

Abstract
The metabolic functions of androgen receptor (AR) in normal prostate are circumvented in prostate cancer (PCa) to drive tumor growth, and the AR also can acquire new growth-promoting functions during PCa development and progression through genetic and epigenetic mechanisms. Androgen deprivation therapy (ADT, surgical or medical castration) is the standard treatment for metastatic PCa, but patients invariably relapse despite castrate androgen levels (castration-resistant PCa, CRPC). Early studies from many groups had shown that AR was highly expressed and transcriptionally active in CRPC, and indicated that steroids from the adrenal glands were contributing to this AR activity. More recent studies showed that CRPC cells had increased expression of enzymes mediating androgen synthesis from adrenal steroids, and could synthesize androgens de novo from cholesterol. Phase III clinical trials showing a survival advantage in CRPC for treatment with abiraterone (inhibitor of the enzyme CYP17A1 required for androgen synthesis that markedly reduces androgens and precursor steroids) and for enzalutamide (new AR antagonist) have now confirmed that AR activity driven by residual androgens makes a major contribution to CRPC, and led to the recent Food and Drug Administration approval of both agents. Unfortunately, patients treated with these agents for advanced CRPC generally relapse within a year and AR appears to be active in the relapsed tumors, but the molecular mechanisms mediating intrinsic or acquired resistance to these AR-targeted therapies remain to be defined. This review outlines AR functions that contribute to PCa development and progression, the roles of intratumoral androgen synthesis and AR structural alterations in driving AR activity in CRPC, mechanisms of action for abiraterone and enzalutamide, and possible mechanisms of resistance to these agents.
Keywords: androgen, androgen receptor, prostate cancer, castration-resistant prostate cancer
INTRODUCTION
The standard systemic treatment for prostate cancer (PCa) since the 1940s has been androgen deprivation therapy (ADT, surgical or medical castration) to suppress androgen receptor (AR) transcriptional activity, but most patients relapse within several years with disease that is generally more aggressive and is currently referred to as castration-resistant PCa (CRPC). The past several years have seen a paradigm shift in therapy for CRPC, as it is now widely accepted that the AR is active and stimulating the growth of these cancers that relapse despite castrate levels of androgens (testosterone and dihydrotestosterone). Historically, studies from many groups had indicated that AR was reactivated in CRPC, and that it was driven at least in part by residual steroid hormones from the adrenal glands. Indeed, during the late 1940s and 1950s adrenalectomy or hypophysectomy were shown to be effective secondary hormonal therapies for men who relapsed after orchiectomy.1 These surgical approaches were subsequently replaced by medical therapies such as prednisone and ketoconazole that suppressed adrenal androgen synthesis, which were similarly effective in a subset of patients based on clinical and biochemical criteria (decrease in serum prostate-specific antigen (PSA)).2–4 Based on the hypothesis that residual androgens were still driving AR after castration, clinical trials were conducted in the 1980s and 1990s to determine whether the efficacy of castration (orchiectomy or GnRH agonist) could be improved by addition of a direct AR antagonist (flutamide, nilutamide or bicalutamide) to block the effects of residual androgens.5,6 While meta-analyses of these studies showed a survival benefit for the combined therapies, the magnitude of the benefit was very small and enthusiasm for further therapies beyond castration to suppress AR waned.
