

Abstract
The mechanisms of SUDEP have been difficult to define, as most cases occur unwitnessed, and physiological recordings have been obtained in only a handful of cases. However, recent data obtained from human cases and experimental studies in animal models have brought us closer to identifying potential mechanisms. Theories of SUDEP should be able to explain how a seizure starting in the cortex can sometimes lead to changes in brainstem cardiorespiratory control mechanisms. Here we focus on three major themes of work on the causes of SUDEP. First, evidence is reviewed identifying postictal hypoventilation as a major contributor to the cause of death. Second, data are discussed that brainstem serotonin and adenosine pathways may be involved, as well as how they may contribute. Finally, parallels are drawn between SIDS and SUDEP, and we highlight similarities pointing to the possibility of shared pathophysiology involving combined failure of respiratory and cardiovascular control mechanisms. Knowledge about the causes of SUDEP may lead to potential pharmacological approaches for prevention. We end by describing how translation of this work may result in future applications to clinical care.
This manuscript focuses on mechanisms of SUDEP, as revealed by animal and human studies. Analysis of data from monitored human cases has refined our understanding of the final events leading to death. It is now clear that seizures immediately preceding SUDEP often lead to rapidly developing hypoventilation and bradycardia. More extensive use of animal models, including ones that more closely replicate the human condition, provide a detailed understanding of the pathophysiology of seizure-induced changes in cardiorespiratory function. Increasing our knowledge of the mechanisms of SUDEP is our best hope for developing pharmacological approaches for prevention, and for guiding targeted translation of research to clinical care. We will address three main themes.
First, recent evidence supports the hypothesis that apnea/hypoventilation plays a significant role in SUDEP, and in some cases may be the primary cause of death.1 Some investigators previously assumed SUDEP to be exclusively due to cardiovascular mechanisms (asystole, bradycardia, arrhythmia, hypotension, etc.), but data from monitored cases of SUDEP do not support the conclusion that the initial inciting event is usually cardiac.1 The relative importance of cardiovascular versus respiratory mechanisms is unknown, but both appear to be involved, possibly reflecting a global neurovegetative dysfunction induced centrally by seizures.1 In addition, impaired arousal mechanisms have not received much attention, and yet the deep post-ictal unresponsiveness, of which post-ictal generalized EEG suppression (PGES) may be a correlate,2 may also play an important role by preventing protective reflexes.3–5
Second, serotonin and adenosine pathways are promising targets for pharmacological therapy4,6–9. It is unclear whether either one is central to the pathophysiology, but drugs targeting one or both of these two systems might reduce the risk of SUDEP in high-risk populations. Sites of interaction between these two neurotransmitter pathways may be particularly effective targets for treatment. Two other neurotransmitter pathways of potential interest are that of endogenous opioids and γ-aminobutyric acid (GABA).
Third, and separate from any possible cardiac susceptibility to sudden death, there are similarities between SUDEP and SIDS that may lead to a better understanding of the mechanisms of both, and may help guide preventive measures.4, 5, 10–12 Recent data suggest that some cases of SIDS may be due to seizures that go unrecognized.13, 14 SIDS and SUDEP have both been linked to the 5-HT system,4, 11, 15–20 and together with other similarities in presentation and diagnostic criteria, this has led to the suggestion that they may share a final common pathway leading to death.4,5,12
Respiratory dysfunction in SUDEP
It has long been known that changes in cardiovascular function occur during and after seizures. Since most sudden deaths in non-epileptic patients without structural cardiac disease are due to arrhythmias, this led some to assume that SUDEP is usually a result of seizure-induced cardiovascular dysfunction, such as tachyarrhythmias, asystole or parasympathetic vasodilation and hypotension.21 The possible contribution to SUDEP of genetic susceptibility to sudden cardiac death is discussed in detail by Goldman et al in this supplement,22 but it is nevertheless worth considering that some genes implicated in cardiac arrhythmias may also be expressed in brainstem respiratory nuclei.
In some witnessed SUDEP cases, respiratory difficulties were seen prior to death.1, 23–25 For more than a century, it has been known that seizures can induce significant hypoventilation.26 Recently it has been found that apnea and O2 desaturation are much more common than previously realized after generalized convulsive or partial seizures.27,28
Direct observations of SUDEP are limited. Until 2013 there were only eight published cases of SUDEP monitored at the time of death,2, 23, 29–33 and in that year the MORTEMUS study added another six while systematically reanalyzing five previously published cases.1 In none of these 14 cases were oronasal airflow, impedance plethysmography, tidal volume, ventilation, end tidal CO2, O2 saturation or blood gases recorded. In 10 cases, respiratory rate was estimated by watching chest and abdominal movements on video, together with respiration-induced artifact on EEG,1 but this method is not able to rule out airway obstruction, paradoxical breathing or shallow breathing. Measuring respiratory rate alone does not allow an estimate of alveolar ventilation, which requires knowledge of tidal and dead space volumes.
Data from patients who died of SUDEP while being monitored in epilepsy monitoring units (EMUs) have yielded highly valuable information about pathophysiological mechanisms. These data, such as from the MORTEMUS study,1 reveal that apnea/hypoventilation and bradycardia/asystole both occur as terminal events. Methodological limitations have made it difficult to draw firm conclusions from these observations. However, among the many important results from this study, one of the most valuable was that central apnea occurred prior to death, and changes in breathing were not those that occur in response to global hypoperfusion.34 Another was that every case of monitored SUDEP occurred after a generalized tonic-clonic seizure (GTCS). Therefore, it is reasonable to conclude that changes in respiratory and cardiac physiology induced by GTCS are relevant to SUDEP, and these can be studied in patients who do not die.
It is now widely believed that at least some cases of SUDEP are due to hypoventilation induced by seizures. However, it remains necessary to explain why when apnea occurs in around 50% of seizures, whether partial or generalized,27, 28 SUDEP only occurs once out of thousands of seizures and usually only after GTCS.1
Role of 5-HT in SUDEP
Normal function of 5-HT neurons
There is a relatively small number of 5-HT neurons in the brain, located primarily in the brainstem raphe nuclei.35, 36 Some 5-HT neurons are involved in thermoregulation, others in arousal, mood, appetite or other brain functions.36, 37 A subset are sensors of arterial PCO237–41 and respond indirectly through a decrease in intracellular pH.42 They are proposed to be central respiratory chemoreceptors that stimulate breathing to restore CO2 back to normal.37, 43–47 A separate subset of serotonin neurons in the midbrain is also sensitive to an increase in blood PCO2,48 but instead of stimulating breathing they cause cortical arousal,3 an important component of the protective response to hypercapnia. If a person is asleep or postictal, and a mattress, blanket or pillow obstructs their mouth and nose, it is important to become more alert so they can move and relieve the airway obstruction.
Rodent 5-HT neurons in slices or in culture increase their firing rate by an average of 3-fold when pH is decreased from 7.4 to 7.2,39, 40–42 well within the range that stimulates breathing in vivo. These neurons are closely associated with the basilar artery and its major branches where they can accurately monitor arterial PCO2.48, 49 5-HT neurons project to all major respiratory nuclei, and release serotonin, thyrotropin releasing hormone (TRH) and substance P.37 These neurotransmitters stimulate respiratory neurons,37 and in some cases induce bursting pacemaker activity in neurons that are rhythmically active in vivo.50, 51 Stimulation of 5-HT neurons in vivo causes an increase in ventilation.52
Studies of 5-HT neurons have been facilitated by genetically engineered mice deficient in 5-HT neurons (Lmx1bf/f/p). Pet1 is a transcription factor expressed in all 5-HT neurons and only 5-HT neurons.53 Lmx1b is another transcription factor required for survival of many brainstem neurons.54 To specifically delete all 5-HT neurons55 one mouse strain that expressed the virus protein cre recombinase (cre) under control of the promotor for Pet1 (PET1-Cre) was mated with a second mouse strain that contained LoxP sites flanking the LMX1B gene (floxed LMX1B). When bred to produce offspring with two alleles (homozygous) of floxed LMX1B and at least one allele for PET1-CRE, the LMX1B gene was deleted in all embryonic 5-HT neurons.55 As 5-HT neurons first differentiate during embryonic development, they express cre, the LMX1b gene is then excised, and this causes them to die. Other neurons are unaffected.
The absence of 5-HT neurons causes neonatal Lmx1bf/f/p mice to have severe apnea lasting as long as 55 seconds and 30% die in the first few days of life.56 In those that survive, breathing normalizes after 12 days of age, except that their ventilatory response to inhaled CO2 is decreased by about 50%.45 They also no longer wake up from sleep in response to 3–7% CO2 in the air, and thus lack this important protective reflex.3 These and other data support the conclusion that some 5-HT neurons in the medulla are central respiratory chemoreceptors, whereas other 5-HT neurons in the midbrain are “central arousal chemoreceptors” that are a component of the ascending reticular activating system.
Evidence that 5-HT mechanisms contribute to SUDEP
The first clue that the 5-HT system might be involved in SUDEP was that mice in which the 5-HT2c receptor is genetically deleted have spontaneous generalized seizures, and during those seizures that progress to a tonic extension phase, these HTR2C−/− mice rapidly die8, 57 due to respiratory arrest. These mice also provided early evidence that 5-HT can raise the seizure threshold, a conclusion now supported by data from many other approaches.
