele. The D50A mutant allele however, exhibited poor expression at both protein and mRNA levels and hence, loss of kinase activity. Despite this, it showed normal poly I:C responsiveness in fibroblasts as demonstrated by normal IRF3 activation and IFNβ, IFNλ, and IL-6 production [6]. It was therefore concluded that the D50A allele is dominant due to haploinsufficiency. Autophagy function was not tested in these patients. Of note, both patients’ fibroblasts presented intact RLR-mediated IFN production, suggesting that TBK1 function was unaffected downstream of the cytosolic PRRs [6]. However, fibroblasts from both patients were unable to control HSV1 or VSV infections, indicating that a functional TBK1-dependent TLR3-IFN pathway was necessary for limiting viral replication [6]. It should be noted that it is currently not possible to rule out the involvement of other, yet-to-be defined mechanisms potentially contributing to HSE pathology (Box 2).
Table 1.
Molecular Characterization of TBK1 Variants Reported in Human Diseases.
| Type of variant |
Mutation (location in a.a. or kbps) |
Premature STOP |
Expression | Function | Disease | References | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| mRNA level | Protein level | Optineurin binding |
IFN | |||||||
| Allele- specific |
Patient cells |
Allele- specific |
Patient cells |
|||||||
| Frameshift | p.T77WfsX4 | Yes | – | Reduced | – | Reduced | – | – | ALS-FTD | [8] |
| p.T320QfsX40 | Yes | – | – | No | – | Impaired | Impaired | ALS-FTD | [8] | |
| p.S398PfsX11 | Yes | – | Reduced | – | Reduced | – | – | ALS | [11] | |
| p.I450KfsX15 | Yes | – | Reduced | Truncated | Reduced | Impaired | Impaired | ALS-FTD | [8] | |
| p.V479EfsX4 | Yes | – | – | Truncated | – | Impaired | Impaired | ALS-FTD | [8] | |
| p.S518LfsX32 | Yes | – | Reduced | – | Reduced | – | – | ALS | [11] | |
| Deletion | p.D167del | No | – | Normal | – | Normal | – | – | ALS | [11] |
| p.G272_T331del | No | – | Reduced | – | Reduced | – | – | FTLD | [11] | |
| p.E643del | No | – | Normal | – | Reduced | – | – | ALS; FTD; ALS-FTD | [8] [11] | |
| p.690-713del | No | – | Normal | Normal & truncated | Normal & truncated | Impaired | Normal | ALS-FTD | [8] | |
| Nonsense | p.Y185X | Yes | – | Reduced | – | – | – | – | ALS-FTD | [8] |
| p.R117X† | Yes | – | Reduced | – | Reduced | – | – | FTD | [9] | |
| p.A417X | Yes | – | Reduced | – | Reduced | – | – | ALS-FTD | [8] | |
| Missense | p.R47H | No | – | – | Normal | Normal | Normal | Impaired* | ALS-FTD | [8] |
| p.D50A | No | – | Reduced | No | Reduced | – | Normal | HSE | [6] | |
| p.G159A | No | – | Normal | Normal | Normal | – | Impaired | HSE | [6] | |
| p.R271L | No | – | Normal | – | Normal | – | – | FTD | [11] | |
| p.K291E | No | – | Normal | – | Normal | – | – | FTD | [11] | |
| p.L306I | No | – | – | – | Normal | – | – | FTD | [9] | |
| p.R308Q | No | – | – | Normal | – | Normal | Normal** | ALS-FTD | [8] | |
| p.H322Y | No | – | Normal | – | Normal | – | – | ALS | [11] | |
| p.R357Q | No | – | – | Normal | – | Reduced | Impaired*** | ALS-FTD | [8] | |
| p.K401E | No | – | – | – | Reduced | – | – | FTD | [9] | |
| p.I515T | No | – | Normal | – | Normal | – | – | ALS | [11] | |
| p.A535T | No | – | Normal | – | Normal | – | – | FTD | [11] | |
| p.M559R | No | – | – | Normal | – | Impaired | Impaired | ALS-FTD | [8] | |
| p.M598V | No | – | – | – | Normal | – | – | ALS-FTD | [8] | |
| p.E696K | No | – | – | Normal | Reduced | Impaired | Normal | ALS-FTD; FTD | [8] [9] | |
| Duplication | 12:62, 980 – 63, 670 kbp | – | – | Elevated | – | – | – | – | NTG | [10] |
All variants are either novel or have allele frequency of <0.0005% in general population.
Expression: assessed either allele specifically (in transfected cells) or in patient cells (expression of combined WT/mutant levels).
Function: autophagy function was tested by optineurin binding; IFN activation was tested by either IRF3 binding, phosphorylation, or IFNβ promoter induction.
normal IRF3 binding but impaired IRF3 phosphorylation/IFNβ induction.
normal IRF3 binding, phosphorylation but reduced IFNβ induction.
no IRF3 binding but reduced IRF3 phosphorylation/IFNβ induction.
patient also carried a heterozygous deletion in OPTN exons 13–15
“–” = not determined.
Box 2. Putative TBK1 Aberrations: Dysfunctional Autophagy in HSE? IFN Impairment in ALS-FTD?
HSE in patients with AD TBK1 deficiency has been attributed to impaired type-I and type-III IFN production, similar to other HSE-causing genes of the TLR3-IFN signaling pathways [46]. It would be of interest to further test whether autophagy defects are observed in HSE patients. Although no mutations occur in the CCD2 domain of TBK1, which is particularly important for autophagy, the dominant negative HSE mutation has shown overall functional reduction in TBK1, presumably affecting its autophagic function. Furthermore, the haploinsufficient TBK1 mutation in HSE, despite exhibiting moderate reduction (~50%) in protein levels in heterozygous cells, has not shown an impairment in the IFN pathway, even though patient cells were shown to be susceptible to viral infection [6]. This might suggest that other TBK1 pathways could be affected. Assessing the role of autophagy is particularly relevant in the context of HSV1 infections because the process has been shown to be critical in controlling HSV infection in post-mitotic neuronal cells [90]. Beyond TBK1-deficient HSE patients, whether or not autophagy reveals a general feature of HSE disease remains to be explored.
