

Abstract
Different technologies, such as quantitative real-time PCR or microarrays, have been developed to measure miRNA expression levels. Quantification of miRNA transcripts implicates data normalization using endogenous and exogenous reference genes for data correction. However, there is no consensus about an optimal normalization strategy. The choice of a reference gene remains to be problematic, and can have serious impact on the actual available transcript levels and consequently, on the biological interpretation of data. In this review article we intend to discuss the reliability of use of small RNAs commonly reported in the literature as miRNAs expression normalizers and to compare different normalization strategies used, setting the base for establishing a global standard of miRNAs expression data normalization.
Introduction
MicroRNAs (miRNAs) are small non-coding RNA transcripts of ~22 nucleotides length that exert an important regulatory role of cell activity at the transcriptional level. miRNAs are initially transcribed as immature pri-miRNAs, being processed in the cell nucleus and cytoplasm by Drosha and Dicer, respectively, and loaded into Ago proteins to form the RISC (RNA-induced silencing) complex (1). Mature functional miRNAs loaded in RISC interact with complementary sequences usually located at the 3′-UTR of mRNAs present in the cell cytoplasm, promoting mRNA cleavage or repressing their translation (2). Ultimately, this regulates gene expression by decreasing the production of effector proteins. MiRNAs can repress multiple targets within the same pathway resulting in amplification of their biological effects (3).
In physiological conditions, miRNAs regulate cell differentiation, cell proliferation and survival, metabolism, among many other functions (2). Additionally, disruption of their expression patterns implicates miRNAs in disease onset and progression (8), such as cancer (9), and their potential role as prognostic and predictive biomarkers in patient management has been described (10). Beyond the functions they exert in the cells that produce them, miRNAs may also be secreted and transferred to other cells, circulating in virtually all body fluids, either in protein complexes or enclosed inside extracellular vesicles, such as microvesicles and exosomes (11).
Since the collection of miRNAs produced by cells reflect their physiological state, these non-coding RNAs have been greatly explored as disease biomarkers (12). In this way, different methodologies have been applied to characterize qualitatively and quantitatively the miRNAs expression patterns associated with pathological versus normal conditions, such as quantitative real-time PCR (qPCR), microarray screening, northern blotting, ultra-high throughput miRNA sequencing (e.g., small RNA-seq, next generation sequencing), in situ hybridization with locked nucleic acids probes and hybridization in solution with tagged probes (e.g., nCounter® nanoString technology), among many others (13). To accurately determine the levels of analyzed miRNAs, their expression data is usually normalized relatively to endogenous and/or exogenous reference genes. However, different studies use different normalization strategies to report miRNA expression data. This leads to ambiguous data interpretation, misleading conclusions, and erroneous biological predicted effects, impairing comparisons between studies, and consequently none optimal normalization strategy seems to be consensual for the scientific community so far.
Technical challenges
The outcomes of miRNA analysis depend on several aspects of the whole process, beginning with the nature of the sample, the way it is collected, preserved and processed, the technical method applied for miRNA detection and the strategy followed for data normalization and analysis (14).
The accurate comparison of miRNA expression between samples requires that equal amounts of miRNAs are used as input for the detection method applied. Usually, the determination of the quantity of sample input for miRNA detection techniques is based on total RNA quantification, but the real proportion of miRNAs may vary from sample to sample, especially if they have a different origin. Besides, miRNAs integrity is hardly determined, with microfluidic capillary electrophoresis being currently the best method to assess miRNA quality, even with results being compromised by mRNA degradation in the samples (15).
miRNAs may be isolated by different methods from cultured cells, fresh tissues, frozen and fixed tissues, or as cell-free circulating RNAs from conditioned cell culture media and body fluids, for instance whole blood, plasma, serum, urine, cerebrospinal fluid, etc. (16–20). The collection of fresh human samples requires their preservation by different methods, as coagulation prevention, freezing, fixing and paraffin-embedding. The preservation process induces molecular changes that may lead to global miRNA instability or enrichment/depletion of specific miRNAs in the samples (21). As an example, Kim et al. demonstrated that the capacity to detect endogenous and exogenous miRNAs in plasma samples strongly depends on the method used to prevent blood coagulation (22). In the same way, Farina et al. showed that freeze and thawing cycles differently affected the levels of specific miRNAs in serum samples (23). Furthermore, the time selected for sample collection is also important when analyzing circulating miRNAs, since their physiological levels may vary according to the circadian rhythm, meal ingestion and overall lifestyle (for instance, smoking and drug consumption) (24, 25). Another aspect to take into account when analyzing the levels of miRNAs in pathological conditions is the variation of their levels according to disease stage and progression (26, 27), as well as medical interventions and treatments in course (28, 29).
Currently, the study of circulating miRNAs strongly focuses on RNA isolated from microvesicles and exosomes actively secreted by cells. For the particular case of exosomes, different methods can be employed for their isolation, being the most common ultracentrifugation (using or not a density gradient), filtration, size-exclusion chromatography and precipitation (using polymeric solutions or beads with immunoaffinity to a exosomal protein marker). Considering their working principle, the different methods lead to an enrichment of specific vesicle subpopulations that likely carry different cargo (25, 30). Of note, large and dense complexes of proteins associated with other biomolecules, such as RNA, have been detected as co-precipitants in exosome pellets isolated by ultracentrifugation. Consequently, miRNA analysis on these samples does not reflect the real intra-exosomal content. Size-exclusion chromatography has been arising as a good isolation alternative to circumvent this issue (31). Immunoaffinity-based methods have been widely used to isolate exosomes from body fluids, targeting proteins described as disease specific biomarkers and carried on vesicles surface. This strategy may bias the definition of specific circulating miRNAs as a disease biomarker, when relatively compared to miRNAs expression in exosomes isolated in healthy conditions by a method of diminished specificity (32). Conversely, disease-specific miRNAs biomarkers may be missed due to their absence in vesicles selected by a particular protein biomarker chosen (33).
Ultimately, the global analysis of miRNA expression, especially for the confident discovery and validation of disease biomarkers, strongly depends on the size of cohorts/sets of samples analyzed. Very frequently, only small-sized test populations are studied, leading to an erroneous or biased misidentification of biomarkers. In a recently published meta-analysis reviewing miRNA biomarker discovery in non-neoplasic diseases, it was revealed that the majority of the studies published rely on populations under 100 individuals (median size = 69 subjects) (34).
The methodology chosen for miRNA detection also influences the outcomes of miRNA quantification. Nowadays, the most used methodologies are qPCR and hybridization on microarray platforms, with the former being the gold standard for detection of specific sets of miRNAs of interest and the later mostly applied for large scale profiling.
Measurement of miRNAs by qPCR is very specific and sensitive, allowing the detection of very small quantities of miRNAs, is relatively inexpensive and commercial ready-to-use kits are widely available. The most common qPCR detection techniques are stem-loop shaped RT-primer Taqman assays (Applied Biosystems), assays using locked nucleic acid primers (Exiqon), and assays with poly-A tailing primers (QIAGEN, Stratagene). With these approaches, only a limited set of miRNAs can be tested in a single reaction, and detection is greatly influenced by the specificity of the primers designed (35).