Despite these disappointing clinical results, immunohistochemical and molecular studies on patient samples in the 1990s showed that AR was highly expressed and transcriptionally active (based on high-level expression of AR-regulated genes such as PSA) in CRPC.7–10 Moreover, AR gene amplification in CRPC,11 and the identification of ARs with gain of function mutations in AR antagonist-treated patients that could be strongly activated by the antagonists,8,12 showed that these tumors were under strong selective pressure to maintain AR activity. Subsequent studies in xenograft models similarly showed increased AR and restoration of AR activity in tumors that relapsed after castration,13–15 and RNA interference and related approaches established that AR was still required for growth in these CRPC models.16,17 Studies showing relatively high levels of androgens in CRPC samples from patients,18–20 in conjunction with studies showing that these tumors had increased expression of androgen synthetic enzymes,10,20 established androgen synthesis by tumor cells as a mechanism for AR reactivation in CRPC.21
Most recently, phase III clinical trials of abiraterone (inhibitor of the enzyme CYP17A1 required for androgen synthesis) and enzalutamide (more effective direct AR antagonist) in CRPC established that further AR suppression can extend patient survival, and led to Food and Drug Administration approval of these agents.22–24 Unfortunately, while the majority of patients who have relapsed after castration respond initially to these agents, the overall survival advantage in advanced disease (post chemotherapy) is still modest (4–6 months), and most responding patients relapse within 1–2 years with evidence of renewed AR activity. In order to build on these recent advances in AR-targeted therapies for PCa, it is clearly critical to better understand the critical functions of AR and mechanisms mediating its reactivation, and to develop strategies that can overcome these mechanisms. This review focuses on AR functions in PCa and mechanisms of action and resistance to agents targeting AR in CRPC.
AR STRUCTURE AND NORMAL FUNCTION AS A TRANSCRIPTIONAL ACTIVATOR
The AR is a transcription factor with a large N-terminal transactivation domain (NTD) (exon 1), a C-terminal ligand-binding domain (LBD) (exons 4–8), a central DNA-binding domain (DBD) (exons 2–3), and a hinge region between the DNA-binding domain and LBD that contributes to nuclear localization and degradation (Figure 1). The unliganded AR associates with an HSP90 chaperone complex in the cytoplasm and undergoes proteasome-mediated degradation in the absence of ligand. Similarly to other nuclear receptors, binding of agonist ligands (testosterone or dihydrotestosterone) causes a shift in the position of helix 12 in the AR LBD towards helices 3–5, which stabilizes ligand binding and generates a hydrophobic cleft for binding of leucine-x-x-leucine-leucine (LxxLL) motifs found in many transcriptional coactivator proteins.25,26 A unique feature of AR is that an LxxLL-like motif in the AR N terminus (amino acids 23–27, FQNLF) binds to this hydrophobic cleft, which further stabilizes helix 12 and ligand binding (AR–N–C terminal interaction).27,28 Fluorescence resonance energy transfer studies show that this N–C interaction is initially intramolecular in the cytoplasm, but shifts towards intermolecular in the nucleus and may have some role in nuclear localization, although its precise function is not clear.29–32 Interestingly, fluorescence resonance energy transfer data also suggest that the N–C interaction may be disrupted when AR binds chromatin, possibly in order to allow for coactivator binding.30
Figure 1.

AR structure and responses to binding agonist and antagonist ligands. Androgen binding mediates a conformational change in the position of helix 12 in the LBD. Binding to an FQNLF peptide in the NTD mediates an initial intramolecular N–C interaction, and a subsequent intermolecular interaction may contribute to nuclear localization. AR then binds to androgen-responsive elements at sites that are generally bound initially by the FOXA1 transcription factor, which has been termed a ‘pioneer transcription factor,’ as it opens chromatin locally so AR can access the ARE. These sites also are generally marked by H3K4me2 containing nucleosomes. AR binding displaces a weakly associated central nucleosome, and initiates the assembly of multiple coactivator and chromatin-modifying proteins that loop to the promoter to initiate transcription. LSD1 functions as a critical coactivator for androgen-stimulated genes that is associated with H3K9me2 demethylation, and corepressor for androgen-repressed genes that is associated with H3K4me2 demethylation, but the precise LSD1 mechanisms of action on androgen-stimulated versus -repressed genes remain to be established. The bicalutamide-liganded AR associates more transiently with chromatin, does not effectively mediate coactivator recruitment, and has increased corepressor recruitment. The MDV3100 (enzalutamide) liganded AR localizes in both the nucleus and cytoplasm, but does not detectably associate with chromatin, which may reflect further displacement of helix 12 and abrogation of the N–C interaction.