Select TBK1 mutations in cells from ALS-FTD patients have been assessed for IFN signaling, presenting either complete impairment (T320QfsX40, I450KfsX15, V479EfsX4, R47H, M559R) or reduced IFNβ induction (R357Q) (Table1). This suggests that these mutations may affect antiviral responses, although this parameter has not been specifically tested. These mutations have been examined in an allele-specific manner, overexpressing tagged mutants in HEK293T cells, but not in the context of an endogenous WT allele. Hence, the true effect of a mutation in heterozygosity has not been determined. Whether such IFN impairment might also contribute to ALS-FTD pathogenesis is a possibility that remains unexplored. Nevertheless, other studies using either mouse models of ALS or in vitro assays examining the expression and effects of type I IFNs on CNS-resident cells, have demonstrated a pleiotropic role of type I IFNs in neuronal survival. The cytokines could either confer protection or be detrimental to the cells, suggesting that IFNs may be relevant in ALS pathogenesis [102–104].
A possible crosstalk between impaired TBK-1 mediated autophagy and IFN signaling in disease has been suggested with the implication that optineurin-TBK1 complexes regulate IRF3-IFN responses following dsRNA or viral infections [20,39,105,106]. Of note, these TBK1 variants have not been tested for their role in NF-κB signaling, which may also potentially affect neuroinflammation [107]. In NTG, the role of abnormal NF-κB signaling due to TBK1 duplications has been indeed proposed as a pathogenic mechanism [10]. In the context of heterozygous mutations, it is possible that a putative effect of TBK1 on other pathways exists, potentially further contributing to disease pathogenesis.
TBK1 Variants Can Predispose Individuals to ALS, ALS-FTD, or FTD: Implications of Aberrant Autophagy
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease or Charcot’s disease, is a typically adult-onset neurodegenerative disease characterized by progressive muscle wasting which is usually fatal [59]. First described in 1869 by Jean-Martin Charcot [60], it has a an incidence of 1–2 per 100, 000 adults per year typically affecting individuals of 50–60 years-olds [59]. Approximately 90% of ALS cases are sporadic and the remaining 10% are familial [59]. Parental consanguinity does not seem to be higher than in the general population. ALS is associated with progressive loss of upper and lower motor neurons that lead to weakening and atrophy of muscles, paralysis and eventually death, mostly due to respiratory failure typically within 2 to 3 years after diagnosis [61]. Neuropathological features include extensive degeneration of motor neurons in anterior roots of the spinal cord and brainstem, corticospinal tract and loss of large pyramidal neurons residing in the primary motor cortex. Another hallmark feature is the presence of protein aggregates in degenerating neurons, most of which are ribonuclear proteins, such as transactive response (TAR) DNA-binding protein 43 (TDP-43). Proposed pathophysiological mechanisms of ALS include oxidative stress [62], impaired mitochondrial functions [63] and perturbed axonal transport leading to accumulation of organelles [64]. In addition, ALS is associated with neuroinflammation that is triggered by motor neuron degeneration [65]. Furthermore, 15% of ALS patients develop cognitive abnormalities reminiscent of frontotemporal lobar dementia (FTD), and 15% of FTD patients have features of ALS [66]. Recent studies examining CNS tissues from FTD and ALS patients have proposed that ALS and FTD form part of the same disease spectrum with common underlying features such as the presence of the TDP-43 proteins that accumulate in the cytoplasm of neurons [67]. Currently, there are no effective therapies available for this debilitating and lethal disease.
Many genes associated with ALS pathogenesis have been identified, including superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), FUS RNA-binding protein (FUS), Alsin (ALS2), Ubiquilin-2 (UBQLN2), Optineurin (OPTN), Sequestosome 1 (SQSTM1), Valosin-containing protein (VCP), and chromosome 9 open reading frame 72 (C9orf72) among many others, although these collectively account for less than one third of all ALS cases [68–71] (reviewed in[72]). These genes were all identified initially in familial forms of ALS through linkage studies, and then, further found in sporadic cases. In ALS patients, these mutations have all been found to be typically mono-allelic, with the exception of some forms of disease presenting SOD1, OPTN, FUS or ALS2 mutations, among others [70,73–76]. Protein aggregates are a hallmark of the disease, comprised by proteins which are encoded by genes linked to causing ALS, e.g. SOD1, TARDBP, FUS. These protein aggregates can be stained with antibodies against two previously mentioned autophagy receptors, p62 and optineurin, which have also been implicated in the pathogenesis of ALS (Key Figure, Figure 2) [73,77]. Further evidence identifying autophagy as a putative pathogenic mechanism for ALS, came from a report by Cirulli et al. characterizing for the first time, TBK1 as a new ALS-susceptibility gene in a whole exome sequencing (WES) study of 2869 ALS patients and 6405 controls, along with two other autophagy genes OPTN and SQSTM1 (encoding optineurin and p62, respectively) [7]. Although none of the TBK1 variants found were functionally assessed in this study, heterozygous TBK1 mutations were found to be significantly enriched in patients when compared to controls (1.099% of cases and 0.194% of controls). In particular, TBK1 mutations were bioinformatically predicted to constitute ‘loss-of-function’ (LoF) mutations, including nonsense, splice site, frameshift, and deletions in TBK1, (the latter were 10-fold more prevalent in patient cases) [7] (Table 2).
Table 2.
TBK1 Variants of Unknown Pathogenicity Reported in Human Diseases.