The DBA/1 and DBA/2 mouse strains with audiogenic seizures often die from respiratory arrest after generalized seizures with tonic extension.58, 59 Death of DBA/1 & DBA/2 mice can be prevented by brief postictal mechanical ventilation.9, 59 Pretreatment with fluoxetine, a selective serotonin reuptake inhibitor (SSRI), also prevents respiratory arrest.9
The lack of 5-HT neurons in Lmx1bf/f/p mice makes them more susceptible to maximal electroshock and to pilocarpine-induced seizures, and when they have seizures they are more prone to respiratory arrest.60 Thus, defects in the 5-HT system in mice can increase susceptibility to seizures and to seizure-induced death. Seizure-induced death can be reduced in Lmx1bf/f/p mice by agonists of 5-HT2A receptors. As would be predicted, SSRIs have no effect in these mice that lack 5-HT neurons.60
Similarities between SUDEP and SIDS
SIDS is defined as the sudden and unexpected death of an infant under 12 months of age, usually associated with sleep, that remains unexplained after a complete autopsy, death scene investigation, and review of the clinical history.61 In the U.S., SIDS is the leading cause of death between 1 and 12 months of age, with an incidence of 0.6 per 1000 live births, or 6 deaths per day in the U.S.A.11 The risk of SIDS increases (>3 fold) when an infant is prone in bed,62 an important observation that led to the successful public health campaign to place babies to sleep on their back as discussed by Tomson et al in this supplement.63
Even more than SUDEP, it has been exceedingly rare to capture physiological monitoring data from an actual SIDS event. Based on a very small number of monitored cases, a handful of witnessed cases, observations of death scenes, epidemiology, and other sources of information, the leading theories of pathophysiology have focused on paroxysmal events leading to cardiovascular or respiratory dysfunction made worse by abnormal arousal mechanisms.11, 64 These events are thought to include homeostatic stressors such as airway obstruction, overheating, activation of the laryngeal chemoreflex, etc.11
A major step forward in SIDS research resulted from studies of brainstems of infants who died of SIDS. Using non-routine methods (e.g. receptor autoradiography, immunohistochemistry, HPLC, etc) to examine regions of the brainstem thought to be involved in chemosensitivity,65 a number of abnormalities were found in medullary nuclei that contain 5-HT neurons, such as the raphe and arcuate nuclei. These abnormalities include atrophy66 and decreased muscarinic receptor binding in the arcuate nucleus,17 decreased LSD binding,67 decreased 8-OH-DPAT binding,18 an increase in immature forms of 5-HT neurons visualized using immunohistochemistry for tryptophan hydroxylase,18 and a decrease in 5-HT content in brainstem nuclei using HPLC.68 In addition there have been a number of other studies reporting various abnormalities of the 5-HT system.11 It is not clear how these occur, but the link between cigarette smoking and SIDS11 suggests that environmental exposures could contribute.
There are many similarities between SUDEP and SIDS including their definitions.61, 69 In both it is common for death to occur in the prone position in bed, and both are related, at least at times, to periods of sleep.70 Theories of pathophysiology for both center around cardiac, respiratory and arousal defects, and both have been linked to the 5-HT system. SIDS and SUDEP are heterogeneous syndromes, but there may be shared terminal mechanisms in many cases in both disorders. The many similarities suggest that SIDS and SUDEP may have similar causes or share a final common pathway.
Recent data suggest that some cases of SIDS may be due to unrecognised seizures;13,14 thus, some cases of SIDS could be SUDEP in infants not yet diagnosed with epilepsy. It is common for seizures in infants to manifest only with apnea.71 An 8-month infant died soon after he was found prone having seizures and in respiratory distress after a sleep period.13 Had he been found after death, he would have been diagnosed with SIDS. Another infant who died prone at 10 months of age after a sleep period would have been diagnosed with SIDS had he not had hippocampal asymmetry and microdysgenesis on autopsy.14 Routine autopsies rarely include such detailed analysis of hippocampal pathology, so there may be other infants diagnosed as SIDS with similar pathology.
By definition, SIDS only occurs before the age of 12 months, but toddlers can die from sudden unexpected death in childhood (SUDC) with a similar definition to SIDS and SUDEP and some common features such as an increase in risk when toddlers are asleep prone.72, 73 Some of these children have hippocampal and temporal lobe anomalies, and the risk of these anomalies is increased to 82% if they or a family member have a history of febrile seizures.73
Taken together, the above suggests that a subset of SIDS and SUDC may actually be SUDEP in infants with occult seizures or their first seizure. If true, this informs research in both fields. SIDS research, for example, should examine a link to seizures, while SUDEP research should focus on brainstem pathology looking for evidence of abnormalities of 5-HT or other neurotransmitter systems.
Role of adenosine in SUDEP
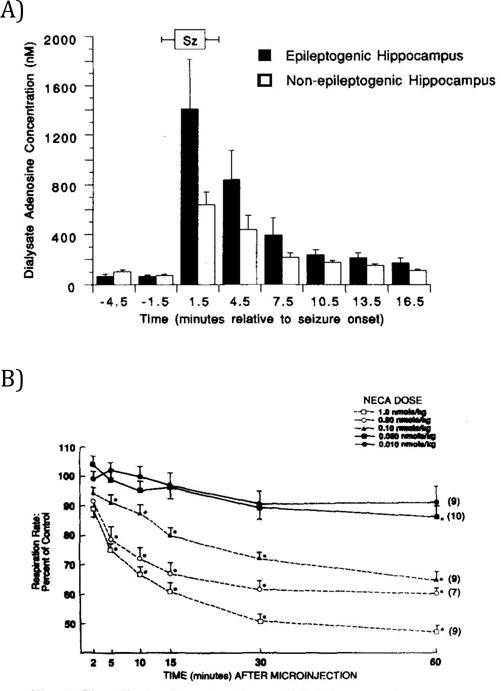
The purine ribonucleoside adenosine is an endogenous homeostatic regulator of network activity.74–76 Adenosine exerts its action via four different receptor subtypes that exhibit differential distributions within the brain, with some of these receptors only activated by the high levels of adenosine observed during seizures.77–82 Conditions of excessive energy consumption as occur during a seizure or after an injury to the brain can trigger a surge in adenosine,78, 83 considered an adaptive response to conserve energy84 and limit the extent of injury.85 Accordingly, increased adenosine suppresses neuronal activity as an innate mechanism to stop seizures.86 Increased levels of adenosine and its metabolites were observed in human TLE patients following a seizure78 and in the rodent hippocampus with seizures induced by several different convulsant drugs.77 Changes in adenosine receptor density have also been observed in epilepsy patients and in animal models of epilepsy.87–89 Importantly, adenosine is also involved in the control of respiration in the brainstem,90, 91 and suppresses cardiovascular and respiratory functions through increased activation of brainstem adenosine receptors.92–94 Paradoxically, a decrease in pH in hippocampal slices causes a rise in adenosine, which causes inhibition of glutamatergic excitatory neurotransmission through actions on A1 adenosine receptors and ATP receptors.87 However, it has been proposed that a decrease in pH in the brainstem causes release of ATP from a subset of respiratory chemoreceptors, and subsequently stimulates respiratory neurons,95 leading to the opposite effect on excitability as occurs in the hippocampus. Adenosine also induces sedative actions,96 which may be relevant to SUDEP. Adenosine agonists prolong post-ictal sedation and adenosine antagonists shorten it.97 The ‘adenosine hypothesis of SUDEP’98 predicts that a seizure-induced adenosine surge in combination with impaired metabolic clearance can trigger lethal apnea or cardiac arrest. If excessive adenosine triggers SUDEP, then adenosine receptor antagonists, such as caffeine or theophylline, might prevent SUDEP.
Clinical relevance of adenosine signalling in SUDEP
Dietary factors have received little attention in the study of SUDEP. Caffeine use is common among the general population; it is thus fair to predict that a proportion of patients with epilepsy regularly consume caffeine. While the chronic use of methylxanthines may increase seizure risk,99 we suggest that the same stimulants might also reduce the risk for SUDEP. To our knowledge, caffeine consumption has not been studied in case control studies of SUDEP. SUDEP often occurs at night100, 101 during a physiological time span when plasma caffeine levels are lowest and brain adenosine levels are highest.102, 103
Role of metabolic adenosine clearance
Although a seizure induced adenosine surge limits the spatial and temporal extent of a seizure,85 the resulting excess in adenosine needs to be metabolized effectively to avoid excessive postictal depression. In the adult brain, the metabolic clearance of adenosine under baseline conditions is largely under the control of astrocytes expressing equilibrative nucleoside transporters, followed by metabolism by two adenosine removing enzymes: 1) adenosine kinase (ADK), and; 2) adenosine deaminase (ADA).7, 84, 104, 105 A seizure-induced adenosine surge, in combination with deficits in the metabolic clearance of adenosine constitutes a candidate mechanism for the development of lethal cardiorespiratory shutdown in SUDEP.7 The following experimental evidence supports the adenosine hypothesis of SUDEP:
The role of adenosine in a mouse model of postnatal central apnea
Mice with genetic deletion of ADK (Adk−/− mice) have been generated,106 and 35% of pups die within 4 days of birth. In most cases, death was accompanied by lethal apnea, leading to the suggestion that Adk−/− mice could be a model of SIDS.106 However, these mice differ from human SIDS in that the latter are normal prior to death and on autopsy, whereas Adk−/− mice have long apneas at baseline and a variety of postnatal abnormalities. These findings, however, provide a direct mechanistic link between impairment of metabolic clearance of adenosine and periods of respiratory distress.
Caffeine prevents lethal apnea after traumatic brain injury
Like seizures, traumatic brain injury (TBI) is a known trigger for an adenosine surge.83 Importantly, in human subjects exposed to severe TBI, high CSF levels of adenosine are associated with lethal apnea.83 If this is due to excessive adenosine receptor activation in the brainstem, then caffeine should prevent lethal apnea after TBI. Severe TBI induced by lateral fluid percussion injury (FPI) in rats results in prolonged periods of apnea and a mortality rate of 47%,107 with apnea significantly longer in animals that died. In contrast, injection of caffeine within 1 minute after the injury prevented extended apnea and lethal outcome.107
Deficiency in metabolic clearance of adenosine triggers lethal outcome after seizures
To evaluate whether seizure-induced adenosine release in combination with deficient adenosine clearance might increase the risk of SUDEP, mice were treated with erythro-9-(2-hydroxy-3- nonyl) adenine (EHNA) and 5-iodotubercidin (ITU), inhibitors of the two major adenosine degradation pathways, 15 min prior to the induction of acute seizures with kainic acid (KA, 35 mg/kg, i.p.). KA-injected animals pretreated with saline instead of EHNA and ITU developed seizures of progressive intensity within 5 minutes after KA. Seizures reached a Racine severity score of about 3 and continued for > 60 min; however, none of these animals died. In contrast, animals treated with EHNA/ITU showed a delayed onset of seizures. However, once seizures developed, all animals rapidly progressed to stage 5 seizures and died within 20 minutes.98 These data suggest that deficiency in the metabolic clearance of adenosine might reduce seizures but contribute to lethal outcome after seizures.