Conversely, microarrays allow the probing of a large set of miRNAs simultaneously, nowadays at much more competitive costs, since several off-the-shelf platforms are commercially available from companies as Affimetrix, Agilent and Exiqon, among others. This technique requires an input of higher amounts of RNA, and assay performance may be sometimes compromised by hybridization conditions that are not optimal to the whole probes in test. Likewise, the design of the probes to include in the platform may reveal troublesome, since they have to be specific enough for the target miRNAs and, at the same time, share similarities in the conditions required for hybridization (36).
Both approaches have advantages and drawbacks associated, but one of the most striking pitfalls is the low correlation between different techniques. In a comparative study by Sato et al. (37), miRNAs in liver and prostate human tissues were profiled using microarray platforms from different companies and expression log-ratios were ranked. Surprisingly, the median rank correlation across platforms was only 0.55, being the highest correlation found of 0.87. Interestingly, the correlation between microarray- and Taqman-based expression data was higher, with a median correlation coefficient of 0.7, and only one of the platforms presenting a correlation coefficient lower than 0.5, supporting the common practice of using qPCR-based techniques as a confirmatory method for the validation of microarray expression data.
In an attempt to minimize the effect imposed by the factors previously described on miRNA expression levels, an accurate data analysis should be performed, using appropriate reference genes for external and internal variations correction (38).
Relative data normalization to endogenous reference genes
Quantification of miRNAs has come to the fore, and particularly produced a wealth of literature during the last five years (39). Here, we will focus on the issues that may arise with the data normalization in the gold-standard method RT-qPCR. There are two most commonly used methods to analyze data from real-time qPCR: absolute and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control (40). For the relative miRNA quantification, the PCR-derived cycle threshold (Cq) of target miRNAs is compared with that of a stably expressed endogenous miRNA from the same sample. The difference between these values is the ΔCq value (41). This normalization approach aims at removing differences due to sampling and quality of the samples. The guidelines for quality control and standardization of RT-qPCR imply the use of an optimal normalization method (42), however there are no universally accepted reference genes or so-called “housekeeping” transcripts for miRNA data normalization. This lack of consensus has resulted in various normalization strategies (43). Some studies had even the erroneous notion of completely abandon an endogenous normalization (44–48), and should of course be approached with some caution. Actually, it was reported that RT-qPCR can be used without endogenous controls to analyze miR-371-3 in the serum of patients with testicular cancer, if the technical procedure is performed under controlled conditions. This study compared the Cq and ΔCq values of miR-371a-3p, miR-372 and miR-373-3p by real-time PCR with and without 18S rRNA (ribosomal RNA) as a reference gene for data normalization. The miRNA transcript levels were measured in 25 testicular germ cell tumor patients, 4 non-testicular germ cell tumor patients and 17 age-matched male controls. A highly positive correlation between Cq and ΔCq values was found in all samples. The highest correlation was found for miR-371a-3p (R2: 0.956) (49). In spite of this positive correlation between Cq and ΔCq values detected in all serum samples by those authors (49), relative data quantification is absolutely indispensable to substantiate robust miRNA data (50). In this study (49), the detected significant correlation is related to the similar (good) quality and handling of the serum samples, but nobody can be sure that this is always the case. There is no evidence for a good sample quality, since total RNA can be partly degraded and bias the results. Therefore, miRNA quantifications without an internal control should be considered critically.
Fortunately, the majority of studies have carried out a relative quantification that is adapted to compare the expression levels of target genes with the expression of an endogenous reference gene. The choice of an endogenous reference gene is a critical step before starting an experiment, in order to avoid misinterpreted data and to identify true changes in miRNA expression levels. Usually, researchers select their endogenous reference gene for miRNA quantification according to reports in the literature or based on a distinguishable low standard deviation in miRNA microarrays. Considering the most recent and significant reports (Table 1), numerous reference genes, such as small nuclear/nucleolar RNAs, have been commonly used for miRNA quantification (51–54). These small RNA molecules share similar properties, such as RNA stability and size and are abundantly expressed. Although these transcripts display constant expression in single analyses, their expression levels can change under different experimental conditions and may be affected by specific types of disease (53). Therefore, the different normalizers should be first established for different sample types, and the combination of several normalizers might be more appropriate than a single universal normalizer (55).
RNU6 and other RNUs as normalizers
The small non-coding RNAs RNU6 (RNU6A) and RNU6B are the most frequently used reference genes as normalizers (Table 1). However, RNU6 is not a miRNA, and consequently, does not reflect the biochemical character of miRNA molecules in terms of their transcription, processing and tissue-specific expression patterns (56, 57). In addition, the efficiency of their extraction, reverse transcription, and PCR amplification may differ from that of miRNAs. It has been argued that it is best to normalize miRNAs with reference genes belonging to the same RNA class (43). In this regard, Gee et al. showed that the use of snRNAs (small nuclear RNAs) as reference genes can introduce bias when quantifying miRNA expression and that they are rather important in cancer prognosis. This study (57) measured RNU6B, RNU44, RNU48 and RNU43 for the data normalization in RT-qPCR analyzing breast cancer and head and neck squamous cell carcinoma (HNSCC) patients. The expression of these snRNAs was as variable as the expression of the target miRNAs miR-21, miR-210 and miR-10b, and data normalization to these recommended reference snRNAs introduced bias in the associations between miRNA and pathology or outcome (57). Using microarray-based serum miRNA profiling followed by RT-qPCR, Xiang et al. screened and compared the expression levels of reference RNAs in patients with different tumors and healthy controls. They found large fluctuations in RNU6 expression and a relatively stable expression of miR-16. The difference of ΔCq values of RNU6 between the highest and lowest expression level was 3.29, while that of miR-16 was 1.23. They also subjected the serum samples to different freeze-thaw cycles, and showed that RNU6 expression gradually decreased after 1, 2 and 4 cycles of freezing and thawing, while the expression of miR-16 and miR-24 remained relatively stable (58). Lamba et al. compared the stability of RNU6 and RNU6B in hepatic tissue, and found that both snRNAs were not suitable for the use as endogenous controls for normalizing miRNA data in this tumor type. They used Taqman-based RT-qPCR to quantify the expression levels of 22 miRNAs along with RNU6 and RNU6B in 50 human liver samples. Both software programs NormFinder (59) and GeNormplus identified RNU6 to be among the least stable of all candidate snRNAs analyzed, and RNU6B was also not among the top genes in stability. In their analyses, miR-152 and miR-23b were identified to be the two most stable candidates, and eligible as endogenous controls for data normalization (60). Benz et al. analyzed RNU6B levels in the serum samples of healthy volunteers, intensive care unit patients and patients with liver fibrosis. They demonstrated that serum RNU6B levels displayed a high variability between the cohorts, and consequently, were dysregulated in a disease-specific manner. Most importantly, the expression levels were significantly upregulated in the serum of patients with critical illness and sepsis compared with controls and correlated with established markers of inflammation. In contrast, in patients with liver fibrosis, RNU6B levels were significantly downregulated (56). Furthermore, Ratert et al. also showed that notably, RNU6B is unsuitable for miRNA normalization. Based on miRNA microarray data, a total of 16 miRNAs were identified as putative reference genes. After validation by RT-qPCR, RNU6B, RNU48, miR-101, miR-125a-5p, miR-148b, miR-151-5p, miR-181a, miR-181b, miR-29c, miR-324-3p, miR-424, miR-874 and Z30 were evaluated by the programs geNorm (61), NormFinder, and BestKeeper. These algorithms recommended the combinations of four (miR-101, miR-125a-5p, miR-148b, and miR-151-5p) and three (miR-148b, miR-181b, and miR-874) reference miRNAs for data normalization (62). In miRNA expression studies on renal cell carcinoma, RNU6B was also unsuitable as normalizer. Validation experiments were performed on four miRNAs (miR-28, miR-103, miR-106a, miR-151) together with RNU6B, RNU44, and RNU48. MiR-28, miR-103, miR-106a and RNU48 were proved as the most stably expressed genes, but RNU6B was differentially expressed. If only a single reference gene can be used, miR-28 was recommended as normalizer, while the combinations of miR-28 and miR-103 or of miR-28, miR-103, and miR-106a were preferred (63). Torres et al. investigated the expression of twelve candidate snRNAs (RNU6, RNU44, RNU48, RNU75, RNU54, RNU49, RNU6B, RNU38B, RNU18A, miR-16, miR-26b, miR-92a) in tissue samples of 30 endometrioid endometrial carcinoma patients and 15 normal endometrium samples using RT-qPCR. The stability of candidate endogenous controls was also evaluated using the algorithm programs and an equivalency test. The results were then validated using a larger number of samples. RNU48, RNU75 and RNU44 were identified as most stably and equivalently expressed snRNAs between malignant and normal tissues. Both programs NormFinder and geNorm indicated that these three snRNAs were optimal for RT-qPCR data normalization in endometrioid endometrial tissues. The authors suggested that the average values of these snRNAs could be used as a reliable endogenous control in studies on endometrioid endometrial cancer (64). In a study on Parkinson’s disease, RNU24 ranked top of the list of reference genes, followed by Z30. In contrast, miR-103a-3p was ranked as the worst reference gene so that in combination with other reference genes this miRNA was able to bias results. It is important to underline that miR-103a-3p alone or in combination with the other reference genes reversed the direction of the expression levels of the target miRNAs miR-29a-3p and miR-30b-5p. Also, RNU6B was not considered to be a reliable reference gene for Parkinson’s disease blood samples, because the efficiency, the r2 and the stability values were too low (51).