| Type of variant | Mutation (location in a.a. or kbps) | Mutation prediction | Disease | References |
|---|---|---|---|---|
| Nonsense | p.Q2X | STOP | ALS | [7] [11] |
| p.R117X | STOP | ALS | [7] | |
| p.R357X | STOP | ALS | [7] | |
| p.R440X | STOP | ALS; ALS-FTD; ALS-dementia | [7] [8] [78] | |
| p.R444X | STOP | ALS | [7] | |
| p.Y482X | STOP | ALS-FTD | [78] | |
| p.S499X | STOP | ALS | [7] | |
| p.Q655X | STOP | ALS-FTD | [78] | |
| Frameshift | p.T156RfsX6 | STOP | ALS-FTD | [78] |
| p.T278fs | n.a. | ALS | [7] | |
| p.L399fs | n.a. | ALS | [79] | |
| p.V421fs | n.a. | ALS | [7] | |
| p.T462fs | n.a. | ALS | [7] | |
| p.D500fs | n.a. | ALS | [7] | |
| p.E550fs | n.a. | ALS | [7] | |
| p.Q629fs | n.a. | ALS | [7] | |
| Deletion | p.E640del | n.a. | ALS | [7] |
| Splice | p.180sp | n.a. | ALS | [7] |
| p.331sp | n.a. | ALS | [7] | |
| p.587sp | n.a. | ALS | [7] | |
| c.1960-2A>G; p.653sp | n.a. | ALS-FTD | [78] | |
| Missense | p.T4A | Probably damaging | FTD | [78] |
| p.L11S | Possibly damaging | ALS | [7] | |
| p.N22H* | Probably damaging | ALS | [7] | |
| p.N22D | Probably damaging | ALS | [7] | |
| p.R25H | Probably damaging | ALS | [7] | |
| p.G26E | Probably damaging | ALS | [78] | |
| p.Y105C | Possibly damaging | ALS-FTD | [8] | |
| p.N129D | Possibly damaging | ALS | [7] | |
| p.V132E | Probably damaging | ALS | [7] | |
| p.R134H | Probably damaging | ALS | [7] | |
| p.R143C | Probably damaging | ALS | [78] | |
| p.S151C | Probably damaging | ALS | [7] | |
| p.S151F | Probably damaging | ALS; NTG | [7] [10] | |
| p.G217R | Probably damaging | ALS | [7] | |
| p.R228H | Probably damaging | ALS | [7] | |
| p.I257T | Probably damaging | ALS | [7] | |
| p.L277V | Benign | ALS | [7] | |
| p.I305T | Possibly damaging | ALS-FTD | [8] | |
| p.L306I | Possibly damaging | NTG | [10] | |
| p.T320I | Benign | ALS | [78] | |
| p.T331I | Benign | ALS | [7] | |
| p.T343S | Probably damaging | ALS | [7] | |
| p.Y394D | Possibly damaging | ALS | [7] | |
| p.R440Q* | Probably damaging | ALS | [7] | |
| p.V464A | Benign | NTG | [10] | |
| p.C471Y | Benign | ALS | [7] | |
| p.I522M | Possibly damaging | ALS | [7] | |
| p.A571V | Benign | ALS-FTD | [8] | |
| p.Q565P | Probably damaging | ALS | [7] | |
| p.Q581H | Probably damaging | ALS | [7] | |
| p.M662T | Benign | ALS-FTD | [78] | |
| p.I710N* | Benign | ALS | [7] | |
| Duplication | 12:62, 900 – 63, 680 kbp | n.a. | NTG | [10] |
| 12:62, 760 – 63, 410 kbp | n.a. | NTG | [10] | |
| 12:63, 060 – 63, 360 kbp | n.a. | NTG | [10] | |
| 12:64, 802 – 65, 099 kbp | n.a. | NTG | [85] | |
| 12:64, 830 – 65, 096 kbp | n.a. | NTG | [86] |
Mutation predictions were predicted by online tool PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml).
n.a.= not applicable.
Mutations also found in controls.
This finding was further supported by Freischmidt et al., who identified genome-wide enrichment of TBK1 mutations in 252 familial ALS patients [8]. WES of 13 European Caucasian families diagnosed with ALS or ALS-frontotemporal dementia (FTD) identified 8 heterozygous LoF classes of variants in TBK1. These mutations were assessed for optineurin binding as well as for their ability to induce IFNβ signaling (IRF3 activation and IFN induction) in HEK293T cells (Figure 1, Table 1). These LoF mutations were shown to have no mRNA or protein expression, consistent with haploinsufficiency (Figure 1, Table 1). Specifically, patients’ cells (lymphoblastoid cell lines, keratinocytes, or fibroblasts), heterozygous for four of these variants, (Y185X, I450KfsX15, T77WfsX4, A417X), exhibited 50% reduced expression of TBK1 at the mRNA and/or protein levels [8]. Furthermore, HEK293T cells expressing two of the other mutations (T320QfsX40, V479EfsX4) showed no allele-specific expression of the TBK1 protein. A number of these LoF variants was tested for optineurin binding and IFN induction in HEK293T cells, which showed complete impairment of both TBK1-related functions (Table 1). These variants were therefore reported to exert their effect via haploinsufficiency and determined to be causative. The p.690-713del variant, despite producing a TBK1 protein product, had a 24 amino acid deletion in the C-terminal CCD2 domain, specifically at the optineurin binding site, resulting in impaired binding to optineurin. The causative LoF mutations (Figure 1), p.Y185X, p.I450KfsX15, p.T77WfsX4, p.A417X, p.T320QfsX40, p.V479EfsX4, p.690-713del (R440X was not assessed), resulted in either haploinsufficiency, or loss of CCD2 function, and were found in ALS-FTD patients (approximately 50% of cases presented significant cognitive disabilities, often progressing towards FTD), as seen with other familial forms of the C9orf72 mutation, extending the TBK1 phenotype to include FTD [8]. The clinical penetrance of these mutations was high, with 33 out of 40 carriers beyond 60 years of age, harboring TBK1 mutations and presenting ALS [8].
In addition, Freischmidt et al. reported 9 missense mutations and 1 in-frame deletion in ALS, ALS-FTD patients [8]. Although missense mutations were not found to be enriched in the genetic analysis, in vitro assays on select mutations (optineurin binding and IFN induction in HEK293T cells) showed impaired TBK1 function. However, the authors suggest that further experiments are required to determine pathogenicity of these missense mutations (Figure 1, Tables 1 and 2) [8]. One particular missense variant located in the CCD2 domain, E696K, resulted in a failure of TBK1 to bind optineurin following co-immunoprecipitation in HEK293T cells. This mutation, as well as the E643del mutation [8], have been identified as causative in a separate study of isolated FTD cases, since reduced expression of these variants were described in patient post mortem cerebellar tissue and lymphoblast cells, respectively [9,11]. Additional mutations in French ALS, ALS-FTD, and isolated FTD patient cohorts, as well as a Chinese ALS patient have been subsequently reported (Table 2) [78,79]. Published disease-causing mutations are shown in Figure 1, whereas Tables 1 and 2 list variants of unknown pathogenicity that have been respectively either molecularly characterized, or not. In summary, these studies have now provided a link between TBK1 and other previously identified ALS genes SQSTM1 and OPTN, to autophagy. The data suggest that this link represents an important cellular regulatory mechanism, which, when dysfunctional, can contribute to neurodegeneration, as observed in ALS disease. Full functional characterization of TBK1 mutations in the context of autophagy, will be necessary to unequivocally prove autophagy dysregulation (e.g. using autophagy flux assays, among other techniques). Moreover, it will be interesting to determine if IFN signaling is also impaired in these diseases, since autophagy has also been shown to regulate IFN responses [39,40] (Box 2).