Chronic recurrent seizures cause changes in adenosine metabolism in the brainstem
To assess the role of adenosine homeostasis in a model of pharmacoresistant temporal lobe epilepsy, intra-hippocampal KA was employed.108, 109 Four weeks following KA-injection, when animals typically develop hippocampal sclerosis and spontaneous recurrent seizures, prominent upregulation of ADK immunoreactivity was seen in the nucleus tractus solitarius (NTS). Upregulation of ADK in the NTS might be a compensatory response to enhance the metabolic clearance of adenosine in this critical brainstem area.
SUDEP modelled in heterozygous Adk+/− mice
To further investigate whether impaired metabolic clearance of adenosine could contribute to SUDEP Adk+/− mice were chosen, which express only 50% of ADK.106 Eight Adk+/− mice and 11 wild-type (WT) littermates received intrahippocampal injections of KA to trigger epilepsy. Four weeks after KA injection, intrahippocampal EEG recordings confirmed an epileptic phenotype. Importantly, the epileptic phenotype in Adk+/− mice was less severe than in WT littermates, which experienced more than twice as many seizures. However, Adk+/− mice, despite their attenuated epileptic phenotype, displayed an increased incidence of ‘sudden death’ with three out of eight animals dying within eight weeks of the emergence of epilepsy. In contrast, none of the 11 WT littermates died during the same time span.
Adenosine and seizure-induced respiratory arrest in mice and rats
DBA/2 mice are susceptible to seizure-induced respiratory arrest (SIRA).9, 58, 59, 110, 111 In a small percentage of DBA/2 mice that exhibit seizures without SIRA, administration of adenosine (2 mg/kg) significantly increased the incidence of SIRA. Higher adenosine doses, however, reduced the incidence of seizures in DBA/2 mice, so that SIRA could no longer be evoked. In a second group of DBA/2 mice that exhibited seizures without SIRA, administration of ITU caused a significant increase in SIRA at 30 minutes and partial reduction in SIRA at 24 hours. In a third group of DBA/2 mice that initially exhibited SIRA, administration of caffeine at certain doses significantly reduced the incidence of SIRA. Blocking adenosine metabolism with ITU and EHNA in genetically epilepsy prone rats (GEPR-9s) resulted in a significant decrease in post-ictal SpO2% and a significant increase in the incidence of death between 1 and 24 hours post-treatment, compared to untreated GEPR-9s. These data suggest that a seizure-induced rise in adenosine levels may contribute to seizure-induced respiratory depression observed in patients with epilepsy.27, 28
Translating progress in the pathophysiology and pharmacology of SUDEP into clinical trials in patients with epilepsy
The challenge of studying SUDEP prevention in patients with epilepsy
As discussed by Tomson et al in this supplement,63 any attempt to design a clinical trial targeting prevention of SUDEP needs to fully appreciate the challenges posed by the sample size needed and patient recruitment. If one considers an incidence of SUDEP in patients with refractory epilepsy of about 5/1000 patient-years,112 the sample size required to demonstrate a 50% reduction in SUDEP rate over one year, with a statistical power of 90% and a 5% type-1 error, would be 25,120. Recruiting that many patients with intractable epilepsy in a one-year long double-blind randomized controlled trial (RCT) is unrealistic, both in terms of cost, recruitment capacities, and ethics, taking into consideration the level of evidence regarding currently proposed prevention strategies. Accordingly, and as discussed below, progress in three areas are needed before a RCT can be considered: 1) identification of a population at higher risk of SUDEP than the general population of patients with refractory epilepsy, 2) determining a safe strategy, the potential effectiveness of which in preventing SUDEP is considered high, and 3) seeking further evidence, both in animals and patients with epilepsy, in support of such effectiveness.
Identifying a population at very high risk of SUDEP
Patients with refractory epilepsy demonstrate the highest SUDEP incidence (about 0.5% per year) culminating in a rate of 9.3/1000 patient-years in those undergoing pre-surgical evaluation or having failed epilepsy surgery,113 and 6.9/1000 patient-years in patients enrolled in add-on RCTs of antiepileptic drugs (AEDs) allocated to placebo treatment.114 However, a subgroup of patients, still to be identified, might carry most of the SUDEP burden and suffer an annual risk of up to 2–5% between the age of 20 and 40. Identifying these patients would dramatically reduce the number needed for recruitment in a SUDEP prevention RCT, down to 2420 patients using the same figures as those previously considered (1-year follow-up; 50% reduction of SUDEP rate; α = 0.05, 1−β = 0.9). Selection of patients at very high risk of SUDEP should also promote participation of patients to be recruited in such a trial.
The main risk factor for SUDEP is the presence and frequency of generalized tonic-clonic seizures with an odds ratio of above 15 for patients with ≥3 GTCS per month.115, 116 This figure was calculated from mixed populations of patients seen at epilepsy centres and community-based cohorts of prevalent epilepsy,115 both of which suffer SUDEP rates between 1 and 2/1000 patient-years.117 Considering that 12% of the controls suffered from more than three GTCS per year,115 one can extrapolate that the subgroup of patients with ≥ 3 GTCS/year suffer an annual rate of SUDEP grossly ranging from 5 to 18/1000 patient-years. Thus, we need to identify other biomarkers to select patients with the greatest risk of SUDEP. Those currently considered include the occurrence of nocturnal seizures,118 prolonged post-ictal EEG suppression, ictal/post-ictal hypoxemia, mood disorders and other biomarkers of serotonergic dysfunction, and gene polymorphisms.22, 119 For some of these biomarkers, development of medical devices and biosensors that will collect such information in ambulatory patients might be a prerequisite to the demonstration of their predictive value.
Determining a safe SUDEP prevention strategy with high likelihood of success
Another way to promote the feasibility of RCTs for SUDEP prevention is to increase the effect size of an intervention. Targeting an 80% reduction in SUDEP rate, rather than 50%, would reduce the number of patients to be recruited from 2420 to 760, using the same parameters as above (patients with 5% annual risk; α = 0.05, 1−β = 0.9). While such an ambitious objective might sound unrealistic, it is supported by recently published data. Indeed, a meta-analysis of all double-blind randomised placebo controlled trials performed in adult patients with refractory epilepsy showed that patients receiving an add-on AED had an 87% lower risk of SUDEP (0.9/1000 patient-years) than those receiving placebo on top of their baseline AED treatment (6.9/1000 patient-years) during the brief double-blind phase of clinical trials.114 If simply adding an AED can reduce the risk of SUDEP to such an extent, one can hope that an intervention specifically tackling the pathophysiology of SUDEP might prove as effective.
Potential prophylactic and peri-ictal pharmacological SUDEP intervention strategies
Interventions to be tested in RCTs for SUDEP prevention should first be proved to be highly potent in pre-clinical and proof-of-concept studies. These interventions should also be proved to be reasonably safe, especially regarding their impact on seizure frequency and severity. As discussed in previous sections, targeting mechanisms involved in seizure termination, such as adenosine release, might carry the risk of aggravating seizures. Non-pharmacological interventions are discussed by Rugg-Gunn et al in this supplement120 as is the role of education (Donner et al).121 Here we highlight potential pharmacological interventions, prophylactic and peri-ictal.
SSRIs
A bulk of experimental data suggest that potentiation of serotonergic neurotransmission is associated with antiepileptic properties.122–128 Several studies evaluated the impact of SSRIs on seizure frequency in patients with epilepsy. In a pilot open-label study of fluoxetine in 17 patients with drug resistant focal seizures and no depression, six became seizure-free for a mean period of 14 months, while the remaining patients demonstrated a 30% mean reduction in seizure frequency.129 Three other series evaluated the risk of seizure aggravation in a total of 181 patients with epilepsy and comorbid depression treated with either sertraline, citalopram, or fluoxetine for a mean period of 4, 10 and 25 months.130–132 Only two, both treated with sertraline, reported a significant increase in seizure frequency130, 132. In one of these studies, seizure frequency decreased in 39 of 45 treated patients.131 Overall, using SSRIs in patients with focal epilepsy at risk of SUDEP appears safe, regardless of whether or not patients are depressed.
Adenosine receptor antagonists and promoters of adenosine clearance
Conversely, the therapeutic use of the non-selective adenosine receptor antagonist caffeine may be a double-edged sword. Caffeine (as all methylxanthines) may exacerbate seizures via blockade of the ‘anticonvulsant’ A1Rs99 in the limbic system. In patients with epilepsy, case reports also suggest that caffeine and aminophylline might increase seizure frequency.133 Caffeine was also found to potentiate the length of seizures triggered by electroconvulsive therapy.134 The use of region-selective or receptor subtype selective (e.g. A2AR vs. A1R) agents may capitalize on the anti-SUDEP effects of adenosine antagonists while avoiding any proconvulsant effects. An innovative strategy might be the enhancement of metabolic clearance of adenosine. Human patients with severe combined immunodeficiency syndrome (SCID) lack ADA and receive a stabilized form of the enzyme (PEG-ADA) as an enzyme replacement therapy to prevent this otherwise lethal condition.135, 136 PEG-ADA therapy might be sufficient to enhance the metabolic clearance of adenosine to avoid overactivation of adenosine receptors in the brainstem. This extremely expensive treatment currently requires intra-muscular injection.
Importantly, adenosine homeostasis in the brain is influenced by factors such as diet, sleep, and exercise.75 It would thus be of considerable interest to investigate the potential role of such lifestyle choices as modifiers of SUDEP risk.