These findings suggest that RNU6 may be unsuitable as an endogenous reference gene in the research of miRNA quantification. In contrast, Han et al. recommended RNU6 as reference gene for the relative quantification of miRNA expression levels in pleural effusion. Following miRNA microarray, the expression levels of candidate reference miRNAs, together with RNU6B, RNU44 and RNU48 were validated in 46 benign pleural effusion samples and 45 lung adenocarcinoma-associated malignant pleural effusion samples by RT-qPCR, and verified using the NormFinder and BestKeeper algorithms. RNU6B, as well as miR-192, were identified as single reference genes and the combination of these genes was preferred for the relative quantification of miRNA expression levels in pleural effusion (65).
miR-16 as normalizer
Moreover, miR-16 is also frequently used as a normalizer, as it is highly expressed and relatively invariant across various samples (66). To normalize RT-qPCR data, McDermott et al. demonstrated that the combined use of miR-16 together with miR-425 generated more reliable results than using either one of these miRNA alone, or the use of RNU6. Following miRNA profiling of approximately 380 miRNAs, RT-qPCR was performed in 40 breast cancer patients and 20 healthy women. The analysis by geNorm and NormFinder algorithms showed that miR-16 and miR-425 were the most stably combination, achieving the lowest V-value of 0.185 (67). Song et al. analyzed the expression levels of miR-16 together with let-7a, miR-93, miR-103, miR-192, miR-451 and RNU6B in the serum samples of 40 gastric cancer patients and 20 healthy volunteers by RT-qPCR. The geNorm, NormFinder and Bestkeeper algorithms were used to select the most stably expressed reference gene from the seven candidates. The algorithms revealed miR-16 and miR-93 to be the most stably expressed reference genes, with stability values of 1.778 and 2.213, respectively, for serum miRNA quantification across all the patients and healthy controls. The effect of different normalization strategies was also compared. When the data were normalized to the serum volume, there were no significant differences of miRNA levels between patients and controls. However, when the data were normalized to miR-16 and miR-93, or the combination of miR-93 and miR-16, significant differences were detected. These results demonstrated that the use of reference gene for RT-qPCR data normalization has a great effect on the study outcome, and that miR-16 and miR-93 can be recommended as suitable reference genes for serum miRNA quantification in gastric cancer patients (54). In contrast, Schaefer et al. reported that data normalization to miR-16 may lead to biased results using tissue and normal adjacent tissue sample pairs from men with untreated prostate carcinoma collected after radical prostatectomy. In this study (68), the expression of four putative reference genes (miR-16, miR-130b, RNU6-2, SNORD7) was examined with regard to their use as normalizer. Candidate miR-130b and RNU6-2 showed no significantly different expression levels between the matched malignant and non-malignant tissue samples, whereas miR-16 was significantly downregulated in malignant tissue. GeNorm and NormFinder algorithms predicted miR-130b and the geometric mean of miR-130b and RNU6-2 as the most stable reference genes (68). To date, the expression of miR-16 has also been described to be deregulated in different diseases by several other studies (69–74). For example, in osteoclast differentiation, the expression of miR-16 is elevated, and miR-16 was characterized as a regulator of osteolytic bone metastasis.(70).