TBK1 Duplications and Predisposition to Glaucoma: Gain-of-function Mutations
Glaucoma is the leading cause of adult-onset blindness with a prevalence of 1.86% in the US in adults over 40 years old; it is a neurodegenerative disease affecting the retinal ganglion cells of the optic nerve, usually resulting in irreversible ocular damage [80,81]. Glaucoma can be classified into two subtypes; primary open angle glaucoma (POAG), characterized by high intraocular pressure causing damage to the optical nerves, and normal tension glaucoma (NTG), associated with normal intraocular pressure (IOP) [82,83]. Single-gene heterozygous mutations underlying both types of glaucoma have been described, and are thought to account for 5% of all cases [84]. Myocilin (MYOC) is a protein found in the trabecular meshwork and the ciliary body of the eye thought to regulate IOP. Heterozygous nonsense mutations in the myocilin gene (MYOC) are known to cause POAG with relatively high penetrance (98.6%), and recent reports describe familial and sporadic NTG patients harboring heterozygous mutations in OPTN [84]. Mutant OPTN (p.E50K) has been shown to form aggregates of insoluble protein in neuronal cells derived from NTG patient’s iPSCs, thus leading to cell death [83]. TBK1 has also been found to interact with mutant E50K OPTN protein, contributing to insolubility of the latter, and consequently, to NTG pathology [83]. Moreover, familial analysis of NTG patients has revealed several highly penetrant copy number variants (CNV) encompassing a region within chromosome 12, and inclusive of TBK1 (Figure 1, Tables 1 and 2) [10]. This heterozygous duplication has been associated with higher TBK1 transcription levels in skin fibroblasts derived from patients, suggesting a TBK1 GoF, underlying the glaucoma [10]. Hence, NTG TBK1 mutations present a different genetic etiology than that which has been observed in TBK1 deficiency models underlying HSE, ALS, ALS-FTD, or FTD. Furthermore, this gene duplication has since been observed in other cohorts of NTG patients (Table 2) [85,86]. The original study also reported three missense heterozygous TBK1 variants in the patient cohort, (p.S151F, p.L306I, p.V464A) although these variants remain of unknown pathogenicity (Table 2) [10].
TBK1: One Gene, Multiple Diseases: Molecular Basis to Disease Pathogenesis
It comes as no surprise that TBK1 would be important for human health, as it is highly conserved evolutionary and is prevalent in the general population (only 1 commonly occurring missense variant has been reported in 66,000 WES individuals in the Exome Aggregation Consortium (ExAC) [87]). HSE, ALS, FTD, and NTG are diverse diseases caused either by infection, or protein aggregate accumulation in neuronal cells. Of course, the identified genes associated with these diseases explain only a proportion of all patient cases, suggesting that further genetic heterogeneity is present. Indeed, these diseases share heterozygous mutations in TBK1, an essential multifunctional kinase participating in two distinct pathways: innate immune inflammatory signaling (TLR3-IFN pathway) and autophagy. So what potential mechanisms render this gene responsible for such clinically-distinct pathological conditions?
TBK1 Domain-specific Mutations
Mutations in a single gene can give rise to different phenotypes due to domain specific mutations which determine modular impairment of a multimeric protein. Examples of this are not uncommon and include the STAT1 deficiencies [88]. Interestingly, none of the HSE and NTG mutations have been found in ALS, isolated FTD or ALS-FTD patients, although identical mutations have been observed in the latter three diseases. The HSE mutations have been shown to occur exclusively in the kinase domain, resulting in allele-specific impairment of IFNβ induction [8]. This may possibly suggest that the kinase domain is particularly important for effective IFN production. (Figure 1, Table 1). In contrast, Freischmidt et al. reported CCD2 domain mutations in TBK1, impairing optineurin binding but maintaining normal IFNβ promoter activation, indicating that TBK1 autophagy function may play a protective role in ALS and FTD [8]. As such, TBK1 mutations affecting domain specificity might represent an underlying factor contributing to differential phenotypes in these diseases.
Subcellular Localization and Tissue Specificity of TBK1
On a similar note, mutations affecting specific protein interactions could affect subcellular localization of TBK1, which might potentially affect disease manifestation. In that regard, the subcellular localization of TBK1 has been shown to determine its role in different pathways [89]. For example, one study reported that TBK1 could interact with each of its adaptors TANK, SINTBAD, and NAP1 in a mutually exclusive manner in HeLa cells, such that TBK1 activation following viral infection was TBK1-TANK-dependent and specifically occurring in perinuclear compartments, whereas TBK1-NAP1 co-localized with autophagosomes [89]. Hence, it is conceivable that mutations which alter the spatial distribution of TBK1 in a cell might be connected to altered cellular phenotypes and manifested as different pathologies. Moreover, despite its ubiquitous expression, TBK1 may have cell type-specific roles favoring specific signaling pathways. This has been difficult to determine, as most functional assays have been carried out on leukocytes or fibroblasts, as opposed to the relevant CNS cells affected in these disease types. In light of the fact that CNS cells selectively utilize autophagy over IFN signaling during viral infections, such putative tissue specificity might play a larger role than previously thought [90]. Consequently, intrinsic spatial localization characteristics combined with domain and tissue specificity might play a role in how various TBK1 mutations within the same gene are manifested in different diseases.
TBK1 Mutation Type
It is possible to consider that the type of TBK1 mutation (LoF, GoF, dominant negativity, haploinsufficiency) might also play a role in determining disease type. NTG is a GoF model of TBK1 pathogenicity, due to TBK1 duplications. In contrast, ALS and FTD have been largely associated with LoF heterozygous mutations resulting in haploinsufficiency. By inference, a moderate reduction of TBK1 expression (~50%) by haploinsufficiency, due to residual expression from the wild type allele, could presumably affect TBK1-dependent autophagy function. On the other hand, a missense HSE-causing TBK1 mutation has been shown to result in a dominant negative effect on the wild type allele, leading to impaired IFN signaling, and suggesting that very low levels of functional TBK1 could impact the IFN signaling pathway [6]. The difference in absolute levels of TBK1 due to its respective mutations might be responsible for, or capable of modulating the outcome for such observed differences in cellular phenotypes. This may suggest that different mutations may have different thresholds of effective TBK1 function, which could result in disparate diseases.