Opioid receptor antagonists
Convincing evidence points to a role for opioids in post-ictal seizure inhibition. Potentiation of endogenous anti-ictal mechanisms by opioids has been shown in animal models of epilepsy.137 Kainic acid-induced seizures elicit dynorphin release in rodent hippocampus and activation of presynaptic kappa-opioid receptors (KOR) inhibits glutamate release and limits the spread of excitability in this region.138, 139 Similarly, several reports indicate that enkephalin release is induced by epileptiform activity140 and that mu-opioid receptor (MOR) activation can induce anticonvulsant effects.141 In humans, ictal and immediate post-ictal modifications of opioid receptor binding have been demonstrated using [11C]diprenorphine positron emission tomography in patients with reading-induced epilepsy,142 absence epilepsy143 and temporal lobe epilepsy,144, 145 suggesting an increase of synaptic opioid levels at the time of seizures. Overall, there are convincing data to conclude that endogenous opioids are released during focal and generalized seizures,146, 147 suggesting that the latter might also contribute to the pathophysiology of SUDEP.119 Indeed, the impact of opioids on the control of respiration is well known148 and respiratory depression is one of the cardinal symptoms of opioid overdose.149 Respiratory patterns in patients exposed to high doses of opioids demonstrate alterations of rhythmic breathing, with irregular respiratory patterns and/or transient apnea reminiscent of that observed in monitored SUDEP.148 This effect is related to direct inhibition of respiratory neurons by activation of opioid receptors.148, 150
Opioid-mediated respiratory distress can be very rapidly reversed by application of naloxone, an antagonist of opioid receptors, leading to patient awakening between 30 seconds and 2 minutes post-injection.149 There is no major safety issue reported with naloxone, including no risk of seizures. Naltrexone is another antagonist of MOR and KOR used in the chronic management of alcohol dependence and opioid dependence since 1994.151, 152 It is usually given orally at a standard dose of 50 to 100 mg/day,151 and an intra-muscular extended-release formulation is also available.153 As with naloxone, no life-threatening side effects have been reported with naltrexone,151 including no epilepsy-related alert in patients with chronic alcoholism who suffer a high risk of seizures.154–156 However, neither naloxone nor naltrexone has been specifically investigated in patients with epilepsy.149, 151
Proof-of-concept studies in patients with refractory epilepsy
The current stage of investigation of SUDEP prevention in patients with epilepsy consists of testing interventions on ancillary biomarkers, in particular peri-ictal hypoxemia. This strategy allows testing the potential of such interventions on much smaller populations. However, the predictive value of peri-ictal hypoxemia on the risk of SUDEP remains to be established.119
The effect of SSRIs on peri-ictal respiratory function was recently examined in epilepsy patients admitted to an EMU. Patients were divided into two groups based on whether they were taking any SSRI on admission. Those on an SSRI had significantly less frequent peri-ictal hypoxemia below 85% than those who were not (6% compared to 20%).6 Interestingly, there was no difference in peri-ictal apnea between groups. However, this was not a randomized trial. Patients on an SSRI probably had an indication for it, such as depression. There is evidence that there is overlap between the pathology of epilepsy and depression, and that both may involve the 5-HT system.12, 157, 158 Thus, there were likely differences between the two groups in 5-HT signaling at baseline. Furthermore, differences in peri-ictal hypoxemia between groups were only observed for partial seizures without secondary generalization, and not for generalized tonic-clonic seizures, which usually trigger SUDEP.6
In order to further investigate the potential of SSRIs in preventing peri-ictal hypoxemia, two double-blind RCTs have been launched, including one in the US that failed to achieve appropriate recruitment (https://clinicaltrials.gov/ct2/show/NCT00986310?term=SUDEP&rank=7), and another led by one of the co-authors (PR) that is still ongoing.
Using a similar approach in patients undergoing video-EEG monitoring coupled with pulse oximetry, a recently approved double-blind RCT will test the impact of naloxone 0.4 mg versus placebo, injected immediately following a generalized tonic-clonic seizure, on the occurrence, duration and intensity of post-ictal hypoxemia (PI S Rheims, Lyon, France). To the best of our knowledge, no clinical study testing the impact of adenosine modulation has yet been performed.
Figure 1.
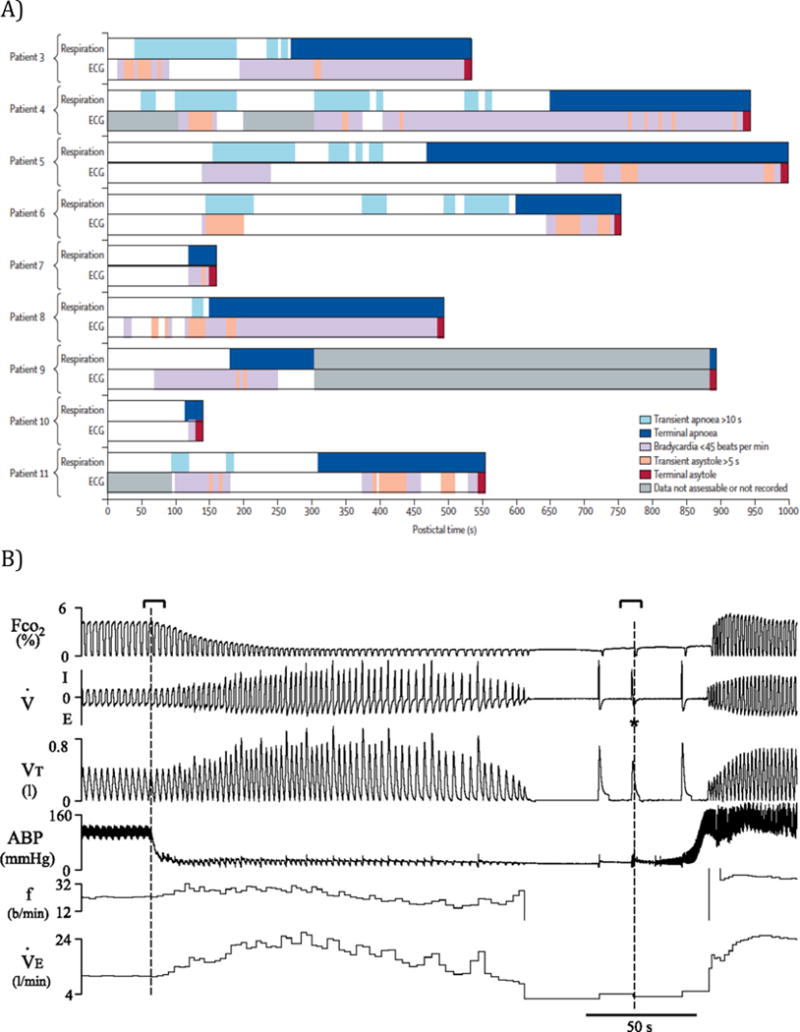
Figure 2.
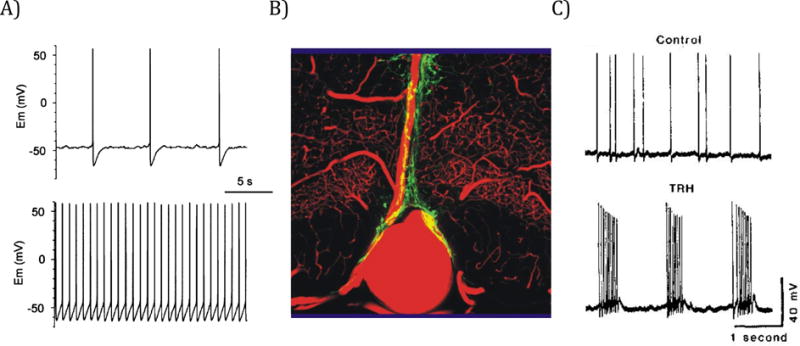
Figure 3.
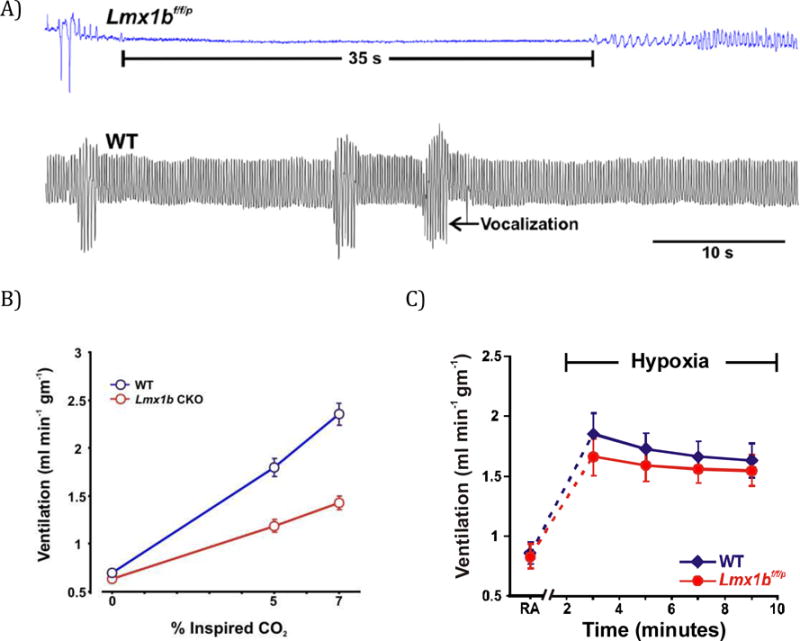
Figure 4.