The agonist-liganded AR then translocates to the nucleus, dimerizes and binds to specific sequences (androgen-responsive elements (AREs)) found in the regulatory regions of AR-target genes. AR binding to these sites displaces a weakly associated central nucleosome and is generally dependent upon prior binding of the transcription factor FOXA1, which has been termed a ‘pioneer transcription factor,’ as it is needed to initially unwind the chromatin around the ARE so it becomes accessible for AR binding.33,34 The AR also undergoes multiple post-translational modifications in response to agonist binding, including phosphorylation, methylation, acetylation, ubiquitylation and sumoylation.35 These modifications have roles in regulating AR stability, cellular localization and transcriptional activity, although the precise mechanisms by which many of these modifications modulate AR structure and interactions with other proteins or DNA remain to be determined (see below). A series of transcriptional coactivator/chromatin-modifying proteins are then recruited through interactions with the AR NTD and LBD. This complex of proteins recruited by AR then interacts by chromatin looping with the promoter regions to stimulate the transcription of a large number of AR-regulated genes.36,37
Consistent with the normal function of androgens in the prostate being to drive the differentiated functions of luminal epithelial cells, these AR-stimulated genes include seminal fluid proteins (such as prostate-specific antigen) and multiple genes in metabolic pathways required to support high levels of protein and lipid synthesis.38,39 Significantly, while androgens can clearly stimulate PCa growth, and androgen deprivation causes a G0/G1 cell cycle arrest,40,41 most genes driving cell cycle progression in response to androgen do not appear to be directly regulated by AR. One mechanism mediating the cell cycle progression in response to androgens is an increase in TORC1 activity and subsequent TORC1-mediated increase in the translation of D-cyclins.39 The decrease in TORC1 activity after androgen deprivation and the increase in TORC1 activity in response to androgen likely reflect the ability of AR to stimulate cellular metabolism by increasing the expression of multiple membrane transporters and other genes driving lipid and protein synthesis (see above).
Androgen deprivation also increases levels of the cyclin-dependent kinase inhibitor p27, while androgen stimulation promotes the rapid degradation of p27.40,42 Recent data indicate that the rapid p27 degradation reflects androgen stimulation of TORC2, the subsequent phosphorylation and activation of AKT, and phosphorylation of p27 by AKT at a site that enhances p27 degradation (threonine 157).43 The androgen-mediated stimulation of TORC2 appears to be independent of transcription, but its mechanism remains to be determined. Interestingly, this AKT site on human p27 is not conserved in the mouse, and a recent study using a tetracycline-inducible myristoylated-AKT indicated that AKT-driven proliferation in mouse prostate epithelium is independent of p27 degradation.44 These observations suggest that studies in mouse models may underestimate the oncogenic activity of PI3 kinase/AKT-pathway activation.
In contrast to p27, the agonist-liganded AR binds to a site on the cyclin-dependent kinase inhibitor p21 gene and directly increases p21 transcription and protein expression.45 As p21 can, in some contexts, stimulate cell cycle progression by increasing assembly of cyclin D/CDK4 complexes, this may be a mechanism that contributes to androgen-stimulated PCa growth. Finally, it should be noted that AR is also weakly expressed by subsets of cells in the prostate stroma, and that AR in these cells can stimulate the expression of growth factors such as KGF/FGF7.46 Through this mechanism, AR in stromal cells may indirectly regulate growth of the epithelium, and loss of these stromal factors likely contributes to prostate involution after castration.