Further Implications for HSE/ALS Pathogenesis
HSE, ALS-FTD and NTG have not been previously proposed as having a similar disease spectrum. However, we propose that their common genetic etiology raises questions about a possible mechanism of shared pathogenesis with implications for new treatment avenues (Figure 3). This may be due to TBK1’s niche role, possibly determined by tissue specificity, in CNS inflammation. Despite sharing ‘TBK1’ features with HSE, ALS and FTD, NTG presents a GoF TBK1 model which sets it apart from the LoF TBK1 model of other diseases. In light of the evidence suggesting a shared disease-causing gene, why do we not see co-occurrence of such diseases? There are no reports on the co-occurrence of HSE and ALS in one same individual. This may be because both HSE and ALS are exceedingly rare events (HSE 1–2/1million/yr; ALS 2/100,000/yr) such that the co-occurrence of disease would be highly unlikely, especially when incomplete penetrance is a feature of both diseases.
Figure 3. Dynamic Interplay between Cells in CNS in ALS, FTD, HSE and NTG.
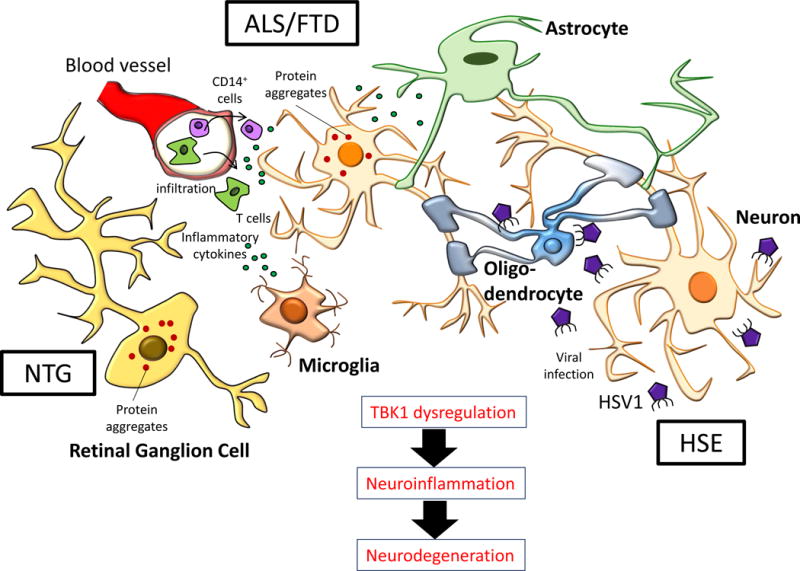
In ALS/FTD, motor neurons accumulate toxic protein aggregates (e.g.: TDP-43 inclusions) which contribute to neurodegeneration. In addition, other cells are known to mediate neuroinflammation, leading to cell death. Activated microglia, infiltrating monocytes and T cells produce inflammatory cytokines; astrocytes downregulate their supportive function, contributing to neurodegeneration [65]. In HSE, studies using iPSCs-derived neurons from a TLR3-deficient patient demonstrated that TLR3-dependent cell-intrinsic immunity was critical in preserving the integrity of neurons and oligodendrocytes during a primary HSV1 infection [54]. In NTG, progressive degeneration of retinol ganglion cells occurs and is poorly understood [114].
Moreover, TBK1’s involvement in ALS is progressive (accumulation of protein aggregates) resulting in the manifestation of disease pathology, whereas in HSE, exposure and infection by HSV1 is necessary to reveal a phenotype. Furthermore, the HSV1 seropositivity rate among adults ranges from 40–87%, contributing to this reduced penetrance [91]. However, as the age of onset for ALS-FTD is much higher than that of HSE – which peaks in early childhood – it would be of interest to carefully follow long-term outcomes of HSE patients, as ALS symptoms may have yet to manifest. Prior to the advent of acyclovir in the 1980s, HSE patients would not have survived. Therefore, presently we do not have any long-term follow-up on these patients. Additionally, the fitness of patients post-HSE is reduced, with mortality rates up to 30% and over 50% suffering from severe sequelae, such that they may never reach age of onset for ALS [42,44]. Testing for the presence of protein aggregates in CNS samples from HSE patients would reveal whether a similar pathology is observed. Of note, HSE patients with TBK1 deficiency have also developed cognitive impairment and/or motor disabilities subsequent to HSE [6]. HSE patients with other TLR3-IFN deficiencies (not TBK1) would probably have a low risk of developing ALS if the molecular defect of HSE was truly restricted to IFN signaling and was autophagy-independent. Testing HSV1 serology in all ALS-FTD patients, in particular those with TBK1 mutations might thus be informative. The reciprocal experiments of testing autophagy and IFN signaling in HSE vs. ALS/FTD patient cells might also help address how similar HSE and ALS/FTD disease states are. Indeed, TBK1 functions in patient cells have not been fully explored in any of the studies discussed here.
Concluding Remarks
Advances in sequencing technologies have begun to reveal a growing number of single gene variants that can underlie a diverse range of diseases [92–94]. Indeed, the concept that mutations in a single gene can cause a broad spectrum of disorders has been well documented [95,96]. Sequencing studies will undoubtedly reveal further novel genetic models to explain disease pathogenesis. In fact, mutations associated with a particular disease, which are found in other atypically presenting diseases, would have been overlooked if it were not for large-scale sequencing analyses. The disparity in disease phenotypes stemming from mutations of a single gene might be mediated through different mechanisms, including i) mutations occurring in domain-specific regions of a given multimeric protein, ii) qualitative differences resulting from a certain type of mutation, or iii) subcellular localization/tissue specificity. Human partial TBK1 deficiency results in neuroinflammatory/neurodegenerative disorders of the CNS such as HSE, ALS, ALS-FTD, whereas TBK1 GoF results in NTG. These conditions are probably a consequence of dysregulated autophagy (ALS, FTD, NTG) or, of impaired IFN signaling (HSE). The surprisingly important role of this protein in the CNS, particularly its role in autophagy, is consistent with other reports indicating that post-mitotic cells such as neurons depend on autophagy to deal with inflammation and cell survival following viral infection [90]. Not only does this suggests a common underlying disease etiology, but it also raises additional questions about the pathogenesis of such diseases (see Outstanding Questions). Despite the exciting and unexpected finding of TBK1’s involvement in several neurological diseases, further studies are warranted in order to validate the pathogenic mechanisms linked to defects in TBK1. Moreover, TBK1’s functional role in neuroprotection and neuroinflammation needs to be further examined so as to fully appreciate its participation in disease (See Outstanding Questions). Since current treatment options for HSE, ALS, FTD and NTG are limited, any further knowledge gained could be applied to ameliorate treatment approaches for these debilitating diseases. Focusing on the neuroprotective aspects stemming from interfering with the TBK1 pathway may prove to be promising (Box 3). Moreover, it may be possible to extend the lessons learnt from one specific TBK1-associated disease to another, potentially contributing to major beneficial advances for many TBK1-associated neuroinflammatory conditions.