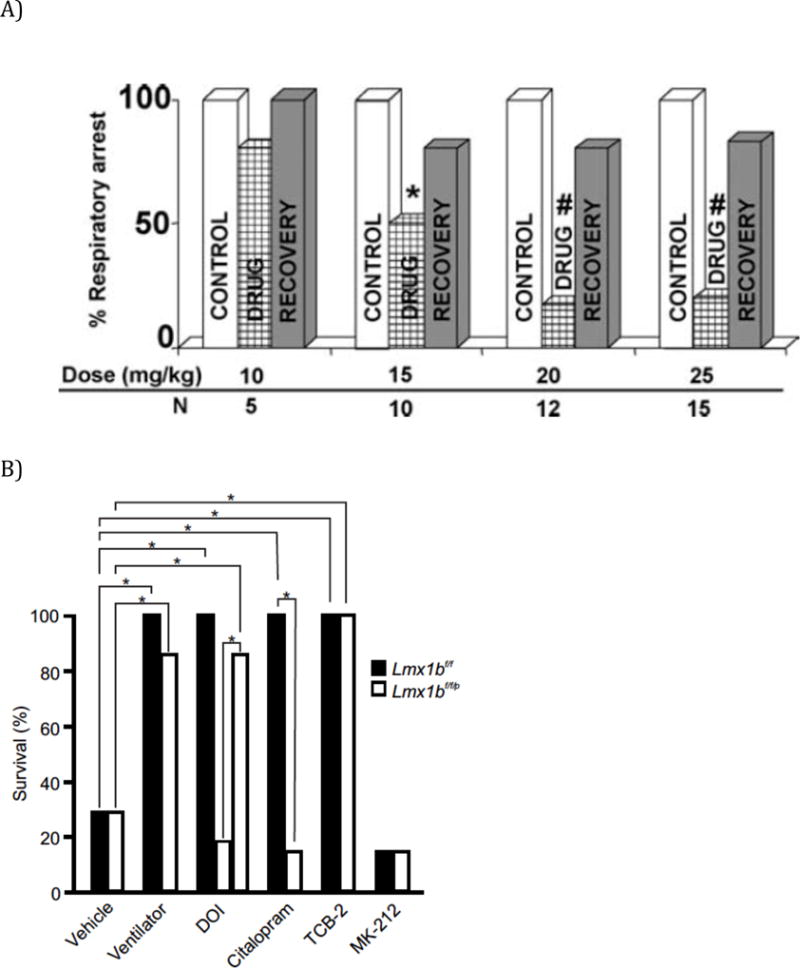
Figure 5.
Key points.
Seizures immediately preceding SUDEP often lead to rapidly developing hypoventilation and bradycardia.
Peri-ictal apnea/hypoventilation plays a significant role in SUDEP. Brainstem serotonin, adenosine and opioid pathways may be involved.
Potential pharmacological prevention strategies influencing the above pathways can be tested in randomised clinical trials.
For such trials to be feasible, identification of biomarkers in high risk individuals is needed.
Interventions tested need to safe with a high likelihood of success as shown by animal studies and surrogate clinical scenarios as in epilepsy monitoring units
Acknowledgments
The authors were supported through grants from the NINDS (DB: NS061844 & NS065957; GBR: P20NS076916 & U54NS041069), the NICHD (GBR: P01HD36379 & R01HD052772) the US Department of the Army (DB: W81XWH-12-1-0283), Citizens United for Research in Epilepsy (CURE; DB) & CURE (CF: Collaborative Grant & Chris Donalty Grant), the Legacy Hospital Foundations (DB), and the Epilepsy Foundation (CF).
CF wishes to thank Marcus Randall, Sri Tupal, and Srinivasa P. Kommajosyula, for their contributions, and Gayle Stauffer for her manuscript assistance.
Footnotes
Disclosure of conflicts of Interest
None of the authors has any conflicts of interest to disclose.
Ethical publication statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Ryvlin P, Nashef L, Lhatoo SD, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12:966–977. doi: 10.1016/S1474-4422(13)70214-X. [DOI] [PubMed] [Google Scholar]
- 2.Lhatoo SD, Faulkner HJ, Dembny K, et al. An electroclinical case-control study of sudden unexpected death in epilepsy. Ann Neurol. 2010;68:787–796. doi: 10.1002/ana.22101. [DOI] [PubMed] [Google Scholar]
- 3.Buchanan GF, Richerson GB. Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci U S A. 2010;107:16354–16359. doi: 10.1073/pnas.1004587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massey CA, Sowers LP, Dlouhy BJ, et al. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol. 2014;10:271–282. doi: 10.1038/nrneurol.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sowers LP, Massey CA, Gehlbach BK, et al. Sudden unexpected death in epilepsy: fatal post-ictal respiratory and arousal mechanisms. Respir Physiol Neurobiol. 2013;189:315–323. doi: 10.1016/j.resp.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bateman LM, Li CS, Lin TC, et al. Serotonin reuptake inhibitors are associated with reduced severity of ictal hypoxemia in medically refractory partial epilepsy. Epilepsia. 2010;51:2211–2214. doi: 10.1111/j.1528-1167.2010.02594.x. [DOI] [PubMed] [Google Scholar]
- 7.Boison D, Chen JF, Fredholm BB. Adenosine signaling and function in glial cells. Cell Death and Differentiation. 2010;17:1071–1082. doi: 10.1038/cdd.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tecott LH, Sun LM, Akana SF, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374:542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- 9.Tupal S, Faingold CL. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia. 2006;47:21–26. doi: 10.1111/j.1528-1167.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 10.Hirsch LJ, Donner EJ, So EL, et al. Abbreviated report of the NIH/NINDS workshop on sudden unexpected death in epilepsy. Neurology. 2011;76:1932–1938. doi: 10.1212/WNL.0b013e31821de7de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinney HC, Richerson GB, Dymecki SM, et al. The brainstem and serotonin in the sudden infant death syndrome. Annual Review of Pathology-Mechanisms of Disease. 2009;4:517–550. doi: 10.1146/annurev.pathol.4.110807.092322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richerson GB, Buchanan GF. The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia. 2011;52:28–38. doi: 10.1111/j.1528-1167.2010.02908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinney HC, McDonald AG, Minter ME, et al. Witnessed sleep-related seizure and sudden unexpected death in infancy: a case report. Forensic Science Medicine and Pathology. 2013;9:418–421. doi: 10.1007/s12024-013-9448-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez ML, McMillan K, Crandall LA, et al. Hippocampal asymmetry and sudden unexpected death in infancy: a case report. Forensic Science Medicine and Pathology. 2012;8:441–446. doi: 10.1007/s12024-012-9367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faingold CL, Randall M, Mhaskar Y, et al. Differences in serotonin receptor expression in the brainstem may explain the differential ability of a serotonin agonist to block seizure-induced sudden death in DBA/2 vs. DBA/1 mice. Brain Res. 2011;1418:104–110. doi: 10.1016/j.brainres.2011.08.043. [DOI] [PubMed] [Google Scholar]
- 16.Faingold CL, Tupal S, Randall M. Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy & Behavior. 2011;22:186–190. doi: 10.1016/j.yebeh.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Kinney HC, Filiano JJ, Sleeper LA, et al. Decreased muscarinic receptor-binding in the arcuate nucleus in sudden-infant-death-syndrome. Science. 1995;269:1446–1450. doi: 10.1126/science.7660131. [DOI] [PubMed] [Google Scholar]
- 18.Paterson DS, Trachtenberg FL, Thompson EG, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA. 2006;296:2124–2132. doi: 10.1001/jama.296.17.2124. [DOI] [PubMed] [Google Scholar]
- 19.Richerson GB. Sudden infant death syndrome: The role of central chemosensitivity. Neuroscientist. 1997;3:3–7. [Google Scholar]
- 20.Uteshev VV, Tupal S, Mhaskar Y, et al. Abnormal serotonin receptor expression in DBA/2 mice associated with susceptibility to sudden death due to respiratory arrest. Epilepsy Res. 2010;88:183–188. doi: 10.1016/j.eplepsyres.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Surges R, Thijs RD, Tan HL, et al. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol. 2009;5:492–504. doi: 10.1038/nrneurol.2009.118. [DOI] [PubMed] [Google Scholar]
- 22.Goldman AM, Cooper PN, Behr ER, et al. SUDEP genetics: Molecular diagnostics and prevention. Epilepsia. 2015 doi: 10.1111/epi.13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bateman LM, Spitz M, Seyal M. Ictal hypoventilation contributes to cardiac arrhythmia and SUDEP: report on two deaths in video-EEG-monitored patients. Epilepsia. 2010;51:916–920. doi: 10.1111/j.1528-1167.2009.02513.x. [DOI] [PubMed] [Google Scholar]
- 24.Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry. 2000;68:211–213. doi: 10.1136/jnnp.68.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lear-Kaul KC, Coughlin L, Dobersen MJ. Sudden unexpected death in epilepsy: a retrospective study. Am J Forensic Med Pathol. 2005;26:11–17. doi: 10.1097/01.paf.0000154453.58795.18. [DOI] [PubMed] [Google Scholar]
- 26.Jackson JH. On asphyxia in slight epileptic paroxysms. Lancet. 1899;153:79–80. [Google Scholar]
- 27.Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. 2008;131:3239–3245. doi: 10.1093/brain/awn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nashef L, Walker F, Allen P, et al. Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry. 1996;60:297–300. doi: 10.1136/jnnp.60.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bird JM, Dembny AT, Sandeman D, et al. Sudden unexplained death in epilepsy: an intracranially monitored case. Epilepsia. 1997;38(suppl 11):S52–S56. [Google Scholar]
- 30.Lee MA. Book EEG video recording of sudden unexpected death in epilepsy (SUDEP) Lippincott, Williams & Wilkins; San Diego, CA: 1998. EEG video recording of sudden unexpected death in epilepsy (SUDEP) pp. S123–124. Editor (Ed)^(Eds) [Google Scholar]
- 31.McLean BN, Wimalaratna S. Sudden death in epilepsy recorded in ambulatory EEG. J Neurol Neurosurg Psychiatry. 2007;78:1395–1397. doi: 10.1136/jnnp.2006.088492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Purves SJ, Wilson-Young M, Sweeney VP. Sudden death in epilepsy: single case report with video-EEG documentation. Epilepsia. 1992;33(suppl 3):123. [Google Scholar]
- 33.Tao JX, Qian S, Baldwin M, et al. SUDEP, suspected positional airway obstruction, and hypoventilation in postictal coma. Epilepsia. 2010;51:2344–2347. doi: 10.1111/j.1528-1167.2010.02719.x. [DOI] [PubMed] [Google Scholar]
- 34.Haouzi P, Ahmadpour N, Bell HJ, et al. Breathing patterns during cardiac arrest. J Appl Physiol. 2010;109:405–411. doi: 10.1152/japplphysiol.00093.2010. [DOI] [PubMed] [Google Scholar]
- 35.Dahlstrom A, Fuxe K. Evidence for existence of monoamine-containing neurons in central nervous system. I. Demonstration of monoamines in cell bodies of brain stem neurons. Acta Physiol Scand. 1964;62:1–55. [PubMed] [Google Scholar]
- 36.Jacobs BL, Azmitia EC. Structure and function of the brain serotonin system. Physiol Rev. 1992;72:165–229. doi: 10.1152/physrev.1992.72.1.165. [DOI] [PubMed] [Google Scholar]
- 37.Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci. 2004;5:449–461. doi: 10.1038/nrn1409. [DOI] [PubMed] [Google Scholar]