Other miRNAs as normalizers
Using geNorm and NormFinder, Peltier and Latham found that miR-191 was the most consistently expressed miRNA across different human tissues, followed by miR-93, miR-106a, miR-17–5p, and miR-25. In contrast, RNU6 and snRNA5S were the least stable. Indeed, the difference in stability between miR-191 and snRNA5S was a standard deviation of nearly 61 Cq or a difference of 62-fold. Normalization to total RNA mass was also evaluated, but this reference approach ranked behind miR-191 and miR-93 in stability (75). Hu et al. designated miR-1228 as a promising stable endogenous control for quantifying circulating miRNAs in cancer patients. In this report circulating miRNAs was quantified in control individuals (healthy subjects and those with chronic hepatitis B and cirrhosis) and cancer patients (hepatocellular, colorectal, lung, esophageal, gastric, renal, prostate, and breast cancer). GeNorm and NormFinder algorithms as well as coefficient of variability (CV) were used to select the most stable endogenous control, whereas ingenuity pathway analysis (IPA) was adopted to explore the signaling pathways involved. MiR-1228 with CV = 5.4% and minimum M and S values presented as the most stable endogenous control across eight cancer types and three controls. IPA showed miR-1228 to be involved extensively in metabolism-related signal pathways and organ morphology, implying that miR-1228 functions as a housekeeping gene. Additionally, functional network analysis found that miR-1228 was associated with hematological system development, explaining its steady expression in the blood (76). Using TaqMan low density array and NormFinder algorithm, Zhu et al. recommended the combination of miR-26a, miR-221, and miR-22* as the most stable set of reference genes for the evaluation of circulating miRNA in hepatitis B virus (HBV)-infected patients and healthy individuals (77). To determine the levels of candidate reference genes (RNU1-4, RNU6-2, SNORD43, SNORD44, SNORD48, SNORA74A, miR-let-7a-1, miR-106a) for urological malignancies, Sanders et al. analyzed cel-miR-39-spiked serum of prostate cancer patients, bladder cancer patients, renal cell carcinoma patients and control subjects in a RT-qPCR. Recovery of cel-miR-39 (mean 11.6%, range 1–56%) was similar in control subjects and cancer patients. SNORD44 and SNORD74A levels were around the detection limit of the assay. Using the NormFinder and geNorm algorithms, SNORD43 was the most stable reference gene. A combination of two genes (SNORD43, RNU1-4) increased somewhat the stability, indicating that SNORD43 may be a suitable reference gene for the quantification of circulating miRNA in uro-oncological patients (78). For uterine cervical tissues, Shen et al. suggested that miR-23a and miR-191 are the optimal reference miRNAs. Following a microarray assay, the stability of candidate reference genes (miR-26a, miR-23a, miR-200c, let-7a, and miR-1979) was assessed in a cohort of 108 clinical uterine cervical samples by RT-qPCR. MiR-23a was identified as the most reliable reference gene followed by miR-191 (79). To screen suitable reference genes for hepatocellular carcinoma, gastric carcinoma, hepatic cirrhosis and hepatitis B, Tang et al. used GeNorm, Normfinder, BestKeeper, and comparative ΔCq algorithms integrated in RefFinder and measured plasma concentrations of RNU6, let-7a, miR-21, miR-106a, miR-155, miR-219, miR-221 and miR-16 in these patients and healthy volunteers. RefFinder revealed miR-106a and miRNA-21 as the most stably expressed reference genes, with comprehensive stability values of 1.189 and 1.861, respectively, whereas RNU6 was the most unstable miRNA (80). Following miSeq sequencing and qPCR, Wang et al. selected 5 genes (miR-193a-5p, miR-16-5p, RNU6, miR-191-5p and let-7d-3p) for stability analysis using geNorm and NormFinder software. These algorithms identified miR-193a-5p and miR-16-5p as the most stably expressed reference genes. One-way analysis of variance indicated that no significant differences were present in the serum levels among patients with non-muscle-invasive bladder cancer, patients with muscle-invasive bladder cancer and healthy controls. The combined use of miR-193a-5p and miR-16-5p demonstrated that normalization of miRNA data may produce reliable and accurate results for the detection of the significant upregulation of serum miR-148b-3p in bladder cancer (81). To find out the control gene for exosomal miRNA normalization, Le et al. evaluated the expression stability of 11 reference genes in circulating exosomes, and found that the combination of miR-221, miR-191, let-7a, miR-181a, and miR-26a can be an optimal gene reference set for normalizing the expression of liver-specific miRNAs. This combination enhanced the robustness of the relative quantification analyses (82).
Upshot of relative data normalization
Taken together, these findings show the endeavors of developing an optimal endogenous miRNA control to normalize miRNA expression levels. The suggested normalizers for target miRNAs are tissue and species specific. So far, the studies also demonstrate that no consensus exists regarding the normalization to a standard reference gene in various diseases making the miRNA results incomparable. On the one hand, the studies have evaluated and suggested convenient miRNAs, snRNA or rRNAs as ideal candidate reference genes for the data normalization in different diseases using specific algorithms, whereas on the other hand, other studies have shown their deregulation, even for the same disease. In this regard, normalization to a standard reference is most in its infancy. Furthermore, the selection of a normalizer should always follow validation screening tests on a subset of the samples under analyses.
However, these studies also demonstrate that the use of more than one reference gene increases the accuracy of quantification compared to the use of a single reference gene. More than ten years ago, Vandesompele et al. evaluated 10 housekeeping genes from different abundance and functional classes in various human tissues, and demonstrated that the conventional use of a single gene for normalization leads to relatively large errors in a significant proportion of samples tested. The geometric mean of multiple carefully selected housekeeping genes was validated as an accurate normalization factor (61). Chugh and Dittmer described the potential pitfalls in microRNA profiling and showed that the best way to approach the analysis of miRNA expression data is through global mean normalization of a set of reference genes that may be tissue-specific. This method takes a minimum of three stable housekeeping genes and takes the geometric mean to provide a reliable normalization factor that can control for outliers and differences in abundance between genes (43).
Relative data normalization to exogenous reference genes
To ensure that miRNA quantification is not affected by the technical variability that may be introduced at different analysis steps, synthetic, non-human spike-in miRNAs are frequently used to monitor RNA purification and reverse transcription efficiencies. The C. elegans miRNA cel-miR-39 is almost exclusively used for normalization to an exogenous reference gene (83–85), but also cel-miR-54 (48), synthetic miRNAs Quanto EC1 and Quanto EC2 (86), as well as the simian virus gene SV40 (87) have been used. Because of the lack of established reference genes, some studies have only carried out miRNA quantification with data normalization to these artificial reference miRNAs. These exogenous miRNAs are usually added to the samples before reverse transcription of RNA, to avoid differences in template quality and warrant efficiency of the reverse transcription reaction. This spike-in method can eliminate some deviations of the experimental process and make the results more reliable, but does not correct for deviations in sampling and quality of samples. A major drawback of using spike-in controls is that only the handling of experiments is considered, but not the quality of tissues, body fluids or extracellular vesicles samples. However, age, body fluids collection, preparation or storing of tissue or fluid samples may result in changes of miRNA levels which may be caused by cell lysis or miRNA degradation. For example, samples with low total RNA quality showed the highest concentrations of miRNA (9, 88). Therefore, the data of this approach should be interpreted with caution. However, when normalization is based on a combination of an endogenous and an exogenous control miRNA, differences in miRNA recovery and differences in cDNA synthesis between samples may be compensated (89).
Absolute data normalization
Some studies have carried out absolute data normalizations, and calculated the miRNA expression using standard curves developed by synthetic miRNAs and melting curves (44–47). Following miRNA expression array, Yau et al. quantified two target miRNAs (miR-221 and miR-18a) in 40 pairs of colorectal carcinoma tissues and 595 stool samples. Their miRNA quantification was based on standard curves and normalized per nanogram of the total input RNA. Derived from standard curves plotted from known amounts of synthetic miR-221 and miR-18a, a technical detection limit of two copies for miR-221 resulted in a Cq value of 42, and a technical detection limit of five copies for miR-18a resulted in a Cq value of 47. All Cq values of >42 for miR-221, and >47 for miR-18a were assigned to ‘0’. Samples with no amplification of miR-221 or miR-18a were also assigned a value of ‘0’ (45). However, this study should be considered critically, since based on the MIQE (Minimum Information for publication of Quantitative real-time PCR Experiments) guidelines for qPCR, Cq values in the order of 40 and more are not reliable (90). Liu et al. examined the expression patterns of serum let-7 in 214 gastric cancer patients, 222 atrophic gastritis patients and 202 controls. The concentration (copy number) of let-7 was calculated using a standard curve. Melting curve analysis was performed to validate the specificity of the expected PCR products (46). Wang et al. normalized five serum miRNAs (miR-487a, miR-502, miR-208, miR-215 and miR-29b) to the serum volumes, because RNU6 and 5S rRNA were degraded in the serum samples and because of the lack of a consensus housekeeping miRNA for RT-qPCR. Additionally, they assessed the detection limits of the RT-qPCR assay and the dynamic range, and calculated the absolute concentration of target miRNAs based on a calibration curve developed by synthetic miRNAs with known concentration (47).