Box 3. Clinician’s Corner: Treatment Avenues for TBK1-associated Neurological Diseases.
Studies on HSE were previously hindered by the fact that this primarily childhood disease is lethal, making it difficult to trace the transmission of the underlying genes, until the advent of acyclovir [42,44]. Acyclovir is a nucleoside analog with proven efficacy in inhibiting HSV-1 DNA replication, significantly reducing mortality [108]. Unfortunately, survivors still suffer from neurological sequelae and neuroinflammation [42,44].
ALS and FTD have no cure, and current treatments involve palliative care with variable success. Riluzole (Rilutek©) is the only FDA-approved drug that can delay ventilator dependence by a few months for ALS patients although its mechanism is unknown [109]. Broad effect treatment such as with the autophagy-inducer rapamycin has been shown to be a promising ALS drug candidate [111].
Glaucoma patients rely on prostaglandin analogs or surgical procedures to relieve their symptoms [110].
Given the implication that TBK1 plays a key role in these diseases, it seems worthwhile to explore TBK1 as a more defined target of novel therapeutics. It may be possible to envisage the use of TBK1 activators to achieve neuroprotection, but pre-clinical validation would be the first step.
As ALS, FTD and glaucoma are progressive debilitating diseases, there is an urgent need to identify viable neuroprotective treatments. From a different angle, HSE might potentially benefit from treatments specifically aimed at decreasing infection-associated neuronal death or neuroinflammation.
Outstanding questions.
What are the respective roles of IFN and autophagy in human TBK1 disorders? Do they work independently or together to resolve/exacerbate inflammation?
Neuronal cells are the common cells affected in human TBK1 disorders. However, what other cell types control TBK1-mediated neuroinflammation?
What is(are) the mediator(s) of tissue damage/neuroinflammation in these human TBK1-associated diseases? Do they point to the same culprit, i.e. protein aggregates? Are protein aggregates a feature of HSE?
Is there evidence of a viral trigger in the development of ALS/glaucoma? Does IFN play a role in the development of ALS, FTD or glaucoma?
Could the dissection of TBK1 function provide us with new therapeutic strategies to treat HSE, ALS, FTD, ALS-FTD or NTG? Common disease pathogenesis focusing on neuroprotective effects or TBK1-mediated pathways may reveal more effective and targeted therapies for these diseases. Treatment to boost autophagy may be helpful, e.g. by using rapamycin to prevent cell toxicity and cell death. Alternatively, it may be possible to use drugs to boost proteasome function in patients with TBK1 deficiency, inducing proteasomal ubiquitination pathways to help clear the toxic buildup of protein aggregates.
Trends Box.
HSE, in a subset of children, is caused by impaired antiviral IFN production due to monogenic mutations in the TLR3-IFN signaling pathway, which may affect TBK1.
Due to advances in sequencing technologies, a number of new amyotrophic lateral sclerosis (ALS) or ALS-frontotemporal dementia (ALS-FTD) genes have been identified, five of which are known to be involved in autophagy, SQSTM1, VCP, OPTN, UBQLN2 and TBK1. These mutations are thought to contribute to disease pathogenesis possibly due to an impaired autophagy process.
The genetic etiology of normal tension glaucoma (NTG) has recently been attributed to copy number variants (CNV) found in chromosome region 12q14, specifically leading to duplications of the TBK1 gene. This duplication has been found to increase TBK1 transcript levels, suggesting a gain of function role for TBK1 in NTG.
Recent developments in the field of selective autophagy have implicated this evolutionarily conserved process in innate immunity and pathogen clearance, including in neuronal cells.
Acknowledgments
We thank Elizabeth T. Cirulli and Tim Harris for providing a detailed list of all TBK1 mutations. We thank Inge A. Meijer, Aubrey J. Cunnington, Serge Mostowy, and Beth Holder for helpful discussions. LA is supported by the Chancellor’s Scholarship of Universiti Brunei Darussalam; VSS, by the Medical Research Foundation and JLC is a member of the Howard Hughes Medical Institute. Our studies are partly funded by NIH grant number 2R01NS072381-05 (JLC and SYZ), ANR award IEIHSEEI (SYZ) and the Medical Research Foundation (VSS).