- 38.Iceman KE, Richerson GB, Harris MB. Medullary serotonin neurons are CO2 sensitive in situ. J Neurophysiol. 2013;110:2536–2544. doi: 10.1152/jn.00288.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richerson GB. Response to CO2 of neurons in the rostral ventral medulla in vitro. J Neurophysiol. 1995;73:933–944. doi: 10.1152/jn.1995.73.3.933. [DOI] [PubMed] [Google Scholar]
- 40.Wang W, Pizzonia JH, Richerson GB. Chemosensitivity of rat medullary raphe neurones in primary tissue culture. J Physiol. 1998;511(Pt 2):433–450. doi: 10.1111/j.1469-7793.1998.433bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W, Tiwari JK, Bradley SR, et al. Acidosis-stimulated neurons of the medullary raphe are serotonergic. J Neurophysiol. 2001;85:2224–2235. doi: 10.1152/jn.2001.85.5.2224. [DOI] [PubMed] [Google Scholar]
- 42.Wang W, Bradley SR, Richerson GB. Quantification of the response of rat medullary raphe neurones to independent changes in pH(o) and P(CO2) J Physiol. 2002;540:951–970. doi: 10.1113/jphysiol.2001.013443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernard DG, Li A, Nattie EE. Evidence for central chemoreception in the midline raphe. J Appl Physiol. 1996;80:108–115. doi: 10.1152/jappl.1996.80.1.108. [DOI] [PubMed] [Google Scholar]
- 44.Hodges MR, Richerson GB. Medullary serotonin neurons and their roles in central respiratory chemoreception. Respir Physiol Neurobiol. 2010;173:256–263. doi: 10.1016/j.resp.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hodges MR, Tattersall GJ, Harris MB, et al. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. Journal of Neuroscience. 2008;28:2495–2505. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ray RS, Corcoran AE, Brust RD, et al. Impaired respiratory and body temperature control upon acute serotonergic neuron inhibition. Science. 2011;333:637–642. doi: 10.1126/science.1205295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teran FA, Massey CA, Richerson GB. Serotonin neurons and central respiratory chemoreception: where are we now? Prog Brain Res. 2014;209:207–233. doi: 10.1016/B978-0-444-63274-6.00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Severson CA, Wang W, Pieribone VA, et al. Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci. 2003;6:1139–1140. doi: 10.1038/nn1130. [DOI] [PubMed] [Google Scholar]
- 49.Bradley SR, Pieribone VA, Wang W, et al. Chemosensitive serotonergic neurons are closely associated with large medullary arteries. Nat Neurosci. 2002;5:401–402. doi: 10.1038/nn848. [DOI] [PubMed] [Google Scholar]
- 50.Dekin MS, Richerson GB, Getting PA. Thyrotropin-releasing hormone induces rhythmic bursting in neurons of the nucleus tractus solitarius. Science. 1985;229:67–69. doi: 10.1126/science.3925552. [DOI] [PubMed] [Google Scholar]
- 51.Ptak K, Yamanishi T, Aungst J, et al. Raphe neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. Journal of Neuroscience. 2009;29:3720–3737. doi: 10.1523/JNEUROSCI.5271-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Depuy SD, Kanbar R, Coates MB, et al. Control of breathing by raphe obscurus serotonergic neurons in mice. J Neurosci. 2011;31:1981–1990. doi: 10.1523/JNEUROSCI.4639-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hendricks TJ, Fyodorov DV, Wegman LJ, et al. Pet-1 ETS gene plays a critical role in 5-HT neuron development and is required for normal anxiety-like and aggressive behavior. Neuron. 2003;37:233–247. doi: 10.1016/s0896-6273(02)01167-4. [DOI] [PubMed] [Google Scholar]
- 54.Ding YQ, Marklund U, Yuan W, et al. Lmx1b is essential for the development of serotonergic neurons. Nat Neurosci. 2003;6:933–938. doi: 10.1038/nn1104. [DOI] [PubMed] [Google Scholar]
- 55.Zhao ZQ, Scott M, Chiechio S, et al. Lmx1b is required for maintenance of central serotonergic neurons and mice lacking central serotonergic system exhibit normal locomotor activity. J Neurosci. 2006;26:12781–12788. doi: 10.1523/JNEUROSCI.4143-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hodges MR, Wehner M, Aungst J, et al. Transgenic mice lacking serotonin neurons have severe apnea and high mortality during development. Journal of Neuroscience. 2009;29:10341–10349. doi: 10.1523/JNEUROSCI.1963-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brennan TJ, Seeley WW, Kilgard M, et al. Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet. 1997;16:387–390. doi: 10.1038/ng0897-387. [DOI] [PubMed] [Google Scholar]
- 58.Purpura DP. Experimental models of epilepsy–a manual for the laboratory worker. Raven Press; New York: 1972. [Google Scholar]
- 59.Venit EL, Shepard BD, Seyfried TN. Oxygenation prevents sudden death in seizure-prone mice. Epilepsia. 2004;45:993–996. doi: 10.1111/j.0013-9580.2004.02304.x. [DOI] [PubMed] [Google Scholar]
- 60.Buchanan GF, Murray NM, Hajek MA, et al. Serotonin neurones have anticonvulsant effects and reduce seizure-induced mortality. J Physiol. 2014;592:4395–4410. doi: 10.1113/jphysiol.2014.277574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krous HF, Beckwith JB, Byard RW, et al. Sudden infant death syndrome and unclassified sudden infant deaths: A definitional and diagnostic approach. Pediatrics. 2004;114:234–238. doi: 10.1542/peds.114.1.234. [DOI] [PubMed] [Google Scholar]
- 62.Willinger M, Hoffman HJ, Hartford RB. Infant sleep position and risk for sudden infant death syndrome: report of meeting held January 13 and 14, 1994, National Institutes of Health, Bethesda, MD. Pediatrics. 1994;93:814–819. [PubMed] [Google Scholar]
- 63.Tomson T, Surges R, Delamont R, et al. Who to target in SUDEP prevention and how? Risk factors, biomarkers, and intervention study designs. Epilepsia. 2015 doi: 10.1111/epi.13234. [DOI] [PubMed] [Google Scholar]
- 64.Hunt CE, Brouillette RT. Sudden infant death syndrome: 1987 perspective. J Pediatr. 1987;110:669–678. doi: 10.1016/s0022-3476(87)80001-x. [DOI] [PubMed] [Google Scholar]
- 65.Filiano JJ, Choi JC, Kinney HC. Candidate cell populations for respiratory chemosensitive fields in the human infant medulla. J Comp Neurol. 1990;293:448–465. doi: 10.1002/cne.902930308. [DOI] [PubMed] [Google Scholar]
- 66.Filiano JJ, Kinney HC. Arcuate nucleus hypoplasia in the sudden infant death syndrome. J Neuropathol Exp Neurol. 1992;51:394–403. doi: 10.1097/00005072-199207000-00002. [DOI] [PubMed] [Google Scholar]
- 67.Panigrahy A, Filiano J, Sleeper LA, et al. Decreased serotonergic receptor binding in rhombic lip-derived regions of the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol. 2000;59:377–384. doi: 10.1093/jnen/59.5.377. [DOI] [PubMed] [Google Scholar]
- 68.Duncan JR, Paterson DS, Hoffman JM, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. 2010;303:430–437. doi: 10.1001/jama.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nashef L, So EL, Ryvlin P, et al. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia. 2012;53:227–233. doi: 10.1111/j.1528-1167.2011.03358.x. [DOI] [PubMed] [Google Scholar]
- 70.Liebenthal JA, Wu S, Rose S, et al. Association of prone position with sudden unexpected death in epilepsy. Neurology. 2015;84(7):643–644. doi: 10.1212/WNL.0000000000001260. [DOI] [PubMed] [Google Scholar]
- 71.Watanabe K, Hara K, Hakamada S, et al. Seizures with apnea in children. Pediatrics. 1982;70:87–90. [PubMed] [Google Scholar]
- 72.Kinney HC, Armstrong DL, Chadwick AE, et al. Sudden death in toddlers associated with developmental abnormalities of the hippocampus: A report of five cases. Pediatric and Developmental Pathology. 2007;10:208–223. doi: 10.2350/06-08-0144.1. [DOI] [PubMed] [Google Scholar]
- 73.Kinney HC, Chadwick AE, Crandall LA, et al. Sudden death, febrile seizures, and hippocampal and temporal lobe maldevelopment in toddlers: A New Entity. Pediatric and Developmental Pathology. 2009;12:455–463. doi: 10.2350/08-09-0542.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boison D. Adenosine and seizure termination: Endogenous mechanisms. Epilepsy Currents. 2013;13:35–U70. doi: 10.5698/1535-7511-13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boison D, Sandau US, Ruskin DN, et al. Homeostatic control of brain function - new approaches to understand epileptogenesis. Frontiers in Cellular Neuroscience. 2013;7 doi: 10.3389/fncel.2013.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Diogenes MJ, Neves-Tome R, Fucile S, et al. Homeostatic control of synaptic activity by endogenous adenosine is mediated by adenosine kinase. Cereb Cortex. 2014;24:67–80. doi: 10.1093/cercor/bhs284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berman RF, Fredholm BB, Aden U, et al. Evidence for increased dorsal hippocampal adenosine release and metabolism during pharmacologically induced seizures in rats. Brain Res. 2000;872:44–53. doi: 10.1016/s0006-8993(00)02441-0. [DOI] [PubMed] [Google Scholar]
- 78.During MJ, Spencer DD. Adenosine - a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol. 1992;32:618–624. doi: 10.1002/ana.410320504. [DOI] [PubMed] [Google Scholar]