AR FUNCTION AS A TRANSCRIPTIONAL REPRESSOR
In addition to its well established function as a transcriptional activator, gene expression studies from many groups have shown that expression of multiple genes is decreased in response to androgen. For many of these androgen-repressed genes, this is likely secondary to transcriptional effects on other genes. However, AR can repress the expression of some genes more directly through interactions with other transcriptional activators or by functioning as a transcriptional repressor. The most studied example of the former mechanism is AR interaction with SP1, which can interfere with SP1-mediated transactivation of genes including the luteinizing hormone β-subunit and c-MET.47,48 Other transcription factors that may be similarly antagonized through interaction with the agonist-liganded AR include RUNX2, JUN and SMAD3.49 Moreover, AR binding of β-catenin, which is a critical coactivator for the T-cell factor transcription factors that mediate canonical Wnt/β-catenin signaling, may suppress this pathway.50–55
The agonist-liganded AR also may function more directly as a transcriptional repressor by recruiting corepressors including ALIEN, DAX1, HEY, AES, PHB and SHP.56–61 However, more studies are needed to address whether these corepressors are targeted to AR on a specific set of genes, and whether any such targeting is determined by features of the ARE or by other mechanisms. The corepressors, NCoR and SMRT, which associate strongly with the LBD of unliganded nonsteroid nuclear receptors and mediate their transcriptional repression functions, can also associate weakly with the agonist-liganded AR (probably through the AR NTD) (Figure 1).62–65 In the absence of ligand, nonsteroid nuclear receptors still bind chromatin and the coactivator-binding site in their LBD is enlarged (due to displacement of helix 12), allowing them to accommodate extended LxxLL-like motifs (CoRNR boxes) in NCoR and SMRT. While the effects of NCoR and SMRT on the agonist-liganded AR are modest, their enhanced recruitment may contribute to the activity of some AR antagonists (see below). Finally, androgen-mediated transcriptional repression has been linked to AR recruitment of EZH2, a histone methyltransferase and component of the polycomb-repressive complex 2, which increases the repressive H3K27me3 mark.66,67
Recent studies have shown that AR also associates with a histone demethylase, lysine-specific demethylase 1 (LSD1, KDM1A), and that LSD1 can function both as a coactivator and corepressor for AR.68–70 LSD1 was identified initially in corepressor complexes and shown to function as a transcriptional corepressor by demethylating mono- and dimethylated lysine 4 on histone 3 (H3K4me1,2) (due to its catalytic mechanism, LSD1 cannot demethylate trimethylated lysines).71 However, it was subsequently shown that LSD1, when associated with AR and possibly other transcription factors including ERa, can function as a transcriptional coactivator and mediate demethylation of repressive mono- and dimethylated H3K9.68,72–75 Indeed, ChIP studies have shown that LSD1 is localized to androgen-responsive elements in multiple androgen-stimulated genes, and that LSD1 inhibition markedly attenuates androgen-stimulated expression of these genes.68,69
In contrast to LSD1 function as a coactivator for androgen-stimulated genes, it functions as an AR corepressor on androgen-repressed genes including the AR gene. A recent study showed that the agonist-liganded AR functions directly as a transcriptional repressor on the AR gene by binding to a site in the second intron of the AR gene, where it recruits LSD1.70 Significantly, androgen-mediated AR and LSD1 binding to this site was associated with demethylation of H3K4me1 and H3K4me2, and LSD1 knockdown or inhibitors prevented this demethylation and downregulation of AR gene expression. Together these findings suggest that a switch in LSD1 substrate specificity from H3K4 to H3K9 may mediate its functions as an AR corepressor versus a coactivator, respectively. Recent studies indicate that phosphorylation of histone 3 on threonine 11 (H3T11ph) by an AR-associated serine/threonine kinase, protein kinase C-related kinase 1 (PRK1, PKN1) enhances the ability of LSD1 to demethylate repressive H3K9me1,2.76 Moreover, phosphorylation of histone 3 on threonine 6 (H3T6ph) by a distinct kinase (protein kinase C β1, PKCβ1) was found to suppress the LSD1-mediated demethylation of H3K4me1,2.77 These results suggest that the dual recruitment of PKN1 and PKCβ1 by AR at androgen-stimulated genes may switch LSD1 substrate specificity from H3K4 to H3K9 (Figure 1). However, it should be emphasized that LSD1 mediates some degree of both H3K4 and H3K9 demethylation at androgen-repressed and -activated genes,70 and that alterations in H3K4 and H3K9 methylation may be the result of alterations in transcription rather than being causal. Therefore, further mechanisms are likely contributing to the activities of AR and LSD1 on androgen-stimulated versus -repressed genes.