Glossary
- TBK1 (TANK-binding kinase 1)
functions downstream of multiple IFN inducing pathways that are activated following pathogen sensing and are mediated by Toll-like receptor 3 (TLR3), RIG-I-like receptors (RLRs) and other cytosolic DNA sensors. Following activation, TBK1 phosphorylates cytosolic IRF3 or IRF7, which then dimerize and enter the nucleus to induce the expression of IFNs
- Trigeminal nerves
innervate the cranium and are responsible for sensory, as well as certain motor facial functions. Following primary infection, HSV1 may take this route to reach the central nervous system and cause acute infection
- Pattern recognition receptors (PRRs)
innate immune receptors acting as a first line of defense against pathogens. They recognize pathogen-associated molecular patterns (PAMPs) which are conserved across different pathogen groups
- TAR DNA-binding protein 43 (TDP-43)
nuclear protein with a role in the regulation of gene expression. Mutations in its gene TARDBP can lead to its accumulation and aggregation in the cytoplasm of motor neurons, a hallmark of ALS and FTD
- Frontotemporal lobar dementia (FTD)
disease characterized by progressive neuronal loss of the frontal and temporal lobes of the brain
- STAT1 deficiencies
Several inborn mutations of human STAT1 exhibit allelic heterogeneity, different modes of inheritance and variable immunological/clinical phenotypes. AR complete and partial deficiencies predispose to bacterial and viral infections due to impaired IFN-γ, -α/β-mediated immunity; AD deficiency selectively underlies mycobacterial disease due to impaired IFN-γ mediated immunity; AD gain of function STAT1 mutations, found exclusively in the coiled-coil domain of STAT1, give rise to autoimmunity and chronic mucocutaneous candidiasis, due to increased IFN-α/β and impaired Th17 responses [96]
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fitzgerald KA, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–6. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 2.Thurston TLM, et al. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–21. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 3.Wild P, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–33. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pilli M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–34. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clément J-F, et al. The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 2008;18:889–99. doi: 10.1038/cr.2008.273. [DOI] [PubMed] [Google Scholar]
- 6.Herman M, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med. 2012;209:1567–82. doi: 10.1084/jem.20111316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cirulli ET, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–41. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freischmidt A, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–6. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- 9.Pottier C, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015;130:77–92. doi: 10.1007/s00401-015-1436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fingert JH, et al. Copy number variations on chromosome 12q14 in patients with normal tension glaucoma. Hum Mol Genet. 2011;20:2482–94. doi: 10.1093/hmg/ddr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gijselinck I, et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology. 2015;85:2116–25. doi: 10.1212/WNL.0000000000002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999;18:6694–704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tojima Y, et al. NAK is an IkappaB kinase-activating kinase. Nature. 2000;404:778–782. doi: 10.1038/35008109. [DOI] [PubMed] [Google Scholar]
- 14.Chau T-L, et al. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem Sci. 2008;33:171–80. doi: 10.1016/j.tibs.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Sharma S, et al. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–51. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 17.Loo Y-M, Gale M. Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–92. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishii KJ, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–8. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 19.Paz S, et al. Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway. Cell Mol Biol (Noisy-le-grand) 2006;52:17–28. [PubMed] [Google Scholar]
- 20.Sun L, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saitoh T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842–6. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–8. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar H, et al. Pathogen recognition in the innate immune response. Biochem J. 2009;420:1–16. doi: 10.1042/BJ20090272. [DOI] [PubMed] [Google Scholar]
- 24.Soulat D, et al. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 2008;27:2135–46. doi: 10.1038/emboj.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 26.Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol. 2014;21:336–45. doi: 10.1038/nsmb.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stolz A, et al. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 28.Kirkin V, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–16. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 29.Matsumoto G, et al. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–89. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 30.Mostowy S, et al. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286:26987–95. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 32.Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 33.Korac J, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126:580–92. doi: 10.1242/jcs.114926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morton S, et al. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008;582:997–1002. doi: 10.1016/j.febslet.2008.02.047. [DOI] [PubMed] [Google Scholar]
- 35.Lussignol M, et al. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol. 2013;87:859–71. doi: 10.1128/JVI.01158-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chou J, et al. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–6. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 37.Orvedahl A, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Ma Y, et al. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J Virol. 2012;86:2188–96. doi: 10.1128/JVI.05376-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang Q, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014;15:228–38. doi: 10.1016/j.chom.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watson RO, et al. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe. 2015;17:811–9. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitley RJ, Gnann JW. Viral encephalitis: familiar infections and emerging pathogens. Lancet (London, England) 2002;359:507–13. doi: 10.1016/S0140-6736(02)07681-X. [DOI] [PubMed] [Google Scholar]
- 42.Abel L, et al. Age-dependent Mendelian predisposition to herpes simplex virus type 1 encephalitis in childhood. J Pediatr. 2010;157:623–9. 629.e1. doi: 10.1016/j.jpeds.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 43.De Tiège X, et al. The spectrum of herpes simplex encephalitis in children. Eur J Paediatr Neurol. 2008;12:72–81. doi: 10.1016/j.ejpn.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 44.Lahat E, et al. Long term neurological outcome of herpes encephalitis. Arch Dis Child. 1999;80:69–71. doi: 10.1136/adc.80.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitley RJ, et al. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med. 1986;314:144–9. doi: 10.1056/NEJM198601163140303. [DOI] [PubMed] [Google Scholar]
- 46.Zhang S-Y, et al. TLR3 immunity to infection in mice and humans. Curr Opin Immunol. 2013;25:19–33. doi: 10.1016/j.coi.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Casrouge A, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–12. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 48.Zhang S-Y, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–7. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 49.Guo Y, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. 2011;208:2083–98. doi: 10.1084/jem.20101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sancho-Shimizu V, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest. 2011;121:4889–902. doi: 10.1172/JCI59259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pérez de Diego R, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33:400–11. doi: 10.1016/j.immuni.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andersen LL, et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med. 2015;212:1371–9. doi: 10.1084/jem.20142274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang S-Y, et al. Mendelian predisposition to herpes simplex encephalitis. Handb Clin Neurol. 2013;112:1091–7. doi: 10.1016/B978-0-444-52910-7.00027-1. [DOI] [PubMed] [Google Scholar]
- 54.Lafaille FG, et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature. 2012;491:769–73. doi: 10.1038/nature11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alexopoulou L, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 56.Weber F, et al. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80:5059–64. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jacquemont B, Roizman B. Ribonucleic acid synthesis in cells infected with herpes simplex virus: characterization of viral high molecular weight nuclear RNA. J Gen Virol. 1975;29:155–65. doi: 10.1099/0022-1317-29-2-155. [DOI] [PubMed] [Google Scholar]