Although these studies show interesting results, the applied absolute normalization does not consider the influence of RNA quality on the performance of RT-qPCR. This normalization method is not optimal for an exact quantification of real miRNA amounts and only reliable for samples with a good RNA quality.
Ideal data normalization models
The lack of consensus on reference gene selection for miRNA expression data normalization has led to the spread of publications screening for suitable normalizers for samples of defined origins and/or implicated in different pathological conditions. New studies performing miRNA detection by qPCR may greatly benefit from this knowledge and thus a strategic experiment workflow is proposed (Figure 1). Prior to qPCR assay performance, researchers are advised to review the literature using samples of the same origin and physiological state, and processed as similarly as possible, in order to find candidate reference genes. Nevertheless, the suitability of these genes for the set of samples under analysis should always be then validated in a sample subset. If the candidate reference genes are stable, qPCR can be performed and data normalized using their expression levels. If not, the specific samples have to be screened for more suitable reference genes. Ideally, good reference genes should have low standard deviations of expression levels across samples, similar mean and median expression values, and be few affected by storage conditions and sample processing, with a high efficiency of extraction. In these cases, the addition of exogenous xenogeneic or synthetic miRNAs to the samples may be beneficial, and their expression levels may be considered simultaneously with endogenous appropriate normalizers for data correction. Considering the advantages and disadvantages of the different methods for miRNA qPCR data normalization, it is evident that the standard approach of relative data normalization with endogenous and exogenous reference genes benefits from a complementary data absolute normalization. Although it should not be used alone, absolute data normalization allows data correction for limitations intrinsic to the qPCR methodology and gives more insights about the overall status of miRNA expression.
Figure 1. Workflow for miRNA qPCR data normalization.
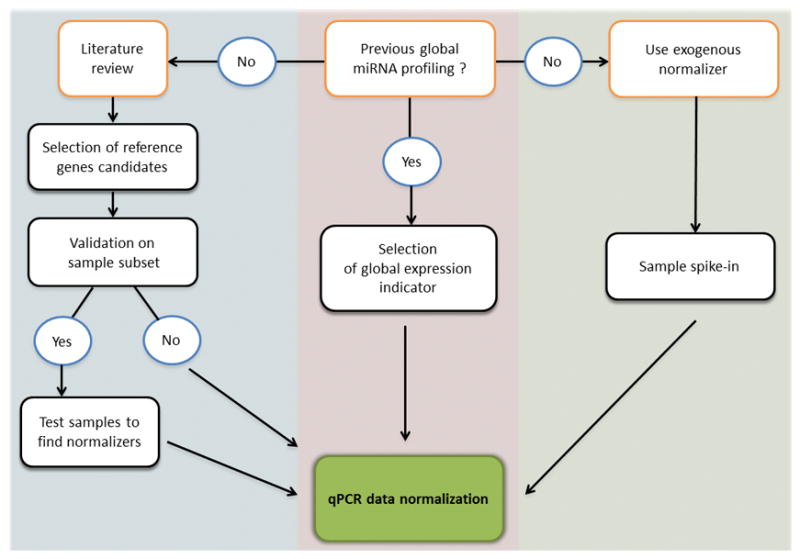
Candidate reference genes may be selected from literature and validated in a subset of the samples under analysis. If not appropriate, samples should be profiled to find adequate endogenous reference genes. Alternatively, exogenous miRNAs may be spiked-in into the samples. Also, mathematical indicators calculated from previous miRNA global expression profiles of the same or similar samples, may be used for normalization.
Alternatively, qPCR data for specific miRNAs expression may be normalized following strategies that take into account the total miRNAs expression in the samples. Very frequently, qPCR analysis of specific miRNAs follows global analysis of total miRNAs expressed in a sample by other technique, as is the case of microarray expression data validation. In this case, information about the whole miRNA content of the sample is available that can be used for global normalization. Mestdagh et al. proposed the use of the mean expression value of whole miRNAs in a sample to normalize miRNA qPCR data (35). In this study, a high-throughput qPCR assay that allows the detection of 430 different human miRNAS and 18 small RNA controls was performed to determine global miRNA expression levels in samples of normal and tumor tissue. The mean miRNA expression value was then calculated considering all the transcripts with a maximal Cq threshold of 35 cycles. A comparative analysis of the stability of mean expression value and common reference genes performed using geNorm clearly showed the adequate application of this strategy for normalization, performing better than genes such as RNU48 and miR-191. Ideally this strategy should be more widespread for qPCR data validation, nevertheless it implies that a large number of genes are always profiled, which may not be cost-effective. To circumvent this issue, the authors propose the selection of reference genes with expression levels similar to values of the global mean expression level previously reported, using their geometric mean for qPCR data normalization. In addition, researchers should bear in mind that, overall, qPCR data normalization may greatly benefit from sample preservation and stability, and thus adequate protocols for sample processing should be standardized at least for samples of the same origin in the same lab.
Conclusion
Normalizing to a reference gene can eliminate differences due to sampling and quality of RNA, and can identify real changes in miRNA expression levels. Therefore, careful validation of reference genes for miRNAs is of crucial importance to obtain accurate miRNA data. Reliability of results reported in the literature, using the wrong reference gene or even performing no data normalization is questionable. It should be emphasized that the applicability of reference genes in some studies does not automatically apply to other studies and that the use of a single reference gene is not sufficient to obtain reliable miRNA data.
Supplementary Material
Footnotes
Disclosures/Conflict of interest
Dr. Calin is The Alan M. Gewirtz Leukemia & Lymphoma Society Scholar. Work in Dr. Calin’s laboratory is supported in part by the NIH/NCI grants 1UH2TR00943-01 and 1 R01 CA182905-01, the UT MD Anderson Cancer Center SPORE in Melanoma grant from NCI (P50 CA093459), Aim at Melanoma Foundation and the Miriam and Jim Mulva research funds, the Brain SPORE (2P50CA127001), the Center for Radiation Oncology Research Project, the Center for Cancer Epigenetics Pilot project, a 2014 Knowledge GAP MDACC grant, a CLL Moonshot pilot project, the UT MD Anderson Cancer Center Duncan Family Institute for Cancer Prevention and Risk Assessment, a SINF grant in colon cancer, the Laura and John Arnold Foundation, the RGK Foundation and the Estate of C. G. Johnson, Jr. Andreia M. Silva is a recipient of a Ph.D. fellowship from FCT–Fundação para a Ciência e a Tecnologia (SFRH/BD/85968/2012).