In addition to the AR gene, androgens similarly repress the expression of at least two enzymes mediating androgen synthesis in prostate cells (AKR1C3 and HSD17B6) by this LSD1-dependent mechanism.70,78 This is consistent with a physiological negative feedback loop that can adjust AR levels and intracellular androgen levels to maintain AR signaling in response to fluctuations in serum androgen levels. By rapidly increasing AR gene transcription and androgen synthesis, this feedback loop may be a mechanism that helps PCa cells initially to adapt to ADT. Significantly, androgen-repressed genes are also highly enriched for genes that mediate DNA synthesis and cell cycle progression, with at least a subset being repressed by an LSD1-dependent mechanism.70 This result is consistent with a normal physiological role of AR being to drive differentiation rather than proliferation, and with previous studies showing that androgens, particularly at higher concentrations, can suppress rather than stimulate growth of PCa cells.70 The androgen repression of these genes mediating DNA synthesis and cell cycle progression is presumably overridden in PCa by other oncogenic signal-transduction pathways that strongly activate these genes.
These dual functions of AR as a transcriptional activator and repressor have important implications for ADT (Figure 2). PCa cells may initially respond to androgen deprivation due to the central role of AR in regulating metabolic functions, so that androgen deprivation essentially starves the cells and represses pathways including TORC1. However, androgen deprivation may also allow cells initially to maintain a low level of AR activity by increasing AR gene transcription and androgen synthesis. Tumor cells then further adapt over time and partially restore AR signaling, based on mRNA levels for AR-regulated genes in CRPC clinical samples.10,79 These levels are presumably adequate to support critical metabolic functions, but not adequate to impair the expression of AR-repressed genes mediating androgen synthesis, DNA synthesis and cell cycle progression. Significantly, high levels of androgen can repress these genes and arrest tumor growth in some CRPC cell line and xenograft models,70 but the efficacy of this approach in patients is unclear and would likely be limited by tumor heterogeneity. Nonetheless, these findings support the development of novel AR antagonists or other agents that can selectively impair AR transcriptional activator functions, and/or enhance its repressor functions, as these may be efficacious in CRPC.
Figure 2.

Increased expression of androgen-repressed genes may contribute to PCa progression after ADT. In the presence of testicular androgen, AR stimulates PCa growth through its positive effects on metabolic genes while its repression of genes mediating DNA synthesis/cell cycle progression is circumvented by activated oncogenic pathways. The initial response to ADT reflects downregulation of metabolic pathways mediating lipid and protein synthesis, but an undesirable effect is to relieve repression of the AR gene, and of genes regulating androgen synthesis and DNA synthesis/cell cycle progression. In CRPC, mechanisms including increased intratumoral androgen synthesis partially restore AR activity and its metabolic functions, but this AR activity is not adequate to decrease expression of the androgen-repressed genes controlling functions that include DNA synthesis/cell cycle progression.