- 79.Fukumitsu N, Ishii K, Kimura Y, et al. Adenosine A1 receptor mapping of the human brain by PET with 8-dicyclopropylmethyl-1-11C-methyl-3-propylxanthine. J Nucl Med. 2005;46:32–37. [PubMed] [Google Scholar]
- 80.Lopes LV, Sebastiao AM, Ribeiro JA. Adenosine and related drugs in brain diseases: present and future in clinical trials. Curr Top Med Chem. 2011;11:1087–1101. doi: 10.2174/156802611795347591. [DOI] [PubMed] [Google Scholar]
- 81.Paul S, Elsinga PH, Ishiwata K, et al. Adenosine A1 receptors in the central nervous system: Their functions in health and disease, and possible elucidation by PET imaging. Current Medicinal Chemistry. 2011;18:4820–4835. doi: 10.2174/092986711797535335. [DOI] [PubMed] [Google Scholar]
- 82.Pedata F, Corsi C, Melani A, et al. Adenosine extracellular brain concentrations and role of A2A receptors in ischemia. Neuroprotective Agents. 2001;939:74–84. doi: 10.1111/j.1749-6632.2001.tb03614.x. [DOI] [PubMed] [Google Scholar]
- 83.Clark RSB, Carcillo JA, Kochanek PM, et al. Cerebrospinal fluid adenosine concentration and uncoupling of cerebral blood flow and oxidative metabolism after severe head injury in humans. Neurosurgery. 1997;41:1284–1292. doi: 10.1097/00006123-199712000-00010. [DOI] [PubMed] [Google Scholar]
- 84.Boison D. Adenosine kinase: Exploitation for therapeutic gain. Pharmacological Reviews. 2013;65:906–943. doi: 10.1124/pr.112.006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fedele DE, Li T, Lan JQ, et al. Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol. 2006;200:184–190. doi: 10.1016/j.expneurol.2006.02.133. [DOI] [PubMed] [Google Scholar]
- 86.Lado FA, Moshe SL. How do seizures stop? Epilepsia. 2008;49:1651–1664. doi: 10.1111/j.1528-1167.2008.01669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dulla CG, Dobelis P, Pearson T, et al. Adenosine and ATP link P-CO2 to cortical excitability via pH. Neuron. 2005;48:1011–1023. doi: 10.1016/j.neuron.2005.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pagonopoulou O, Efthimiadou A, Asimakopoulos B, et al. Modulatory role of adenosine and its receptors in epilepsy: possible therapeutic approaches. Neurosci Res. 2006;56:14–20. doi: 10.1016/j.neures.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 89.Rebola N, Coelho JE, Costenla AR, et al. Decrease of adenosine A(1) receptor density and of adenosine neuromodulation in the hippocampus of kindled rats. European Journal of Neuroscience. 2003;18:820–828. doi: 10.1046/j.1460-9568.2003.02815.x. [DOI] [PubMed] [Google Scholar]
- 90.Vandam RJ, Shields EJ, Kelty JD. Rhythm generation by the pre-Botzinger complex in medullary slice and island preparations: effects of adenosine A(1) receptor activation. BMC Neurosci. 2008;9:95. doi: 10.1186/1471-2202-9-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zwicker JD, Rajani V, Hahn LB, et al. Purinergic modulation of preBotzinger complex inspiratory rhythm in rodents: the interaction between ATP and adenosine. J Physiol. 2011;589:4583–4600. doi: 10.1113/jphysiol.2011.210930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barraco RA, Janusz CA, Schoener EP, et al. Cardiorespiratory function is altered by picomole injections of 5′-N-ethylcarboxamidoadenosine into the nucleus tractus solitarius of rats. Brain Res. 1990;507:234–246. doi: 10.1016/0006-8993(90)90277-i. [DOI] [PubMed] [Google Scholar]
- 93.Evans DB, Schenden JA, Bristol JA. Adenosine receptors mediating cardiac depression. Life Sci. 1982;31:2425–2432. doi: 10.1016/0024-3205(82)90746-9. [DOI] [PubMed] [Google Scholar]
- 94.Herlenius E, Lagercrantz H, Yamamoto Y. Adenosine modulates inspiratory neurons and the respiratory patten in the brainstem of neonatal rats. Pediatr Res. 1997;42:46–53. doi: 10.1203/00006450-199707000-00008. [DOI] [PubMed] [Google Scholar]
- 95.Gourine AV, Kasymov V, Marina N, et al. Astrocytes control breathing through pH-dependent release of ATP. Science. 2010;329:571–575. doi: 10.1126/science.1190721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brown RE, Basheer R, McKenna JT, et al. Control of sleep and wakefulness. Physiol Rev. 2012;92:1087–1187. doi: 10.1152/physrev.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Angelatou F, Pagonopoulou O, Kostopoulos G. Changes in seizure latency correlate with alterations in A1 adenosine receptor-binding during daily repeated pentylentetrazol-induced convulsions in different mouse-brain areas. Neurosci Lett. 1991;132:203–206. doi: 10.1016/0304-3940(91)90302-a. [DOI] [PubMed] [Google Scholar]
- 98.Shen HY, Li T, Boison D. A novel mouse model for sudden unexpected death in epilepsy (SUDEP): role of impaired adenosine clearance. Epilepsia. 2010;51:465–468. doi: 10.1111/j.1528-1167.2009.02248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boison D. Methylxanthines, seizures, and excitotoxicity. Handb Exp Pharmacol. 2011:251–266. doi: 10.1007/978-3-642-13443-2_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lamberts RJ, Tan HL, Leijten QH, et al. Sudden unexpected death in epilepsy: SUDEP. Ned Tijdschr Geneeskd. 2013;157:A6193. [PubMed] [Google Scholar]
- 101.Nobili L, Proserpio P, Rubboli G, et al. Sudden unexpected death in epilepsy (SUDEP) and sleep. Sleep Med Rev. 2011;15:237–246. doi: 10.1016/j.smrv.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 102.Kim Y, Bolortuya Y, Chen LC, et al. Decoupling of sleepiness from sleep time and intensity during chronic sleep restriction: Evidence for a role of the adenosine system. Sleep. 2012;35:861–869. doi: 10.5665/sleep.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Porkka-Heiskanen T, Kalinchuk AV. Adenosine, energy metabolism and sleep homeostasis. Sleep Medicine Reviews. 2011;15:123–135. doi: 10.1016/j.smrv.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 104.Boison D. Adenosine dysfunction in epilepsy. Glia. 2012;60:1234–1243. doi: 10.1002/glia.22285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Etherington LA, Patterson GE, Meechan L, et al. Astrocytic adenosine kinase regulates basal synaptic adenosine levels and seizure activity but not activity-dependent adenosine release in the hippocampus. Neuropharmacology. 2009;56:429–437. doi: 10.1016/j.neuropharm.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Boison D, Scheurer L, Zumsteg V, et al. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci U S A. 2002;99:6985–6990. doi: 10.1073/pnas.092642899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lusardi TA, Lytle NK, Szybala C, et al. Caffeine prevents acute mortality after TBI in rats without increased morbidity. Exp Neurol. 2012;234:161–168. doi: 10.1016/j.expneurol.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gouder N, Fritschy JM, Boison D. Seizure suppression by adenosine A(1) receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia. 2003;44:877–885. doi: 10.1046/j.1528-1157.2003.03603.x. [DOI] [PubMed] [Google Scholar]
- 109.Gouder N, Scheurer L, Fritschy JM, et al. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. Journal of Neuroscience. 2004;24:692–701. doi: 10.1523/JNEUROSCI.4781-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Faingold CL, Randall M, Tupal S. DBA/1 mice exhibit chronic susceptibility to audiogenic seizures followed by sudden death associated with respiratory arrest. Epilepsy & Behavior. 2010;17:436–440. doi: 10.1016/j.yebeh.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 111.Faingold CL, Tupal S, Mhaskar Y, et al. DBA mice as models of sudden unexpected death in epilepsy. In: Lathers CM, Schraeder PL, Bungo MW, et al., editors. Sudden Death in Epilepsy: Forensic and Clinical Issues. CRC Press; Boca Raton, FL: 2010. pp. 659–676. [Google Scholar]
- 112.Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol. 2008;7:1021–1031. doi: 10.1016/S1474-4422(08)70202-3. [DOI] [PubMed] [Google Scholar]
- 113.Dasheiff RM. Sudden unexpected death in epilepsy: a series from an epilepsy surgery program and speculation on the relationship to sudden cardiac death. J Clin Neurophysiol. 1991;8:216–222. [PubMed] [Google Scholar]
- 114.Ryvlin P, Cucherat M, Rheims S. Risk of sudden unexpected death in epilepsy in patients given adjunctive antiepileptic treatment for refractory seizures: a meta-analysis of placebo-controlled randomised trials. Lancet Neurol. 2011;10:961–968. doi: 10.1016/S1474-4422(11)70193-4. [DOI] [PubMed] [Google Scholar]
- 115.Hesdorffer DC, Tomson T, Benn E, et al. Combined analysis of risk factors for SUDEP. Epilepsia. 2011;52:1150–1159. doi: 10.1111/j.1528-1167.2010.02952.x. [DOI] [PubMed] [Google Scholar]
- 116.Hesdorffer DC, Tomson T, Benn E, et al. Do antiepileptic drugs or generalized tonic-clonic seizure frequency increase SUDEP risk? A combined analysis. Epilepsia. 2012;53:249–252. doi: 10.1111/j.1528-1167.2011.03354.x. [DOI] [PubMed] [Google Scholar]
- 117.Shorvon S, Tomson T. Sudden unexpected death in epilepsy. Lancet. 2011;378:2028–2038. doi: 10.1016/S0140-6736(11)60176-1. [DOI] [PubMed] [Google Scholar]