References
- 1.Ha M, Kim VN. Regulation of microrna biogenesis. Nature reviews Molecular cell biology. 2014;15:509–24. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. Micrornas: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Calin GA, Croce CM. Microrna-cancer connection: The beginning of a new tale. Cancer Res. 2006;66:7390–4. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- 4.Montagner S, Deho L, Monticelli S. Micrornas in hematopoietic development. BMC immunology. 2014;15:14. doi: 10.1186/1471-2172-15-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piccoli MT, Gupta SK, Thum T. Noncoding rnas as regulators of cardiomyocyte proliferation and death. Journal of molecular and cellular cardiology. 2015 doi: 10.1016/j.yjmcc.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Sacco J, Adeli K. Micrornas: Emerging roles in lipid and lipoprotein metabolism. Current opinion in lipidology. 2012;23:220–5. doi: 10.1097/MOL.0b013e3283534c9f. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. Micrornas: Target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ardekani AM, Naeini MM. The role of micrornas in human diseases. Avicenna journal of medical biotechnology. 2010;2:161–79. [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarzenbach H, Nishida N, Calin GA, Pantel K. Clinical relevance of circulating cell-free micrornas in cancer. Nature reviews Clinical oncology. 2014;11:145–56. doi: 10.1038/nrclinonc.2014.5. [DOI] [PubMed] [Google Scholar]
- 10.van Schooneveld E, Wildiers H, Vergote I, Vermeulen PB, Dirix LY, Van Laere SJ. Dysregulation of micrornas in breast cancer and their potential role as prognostic and predictive biomarkers in patient management. Breast Cancer Res. 2015;17:21. doi: 10.1186/s13058-015-0526-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X, Liang H, Zhang J, Zen K, Zhang CY. Horizontal transfer of micrornas: Molecular mechanisms and clinical applications. Protein & cell. 2012;3:28–37. doi: 10.1007/s13238-012-2003-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayes J, Peruzzi PP, Lawler S. Micrornas in cancer: Biomarkers, functions and therapy. Trends in molecular medicine. 2014;20:460–9. doi: 10.1016/j.molmed.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Pimentel F, Bonilla P, Ravishankar YG, Contag A, Gopal N, LaCour S, et al. Technology in microrna profiling: Circulating micrornas as noninvasive cancer biomarkers in breast cancer. Journal of laboratory automation. 2014 doi: 10.1177/2211068214561788. [DOI] [PubMed] [Google Scholar]
- 14.Tiberio P, Callari M, Angeloni V, Daidone MG, Appierto V. Challenges in using circulating mirnas as cancer biomarkers. Biomed Res Int. 2015;2015:731479. doi: 10.1155/2015/731479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biassoni R, Raso A, editors. Mrna and microrna purity and integrity: The key to success in expression profiling. Springer; New York: 2014. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Smyth P, Flavin R, Cahill S, Denning K, Aherne S, et al. Comparison of mirna expression patterns using total rna extracted from matched samples of formalin-fixed paraffin-embedded (ffpe) cells and snap frozen cells. BMC biotechnology. 2007;7:36. doi: 10.1186/1472-6750-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monleau M, Bonnel S, Gostan T, Blanchard D, Courgnaud V, Lecellier CH. Comparison of different extraction techniques to profile micrornas from human sera and peripheral blood mononuclear cells. BMC genomics. 2014;15:395. doi: 10.1186/1471-2164-15-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glynn CL, Khan S, Kerin MJ, Dwyer RM. Isolation of secreted micrornas (mirnas) from cell-conditioned media. MicroRNA. 2013;2:14–9. doi: 10.2174/2211536611302010003. [DOI] [PubMed] [Google Scholar]
- 19.Weber JA, Baxter DH, Zhang S, Huang DY, Huang KH, Lee MJ, et al. The microrna spectrum in 12 body fluids. Clinical chemistry. 2010;56:1733–41. doi: 10.1373/clinchem.2010.147405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rani S. Microrna profiling of exosomes isolated from biofluids and conditioned media. Methods in molecular biology. 2014;1182:131–44. doi: 10.1007/978-1-4939-1062-5_12. [DOI] [PubMed] [Google Scholar]
- 21.Ibberson D, Benes V, Muckenthaler MU, Castoldi M. Rna degradation compromises the reliability of microrna expression profiling. BMC biotechnology. 2009;9:102. doi: 10.1186/1472-6750-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim DJ, Linnstaedt S, Palma J, Park JC, Ntrivalas E, Kwak-Kim JY, et al. Plasma components affect accuracy of circulating cancer-related microrna quantitation. The Journal of molecular diagnostics : JMD. 2012;14:71–80. doi: 10.1016/j.jmoldx.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farina NH, Wood ME, Perrapato SD, Francklyn CS, Stein GS, Stein JL, Lian JB. Standardizing analysis of circulating microrna: Clinical and biological relevance. Journal of cellular biochemistry. 2014;115:805–11. doi: 10.1002/jcb.24745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacLellan SA, MacAulay C, Lam S, Garnis C. Pre-profiling factors influencing serum microrna levels. BMC clinical pathology. 2014;14:27. doi: 10.1186/1472-6890-14-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witwer KW, Buzas EI, Bemis LT, Bora A, Lasser C, Lotvall J, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. Journal of extracellular vesicles. 2013;2 doi: 10.3402/jev.v2i0.20360. eCollection 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu R, Chen X, Du Y, Yao W, Shen L, Wang C, et al. Serum microrna expression profile as a biomarker in the diagnosis and prognosis of pancreatic cancer. Clinical chemistry. 2012;58:610–8. doi: 10.1373/clinchem.2011.172767. [DOI] [PubMed] [Google Scholar]
- 27.Vasilescu C, Rossi S, Shimizu M, Tudor S, Veronese A, Ferracin M, et al. Microrna fingerprints identify mir-150 as a plasma prognostic marker in patients with sepsis. PloS one. 2009;4:e7405. doi: 10.1371/journal.pone.0007405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aushev VN, Zborovskaya IB, Laktionov KK, Girard N, Cros MP, Herceg Z, Krutovskikh V. Comparisons of microrna patterns in plasma before and after tumor removal reveal new biomarkers of lung squamous cell carcinoma. PloS one. 2013;8:e78649. doi: 10.1371/journal.pone.0078649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang L, Li H, Wang L, Hu J, Jin T, Wang J, Yang BB. Microrna-17-5p promotes chemotherapeutic drug resistance and tumour metastasis of colorectal cancer by repressing pten expression. Oncotarget. 2014;5:2974–87. doi: 10.18632/oncotarget.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oksvold MP, Kullmann A, Forfang L, Kierulf B, Li M, Brech A, et al. Expression of b-cell surface antigens in subpopulations of exosomes released from b-cell lymphoma cells. Clinical therapeutics. 2014;36:847–62. e1. doi: 10.1016/j.clinthera.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Boing AN, van der Pol E, Grootemaat AE, Coumans FA, Sturk A, Nieuwland R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. Journal of extracellular vesicles. 2014;3 doi: 10.3402/jev.v3.23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathivanan S, Lim JW, Tauro BJ, Ji H, Moritz RL, Simpson RJ. Proteomics analysis of a33 immunoaffinity-purified exosomes released from the human colon tumor cell line lim1215 reveals a tissue-specific protein signature. Molecular & cellular proteomics : MCP. 2010;9:197–208. doi: 10.1074/mcp.M900152-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witwer KW. Circulating microrna biomarker studies: Pitfalls and potential solutions. Clinical chemistry. 2015;61:56–63. doi: 10.1373/clinchem.2014.221341. [DOI] [PubMed] [Google Scholar]