- 58.Balachandran S, et al. Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/Caspase-8 death signaling pathway. J Virol. 2000;74:1513–23. doi: 10.1128/jvi.74.3.1513-1523.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shaw PJ. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry. 2005;76:1046–57. doi: 10.1136/jnnp.2004.048652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Charcot J-M, Joffroy A. Deux cas d’atrophie musculaire progressive avec lésions de la substance grise et des faiseaux antéro-latéraux de la moelle épinière. Arch Physiol Norm Pathol. 1869;2:744–760. [Google Scholar]
- 61.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 62.Barber SC, et al. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta – Mol Basis Dis. 2006;1762:1051–1067. doi: 10.1016/j.bbadis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 63.Shi P, et al. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2010;1802:45–51. doi: 10.1016/j.bbadis.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hirano A, et al. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984;43:461–70. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- 65.Zhao W, et al. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol. 2013;8:888–99. doi: 10.1007/s11481-013-9489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lomen-Hoerth C, et al. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–9. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 67.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 68.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 69.Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kwiatkowski TJ, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 71.Deng H-X, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–5. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Renton AE, et al. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maruyama H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–6. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- 74.Khoris J, et al. Coexistence of dominant and recessive familial amyotrophic lateral sclerosis with the D90A Cu,Zn superoxide dismutase mutation within the same country. Eur J Neurol. 2000;7:207–11. doi: 10.1046/j.1468-1331.2000.00028.x. [DOI] [PubMed] [Google Scholar]
- 75.Hand CK, et al. Compound heterozygous D90A and D96N SOD1 mutations in a recessive amyotrophic lateral sclerosis family. Ann Neurol. 2001;49:267–71. doi: 10.1002/1531-8249(20010201)49:2<267::aid-ana51>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 76.Hadano S, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29:166–73. doi: 10.1038/ng1001-166. [DOI] [PubMed] [Google Scholar]
- 77.Fecto F. <emph type=“ital”>SQSTM1</emph> Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch Neurol. 2011;68:1440. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 78.Le Ber I, et al. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging. 2015;36:3116.e5–8. doi: 10.1016/j.neurobiolaging.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 79.Williams KL, et al. Novel TBK1 truncating mutation in a familial amyotrophic lateral sclerosis patient of Chinese origin. Neurobiol Aging. 2015;36:3334.e1–5. doi: 10.1016/j.neurobiolaging.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 80.Quigley HA. Glaucoma. Lancet (London, England) 2011;377:1367–77. doi: 10.1016/S0140-6736(10)61423-7. [DOI] [PubMed] [Google Scholar]
- 81.Friedman DS, et al. Prevalence of open-angle glaucoma among adults in the United States. Arch Ophthalmol (Chicago, Ill 1960) 2004;122:532–8. doi: 10.1001/archopht.122.4.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quigley HA. Glaucoma. Lancet (London, England) 2011;377:1367–1377. doi: 10.1016/S0140-6736(10)61423-7. [DOI] [PubMed] [Google Scholar]
- 83.Minegishi Y, et al. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum Mol Genet. 2013;22:3559–67. doi: 10.1093/hmg/ddt210. [DOI] [PubMed] [Google Scholar]
- 84.Fingert JH. Primary open-angle glaucoma genes. Eye (Lond) 2011;25:587–95. doi: 10.1038/eye.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kawase K, et al. Confirmation of TBK1 duplication in normal tension glaucoma. Exp Eye Res. 2012;96:178–80. doi: 10.1016/j.exer.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ritch R, et al. TBK1 gene duplication and normal-tension glaucoma. JAMA Ophthalmol. 2014;132:544–8. doi: 10.1001/jamaophthalmol.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Consortium EA, et al. Analysis of protein-coding genetic variation in 60,706 humans. Cold Spring Harbor Labs Journals; 2015. [Google Scholar]
- 88.Casanova J-L, et al. Immunology taught by human genetics. Cold Spring Harb Symp Quant Biol. 2013;78:157–72. doi: 10.1101/sqb.2013.78.019968. [DOI] [PubMed] [Google Scholar]
- 89.Goncalves A, et al. Functional dissection of the TBK1 molecular network. PLoS One. 2011;6:e23971. doi: 10.1371/journal.pone.0023971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yordy B, et al. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe. 2012;12:334–45. doi: 10.1016/j.chom.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Looker KJ, et al. Global and Regional Estimates of Prevalent and Incident Herpes Simplex Virus Type 1 Infections in 2012. PLoS One. 2015;10:e0140765. doi: 10.1371/journal.pone.0140765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang Y, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–11. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee H, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–7. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Casanova J-L. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci U S A. 2015;112:E7128–37. doi: 10.1073/pnas.1521651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu X, et al. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci. 2014;17:773–81. doi: 10.1038/nn.3713. [DOI] [PubMed] [Google Scholar]
- 96.Boisson-Dupuis S, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. 2012;24:364–78. doi: 10.1016/j.coi.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bonnard M, et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 2000;19:4976–85. doi: 10.1093/emboj/19.18.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marchlik E, et al. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J Leukoc Biol. 2010;88:1171–80. doi: 10.1189/jlb.0210071. [DOI] [PubMed] [Google Scholar]
- 99.McWhirter SM, et al. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A. 2004;101:233–8. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miyahira AK, et al. TANK-binding kinase-1 plays an important role during in vitro and in vivo type I IFN responses to DNA virus infections. J Immunol. 2009;182:2248–57. doi: 10.4049/jimmunol.0802466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perry AK, et al. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. 2004;199:1651–8. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang R, et al. Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia. 2011;59:946–58. doi: 10.1002/glia.21167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hofer MJ, et al. The type I interferon-alpha mediates a more severe neurological disease in the absence of the canonical signaling molecule interferon regulatory factor 9. J Neurosci. 2010;30:1149–57. doi: 10.1523/JNEUROSCI.3711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van Boxel-Dezaire AHH, et al. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–72. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 105.Mankouri J, et al. Optineurin negatively regulates the induction of IFNbeta in response to RNA virus infection. PLoS Pathog. 2010;6:e1000778. doi: 10.1371/journal.ppat.1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sakaguchi T, et al. Optineurin with amyotrophic lateral sclerosis-related mutations abrogates inhibition of interferon regulatory factor-3 activation. Neurosci Lett. 2011;505:279–81. doi: 10.1016/j.neulet.2011.10.040. [DOI] [PubMed] [Google Scholar]
- 107.Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–60. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- 108.Elion GB. The biochemistry and mechanism of action of acyclovir. J Antimicrob Chemother. 1983;12(Suppl B):9–17. doi: 10.1093/jac/12.suppl_b.9. [DOI] [PubMed] [Google Scholar]
- 109.Miller RG, et al. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) Cochrane database Syst Rev. 2007 doi: 10.1002/14651858.CD001447.pub2. [DOI] [PubMed] [Google Scholar]
- 110.Sambhara D, Aref AA. Glaucoma management: relative value and place in therapy of available drug treatments. Ther Adv Chronic Dis. 2014;5:30–43. doi: 10.1177/2040622313511286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.de Paula CZ, et al. An Overview of Potential Targets for Treating Amyotrophic Lateral Sclerosis and Huntington’s Disease. Biomed Res Int. 2015;2015:1–7. doi: 10.1155/2015/198612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li J, et al. Crystal structure of the ubiquitin-like domain of human TBK1. Protein Cell. 2012;3:383–91. doi: 10.1007/s13238-012-2929-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang KY, et al. Ubiquilin 2: a component of the ubiquitin-proteasome system with an emerging role in neurodegeneration. Int J Biochem Cell Biol. 2014;50:123–6. doi: 10.1016/j.biocel.2014.02.018. [DOI] [PubMed] [Google Scholar]
- 114.Weinreb RN, et al. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014;311:1901–11. doi: 10.1001/jama.2014.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]