- 118.Lamberts RJ, Thijs RD, Laffan A, et al. Sudden unexpected death in epilepsy: people with nocturnal seizures may be at highest risk. Epilepsia. 2012;53(2):253–257. doi: 10.1111/j.1528-1167.2011.03360.x. [DOI] [PubMed] [Google Scholar]
- 119.Ryvlin P, Nashef L, Tomson T. Prevention of sudden unexpected death in epilepsy: A realistic goal? Epilepsia. 2013;54:23–28. doi: 10.1111/epi.12180. [DOI] [PubMed] [Google Scholar]
- 120.Rugg-Gunn F, Duncan J, Hjalgrim H, et al. From unwitnessed fatality to witnessed rescue. Non-pharmacological unterventions. Epilepsia. 2015 doi: 10.1111/epi.13231. [DOI] [PubMed] [Google Scholar]
- 121.Donner EJ, Osland K, Leach JP, et al. The aftermath of SUDEP – Lessonslearned and the road forward. Epilepsia. 2015 [Google Scholar]
- 122.Browning RA, Wood AV, Merrill MA, et al. Enhancement of the anticonvulsant effect of fluoxetine following blockade of 5-HT1A receptors. Eur J Pharmacol. 1997;336:1–6. doi: 10.1016/s0014-2999(97)01215-6. [DOI] [PubMed] [Google Scholar]
- 123.Dailey JW, Yan QS, Mishra PK, et al. Effects of fluoxetine on convulsions and on brain serotonin as detected by microdialysis in genetically epilepsy-prone rats. J Pharmacol Exp Ther. 1992;260:533–540. [PubMed] [Google Scholar]
- 124.Raju SS, Noor AR, Gurthu S, et al. Effect of fluoxetine on maximal electroshock seizures in mice: Acute vs chronic administration. Pharmacological Research. 1999;39:451–454. doi: 10.1006/phrs.1999.0466. [DOI] [PubMed] [Google Scholar]
- 125.Wada Y, Hirao N, Shiraishi J, et al. Pindolol potentiates the effect of fluoxetine on hippocampal seizures in rats. Neurosci Lett. 1999;267:61–64. doi: 10.1016/s0304-3940(99)00321-3. [DOI] [PubMed] [Google Scholar]
- 126.Wada Y, Nakamura M, Hasegawa H, et al. Microinjection of the serotonin uptake inhibitor fluoxetine elevates hippocampal seizure threshold in rats. Neuroscience Research Communications. 1993;13:143–148. [Google Scholar]
- 127.Wada Y, Shiraishi J, Nakamura M, et al. Prolonged but not acute fluoxetine administration produces its inhibitory effect on hippocampal seizures in rats. Psychopharmacology (Berl) 1995;118:305–309. doi: 10.1007/BF02245959. [DOI] [PubMed] [Google Scholar]
- 128.Yan QS, Jobe PC, Dailey JW. Evidence that a serotonergic mechanism is involved in the anticonvulsant effect of fluoxetine in genetically epilepsy-prone rats. Eur J Pharmacol. 1994;252:105–112. doi: 10.1016/0014-2999(94)90581-9. [DOI] [PubMed] [Google Scholar]
- 129.Favale E, Rubino V, Mainardi P, et al. Anticonvulsant effect of fluoxetine in humans. Neurology. 1995;45:1926–1927. doi: 10.1212/wnl.45.10.1926. [DOI] [PubMed] [Google Scholar]
- 130.Kanner AM, Kozak AM, Frey M. The use of sertraline in patients with epilepsy: Is it safe? Epilepsy & Behavior. 2000;1:100–105. doi: 10.1006/ebeh.2000.0050. [DOI] [PubMed] [Google Scholar]
- 131.Specchio LM, Iudice A, Specchio N, et al. Citalopram as treatment of depression in patients with epilepsy. Clin Neuropharmacol. 2004;27:133–136. doi: 10.1097/00002826-200405000-00009. [DOI] [PubMed] [Google Scholar]
- 132.Thome-Souza MS, Kuczynski E, Valente KD. Sertraline and fluoxetine: safe treatments for children and adolescents with epilepsy and depression. Epilepsy Behav. 2007;10:417–425. doi: 10.1016/j.yebeh.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 133.Chroscinska-Krawczyk M, Radzik I, Miziak B, et al. Safety considerations for patients with epilepsy taking antiepileptic drugs alongside caffeine or other methylxanthine derivatives. Expert Opinion on Drug Metabolism & Toxicology. 2014;10:981–989. doi: 10.1517/17425255.2014.920822. [DOI] [PubMed] [Google Scholar]
- 134.Shapira B, Zohar J, Newman M, et al. Potentiation of seizure length and clinical response to electroconvulsive therapy by caffeine pretreatment: A case report. Convuls Ther. 1985;1:58–60. [PubMed] [Google Scholar]
- 135.Booth C, Gaspar HB. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID) Biologics. 2009;3:349–358. [PMC free article] [PubMed] [Google Scholar]
- 136.Hershfield MS. Peg-Ada replacement therapy for adenosine-deaminase deficiency - an Update after 8.5 Years. Clin Immunol Immunopathol. 1995;76:S228–S232. doi: 10.1016/s0090-1229(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 137.Tortella FC, Long JB, Holaday JW. Endogenous opioid systems: physiological role in the self-limitation of seizures. Brain Res. 1985;332:174–178. doi: 10.1016/0006-8993(85)90403-2. [DOI] [PubMed] [Google Scholar]
- 138.Loacker S, Sayyah M, Wittmann W, et al. Endogenous dynorphin in epileptogenesis and epilepsy: anticonvulsant net effect via kappa opioid receptors. Brain. 2007;130:1017–1028. doi: 10.1093/brain/awl384. [DOI] [PubMed] [Google Scholar]
- 139.Solbrig MV, Adrian R, Baratta J, et al. Kappa opioid control of seizures produced by a virus in an animal model. Brain. 2006;129:642–654. doi: 10.1093/brain/awl008. [DOI] [PubMed] [Google Scholar]
- 140.Rocha L, Maidment NT, Evans CJ, et al. Microdialysis reveals changes in extracellular opioid peptide levels in the amygdala induced by amygdaloid kindling stimulation. Exp Neurol. 1994;126:277–283. doi: 10.1006/exnr.1994.1065. [DOI] [PubMed] [Google Scholar]
- 141.Albertson TE, Joy RM, Stark LG. Modification of kindled amygdaloid seizures by opiate agonists and antagonists. Journal of Pharmacology and Experimental Therapeutics. 1984;228:620–627. [PubMed] [Google Scholar]
- 142.Koepp MJ, Richardson MP, Brooks DJ, et al. Focal cortical release of endogenous opioids during reading-induced seizures. Lancet. 1998;352:952–955. doi: 10.1016/s0140-6736(97)09077-6. [DOI] [PubMed] [Google Scholar]
- 143.Bartenstein PA, Duncan JS, Prevett MC, et al. Investigation of the opioid system in absence seizures with positron emission tomography. J Neurol Neurosurg Psychiatry. 1993;56:1295–1302. doi: 10.1136/jnnp.56.12.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hammers A, Asselin MC, Hinz R, et al. Upregulation of opioid receptor binding following spontaneous epileptic seizures. Brain. 2007;130:1009–1016. doi: 10.1093/brain/awm012. [DOI] [PubMed] [Google Scholar]
- 145.McGinnity CJ, Shidahara M, Feldmann M, et al. Quantification of opioid receptor availability following spontaneous epileptic seizures: correction of [11C]diprenorphine PET data for the partial-volume effect. Neuroimage. 2013;79:72–80. doi: 10.1016/j.neuroimage.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 146.Benarroch EE. Endogenous opioid systems: current concepts and clinical correlations. Neurology. 2012;79:807–814. doi: 10.1212/WNL.0b013e3182662098. [DOI] [PubMed] [Google Scholar]
- 147.Henriksen G, Willoch F. Imaging of opioid receptors in the central nervous system. Brain. 2008;131:1171–1196. doi: 10.1093/brain/awm255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pattinson KT. Opioids and the control of respiration. Br J Anaesth. 2008;100:747–758. doi: 10.1093/bja/aen094. [DOI] [PubMed] [Google Scholar]
- 149.Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367:146–155. doi: 10.1056/NEJMra1202561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Montandon G, Qin W, Liu H, et al. PreBotzinger complex neurokinin-1 receptor-expressing neurons mediate opioid-induced respiratory depression. J Neurosci. 2011;31:1292–1301. doi: 10.1523/JNEUROSCI.4611-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ray LA, Chin PF, Miotto K. Naltrexone for the treatment of alcoholism: Clinical findings, mechanisms of action, and pharmacogenetics. CNS & Neurological Disorders-Drug Targets. 2010;9:13–22. doi: 10.2174/187152710790966704. [DOI] [PubMed] [Google Scholar]
- 152.Tiihonen J, Krupitsky E, Verbitskaya E, et al. Naltrexone implant for the treatment of polydrug dependence: a randomized controlled trial. Am J Psychiatry. 2012;169:531–536. doi: 10.1176/appi.ajp.2011.11071121. [DOI] [PubMed] [Google Scholar]
- 153.Gastfriend DR. Intramuscular extended-release naltrexone: current evidence. Addiction Reviews. 2011;1216:144–166. doi: 10.1111/j.1749-6632.2010.05900.x. [DOI] [PubMed] [Google Scholar]
- 154.Krystal JH, Cramer JA, Krol WF, et al. Naltrexone in the treatment of alcohol dependence. New England Journal of Medicine. 2001;345:1734–1739. doi: 10.1056/NEJMoa011127. [DOI] [PubMed] [Google Scholar]
- 155.Samokhvalov AV, Irving H, Mohapatra S, et al. Alcohol consumption, unprovoked seizures, and epilepsy: A systematic review and meta-analysis. Epilepsia. 2010;51:1177–1184. doi: 10.1111/j.1528-1167.2009.02426.x. [DOI] [PubMed] [Google Scholar]
- 156.Volpicelli JR, Alterman AI, Hayashida M, et al. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- 157.Kanner AM. Can neurobiological pathogenic mechanisms of depression facilitate the development of seizure disorders? Lancet Neurol. 2012;11:1093–1102. doi: 10.1016/S1474-4422(12)70201-6. [DOI] [PubMed] [Google Scholar]
- 158.Lothe A, Didelot A, Hammers A, et al. Comorbidity between temporal lobe epilepsy and depression: a [18F]MPPF PET study. Brain. 2008;131:2765–2782. doi: 10.1093/brain/awn194. [DOI] [PMC free article] [PubMed] [Google Scholar]