- 34.Haider BA, Baras AS, McCall MN, Hertel JA, Cornish TC, Halushka MK. A critical evaluation of microrna biomarkers in non-neoplastic disease. PloS one. 2014;9:e89565. doi: 10.1371/journal.pone.0089565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benes V, Castoldi M. Expression profiling of microrna using real-time quantitative pcr, how to use it and what is available. Methods. 2010;50:244–9. doi: 10.1016/j.ymeth.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 36.Thomson JM, Parker JS, Hammond SM. Microarray analysis of mirna gene expression. Methods in enzymology. 2007;427:107–22. doi: 10.1016/S0076-6879(07)27006-5. [DOI] [PubMed] [Google Scholar]
- 37.Sato F, Tsuchiya S, Terasawa K, Tsujimoto G. Intra-platform repeatability and inter-platform comparability of microrna microarray technology. PloS one. 2009;4:e5540. doi: 10.1371/journal.pone.0005540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F, Vandesompele J. A novel and universal method for microrna rt-qpcr data normalization. Genome biology. 2009;10:R64. doi: 10.1186/gb-2009-10-6-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer SU, Pfaffl MW, Ulbrich SE. Normalization strategies for microrna profiling experiments: A ‘normal’ way to a hidden layer of complexity? Biotechnology letters. 2010;32:1777–88. doi: 10.1007/s10529-010-0380-z. [DOI] [PubMed] [Google Scholar]
- 40.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Rai SN, Ray HE, Yuan X, Pan J, Hamid T, Prabhu SD. Statistical analysis of repeated microrna high-throughput data with application to human heart failure: A review of methodology. Open access medical statistics. 2012;2012:21–31. doi: 10.2147/OAMS.S27907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bustin SA, Beaulieu JF, Huggett J, Jaggi R, Kibenge FS, Olsvik PA, et al. Miqe precis: Practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time pcr experiments. BMC molecular biology. 2010;11:74. doi: 10.1186/1471-2199-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chugh P, Dittmer DP. Potential pitfalls in microrna profiling. Wiley Interdiscip Rev RNA. 2012;3:601–16. doi: 10.1002/wrna.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan W, Li R, Liu Y, Yang P, Wang Z, Zhang C, et al. Microrna expression patterns in the malignant progression of gliomas and a 5-microrna signature for prognosis. Oncotarget. 2014;5:12908–15. doi: 10.18632/oncotarget.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yau TO, Wu CW, Dong Y, Tang CM, Ng SS, Chan FK, et al. Microrna-221 and microrna-18a identification in stool as potential biomarkers for the non-invasive diagnosis of colorectal carcinoma. British journal of cancer. 2014;111:1765–71. doi: 10.1038/bjc.2014.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu WJ, Xu Q, Sun LP, Dong QG, He CY, Yuan Y. Expression of serum let-7c, let-7i, and let-7f microrna with its target gene, pepsinogen c, in gastric cancer and precancerous disease. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014 doi: 10.1007/s13277-014-2967-9. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Pei Y, Zhong Y, Jiang S, Shao J, Gong J. Altered serum micrornas as novel diagnostic biomarkers for atypical coronary artery disease. PloS one. 2014;9:e107012. doi: 10.1371/journal.pone.0107012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuhlmann JD, Baraniskin A, Hahn SA, Mosel F, Bredemeier M, Wimberger P, et al. Circulating u2 small nuclear rna fragments as a novel diagnostic tool for patients with epithelial ovarian cancer. Clinical chemistry. 2014;60:206–13. doi: 10.1373/clinchem.2013.213066. [DOI] [PubMed] [Google Scholar]
- 49.Spiekermann M, Dieckmann KP, Balks T, Bullerdiek J, Belge G. Is relative quantification dispensable for the measurement of micrornas as serum biomarkers in germ cell tumors? Anticancer research. 2015;35:117–21. [PubMed] [Google Scholar]
- 50.Wang X, Gardiner EJ, Cairns MJ. Optimal consistency in microrna expression analysis using reference-gene-based normalization. Mol Biosyst. 2015;11:1235–40. doi: 10.1039/c4mb00711e. [DOI] [PubMed] [Google Scholar]
- 51.Serafin A, Foco L, Blankenburg H, Picard A, Zanigni S, Zanon A, et al. Identification of a set of endogenous reference genes for mirna expression studies in parkinson’s disease blood samples. BMC research notes. 2014;7:715. doi: 10.1186/1756-0500-7-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roth C, Rack B, Muller V, Janni W, Pantel K, Schwarzenbach H. Circulating micrornas as blood-based markers for patients with primary and metastatic breast cancer. Breast Cancer Res. 2010;12:R90. doi: 10.1186/bcr2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mahdipour M, Van Tol HT, Stout TA, Roelen BA. 225 validation of reference micrornas for normalizing expression data generated by quantitative pcr. Reproduction, fertility, and development. 2014;27:202. [Google Scholar]
- 54.Song J, Bai Z, Han W, Zhang J, Meng H, Bi J, et al. Identification of suitable reference genes for qpcr analysis of serum microrna in gastric cancer patients. Digestive diseases and sciences. 2012;57:897–904. doi: 10.1007/s10620-011-1981-7. [DOI] [PubMed] [Google Scholar]
- 55.Shen Y, Tian F, Chen Z, Li R, Ge Q, Lu Z. Amplification-based method for microrna detection. Biosens Bioelectron. 2015;71:322–31. doi: 10.1016/j.bios.2015.04.057. [DOI] [PubMed] [Google Scholar]
- 56.Benz F, Roderburg C, Vargas Cardenas D, Vucur M, Gautheron J, Koch A, et al. U6 is unsuitable for normalization of serum mirna levels in patients with sepsis or liver fibrosis. Experimental & molecular medicine. 2013;45:e42. doi: 10.1038/emm.2013.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gee HE, Buffa FM, Camps C, Ramachandran A, Leek R, Taylor M, et al. The small-nucleolar rnas commonly used for microrna normalisation correlate with tumour pathology and prognosis. British journal of cancer. 2011;104:1168–77. doi: 10.1038/sj.bjc.6606076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang M, Zeng Y, Yang R, Xu H, Chen Z, Zhong J, et al. U6 is not a suitable endogenous control for the quantification of circulating micrornas. Biochemical and biophysical research communications. 2014;454:210–4. doi: 10.1016/j.bbrc.2014.10.064. [DOI] [PubMed] [Google Scholar]
- 59.Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-pcr data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–50. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 60.Lamba V, Ghodke-Puranik Y, Guan W, Lamba JK. Identification of suitable reference genes for hepatic microrna quantitation. BMC research notes. 2014;7:129. doi: 10.1186/1756-0500-7-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative rt-pcr data by geometric averaging of multiple internal control genes. Genome biology. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ratert N, Meyer HA, Jung M, Mollenkopf HJ, Wagner I, Miller K, et al. Reference mirnas for mirnaome analysis of urothelial carcinomas. PloS one. 2012;7:e39309. doi: 10.1371/journal.pone.0039309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wotschofsky Z, Meyer HA, Jung M, Fendler A, Wagner I, Stephan C, et al. Reference genes for the relative quantification of micrornas in renal cell carcinomas and their metastases. Analytical biochemistry. 2011;417:233–41. doi: 10.1016/j.ab.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 64.Torres A, Torres K, Wdowiak P, Paszkowski T, Maciejewski R. Selection and validation of endogenous controls for microrna expression studies in endometrioid endometrial cancer tissues. Gynecologic oncology. 2013;130:588–94. doi: 10.1016/j.ygyno.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 65.Han HS, Jo YN, Lee JY, Choi SY, Jeong Y, Yun J, Lee OJ. Identification of suitable reference genes for the relative quantification of micrornas in pleural effusion. Oncology letters. 2014;8:1889–95. doi: 10.3892/ol.2014.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muller V, Gade S, Steinbach B, Loibl S, von Minckwitz G, Untch M, et al. Changes in serum levels of mir-21, mir-210, and mir-373 in her2-positive breast cancer patients undergoing neoadjuvant therapy: A translational research project within the geparquinto trial. Breast cancer research and treatment. 2014;147:61–8. doi: 10.1007/s10549-014-3079-3. [DOI] [PubMed] [Google Scholar]
- 67.McDermott AM, Kerin MJ, Miller N. Identification and validation of mirnas as endogenous controls for rq-pcr in blood specimens for breast cancer studies. PloS one. 2013;8:e83718. doi: 10.1371/journal.pone.0083718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaefer A, Jung M, Miller K, Lein M, Kristiansen G, Erbersdobler A, Jung K. Suitable reference genes for relative quantification of mirna expression in prostate cancer. Experimental & molecular medicine. 2010;42:749–58. doi: 10.3858/emm.2010.42.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Delfino KR, Rodriguez-Zas SL. Transcription factor-microrna-target gene networks associated with ovarian cancer survival and recurrence. PloS one. 2013;8:e58608. doi: 10.1371/journal.pone.0058608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ell B, Mercatali L, Ibrahim T, Campbell N, Schwarzenbach H, Pantel K, et al. Tumor-induced osteoclast mirna changes as regulators and biomarkers of osteolytic bone metastasis. Cancer cell. 2013;24:542–56. doi: 10.1016/j.ccr.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu Z, Dong J, Wang LE, Ma H, Liu J, Zhao Y, et al. Serum microrna profiling and breast cancer risk: The use of mir-484/191 as endogenous controls. Carcinogenesis. 2012;33:828–34. doi: 10.1093/carcin/bgs030. [DOI] [PubMed] [Google Scholar]
- 72.Liu J, Gao J, Du Y, Li Z, Ren Y, Gu J, et al. Combination of plasma micrornas with serum ca19-9 for early detection of pancreatic cancer. Int J Cancer. 2012;131:683–91. doi: 10.1002/ijc.26422. [DOI] [PubMed] [Google Scholar]
- 73.Xiao G, Tang H, Wei W, Li J, Ji L, Ge J. Aberrant expression of microrna-15a and microrna-16 synergistically associates with tumor progression and prognosis in patients with colorectal cancer. Gastroenterology research and practice. 2014;2014:364549. doi: 10.1155/2014/364549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zuo Z, Calin GA, de Paula HM, Medeiros LJ, Fernandez MH, Shimizu M, et al. Circulating micrornas let-7a and mir-16 predict progression-free survival and overall survival in patients with myelodysplastic syndrome. Blood. 2011;118:413–5. doi: 10.1182/blood-2011-01-330704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peltier HJ, Latham GJ. Normalization of microrna expression levels in quantitative rt-pcr assays: Identification of suitable reference rna targets in normal and cancerous human solid tissues. RNA. 2008;14:844–52. doi: 10.1261/rna.939908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu J, Wang Z, Liao BY, Yu L, Gao X, Lu S, et al. Human mir-1228 as a stable endogenous control for the quantification of circulating micrornas in cancer patients. Int J Cancer. 2014;135:1187–94. doi: 10.1002/ijc.28757. [DOI] [PubMed] [Google Scholar]
- 77.Zhu HT, Dong QZ, Wang G, Zhou HJ, Ren N, Jia HL, et al. Identification of suitable reference genes for qrt-pcr analysis of circulating micrornas in hepatitis b virus-infected patients. Molecular biotechnology. 2012;50:49–56. doi: 10.1007/s12033-011-9414-6. [DOI] [PubMed] [Google Scholar]
- 78.Sanders I, Holdenrieder S, Walgenbach-Brunagel G, von Ruecker A, Kristiansen G, Muller SC, Ellinger J. Evaluation of reference genes for the analysis of serum mirna in patients with prostate cancer, bladder cancer and renal cell carcinoma. International journal of urology : official journal of the Japanese Urological Association. 2012;19:1017–25. doi: 10.1111/j.1442-2042.2012.03082.x. [DOI] [PubMed] [Google Scholar]
- 79.Shen Y, Li Y, Ye F, Wang F, Wan X, Lu W, Xie X. Identification of mir-23a as a novel microrna normalizer for relative quantification in human uterine cervical tissues. Experimental & molecular medicine. 2011;43:358–66. doi: 10.3858/emm.2011.43.6.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tang G, Shen X, Lv K, Wu Y, Bi J, Shen Q. Different normalization strategies might cause inconsistent variation in circulating micrornas in patients with hepatocellular carcinoma. Med Sci Monit. 2015;21:617–24. doi: 10.12659/MSM.891028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang L, Liu Y, Du L, Li J, Jiang X, Zheng G, et al. Identification and validation of reference genes for the detection of serum micrornas by reverse transcriptionquantitative polymerase chain reaction in patients with bladder cancer. Mol Med Rep. 2015;12:615–22. doi: 10.3892/mmr.2015.3428. [DOI] [PubMed] [Google Scholar]
- 82.Li Y, Zhang L, Liu F, Xiang G, Jiang D, Pu X. Identification of endogenous controls for analyzing serum exosomal mirna in patients with hepatitis b or hepatocellular carcinoma. Dis Markers. 2015;2015:893594. doi: 10.1155/2015/893594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang P, Yang D, Zhang H, Wei X, Ma T, Cheng Z, et al. Early detection of lung cancer in serum by a panel of microrna biomarkers. Clinical lung cancer. 2014 doi: 10.1016/j.cllc.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 84.Dong BL, Wang Z, Zhang Z, Wang J. Clinical significance of microrna-24 expression in esophageal squamous cell carcinoma. Neoplasma. 2015;62:250–8. doi: 10.4149/neo_2015_030. [DOI] [PubMed] [Google Scholar]
- 85.Xu T, Liao Z, O’Reilly MS, Levy LB, Welsh JW, Wang LE, et al. Serum inflammatory mirnas predict radiation esophagitis in patients receiving definitive radiochemotherapy for non-small cell lung cancer. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2014;113:379–84. doi: 10.1016/j.radonc.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 86.Liu M, Liu J, Wang L, Wu H, Zhou C, Zhu H, et al. Association of serum microrna expression in hepatocellular carcinomas treated with transarterial chemoembolization and patient survival. PloS one. 2014;9:e109347. doi: 10.1371/journal.pone.0109347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anadol E, Schierwagen R, Elfimova N, Tack K, Schwarze-Zander C, Eischeid H, et al. Circulating micrornas as a marker for liver injury in human immunodeficiency virus patients. Hepatology. 2015;61:46–55. doi: 10.1002/hep.27369. [DOI] [PubMed] [Google Scholar]
- 88.Becker C, Hammerle-Fickinger A, Riedmaier I, Pfaffl MW. Mrna and microrna quality control for rt-qpcr analysis. Methods. 2010;50:237–43. doi: 10.1016/j.ymeth.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 89.Sourvinou IS, Markou A, Lianidou ES. Quantification of circulating mirnas in plasma: Effect of preanalytical and analytical parameters on their isolation and stability. The Journal of molecular diagnostics : JMD. 2013;15:827–34. doi: 10.1016/j.jmoldx.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 90.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The miqe guidelines: Minimum information for publication of quantitative real-time pcr experiments. Clinical chemistry. 2009;55:611–22. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.