NOVEL AR FUNCTIONS IN PCA DEVELOPMENT AND PROGRESSION TO CRPC
The identification of frequent fusions between the strongly AR-regulated TMPRSS2 gene and the Ets family transcription factor ERG gene, as well as additional fusions involving TMPRSS2 or other AR-regulated genes, established that AR acquires new functions in PCa.80 Reported downstream functions of ERG in PCa include increased invasion, increased expression of EZH2 and down-regulation of AR expression and transcriptional activity.81,82 However, while it appears clear that ERG contributes to PCa development, the relevant downstream effectors and functions of ERG in PCa have remained to be established. A recent study found that the SOX9 transcription factor, which regulates ductal morphogenesis in fetal prostate and maintenance of stem/progenitor cells in adult tissues,83–86 is a downstream effector of ERG in TMPRSS2:ERG fusion-positive PCa.87 Specifically, ERG in PCa cells binds to a site 3′ of the SOX9 gene and opens a cryptic AR-binding site, so that androgens strongly stimulate SOX9 in TMPRSS2:ERG fusion-positive PCa cells both by driving ERG expression and directly through this ERG-dependent AR-regulated enhancer in the SOX9 gene (Figure 3). Further studies showed that SOX9 mediates the increased invasion downstream of ERG in vitro, and that SOX9 knockdown could markedly impair the in vivo growth of PCa cells expressing the TMPRSS2:ERG fusion gene.87 In genetically engineered mouse models, SOX9 knockdown impairs PCa development driven by MYC and SV40 T antigen,83 while SOX9 overexpression in prostate on a Pten −/+ background results in high-grade dysplastic lesions that progress to invasive PCa in a subset of mice.87,88
Figure 3.
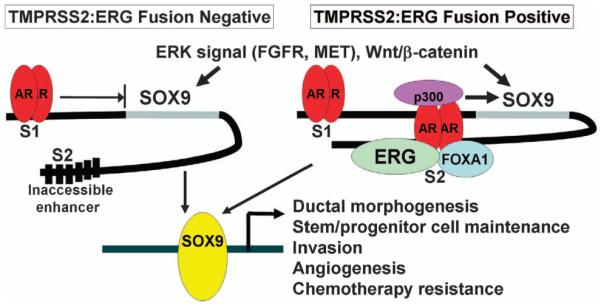
SOX9 is downstream effector of ERG and AR in TMPRSS2:ERG fusion-positive PCa. SOX9 expression can be stimulated by extracellular signals including FGFs and HGF (mediated by their receptors and downstream ERK), and by WNTs though the canonical WNT/β-catenin pathway. AR in fusion-negative cells binds weakly to a site 5′ of the SOX9 gene (S1 site) and can weakly repress basal SOX9 expression (possibly by displacing positive transcription factors). In fusion-positive cells, ERG binds to a cryptic ARE 3′ of the SOX9 gene, with subsequent binding of FOXA1 and strong androgen-stimulated SOX9 expression.
SOX9 is expressed at higher levels in TMPRSS2:ERG fusion-positive versus -negative PCa,87 but is also expressed in fusion-negative tumors where it may be regulated by pathways including MAP kinase and Wnt/β-catenin.89,90 SOX9 expression appears to be further increased in CRPC,90 and higher levels of SOX9 in residual tumor correlated with early relapse in a neoadjuvant clinical trial of androgen deprivation combined with docetaxel and estramustine.91 Significantly, SOX9 expression is repressed by androgens in TMPRSS2:ERG negative PCa cells and in mouse prostate after castration, so that androgen deprivation may increase SOX9 in fusion-negative tumors.87,88 This may have therapeutic implications, as androgen deprivation combined with radiation or cytotoxic chemotherapy may be more effective in fusion-positive tumors expressing high levels of SOX9, that are downregulated by castration, than in fusion-negative tumors. However, further studies are needed to determine whether this increase in SOX9 after androgen deprivation is biologically significant, and whether it affects responses to particular therapies.
Further studies using ChIP-seq methods coupled with gene expression microarrays to comprehensively identify genes that are directly regulated by AR indicate that AR acquires further new functions in CRPC. In particular, AR in CRPC cell lines was found to execute a distinct transcriptional program that included the direct activation of a set of M-phase cell cycle genes such as CDK1 and UBE2C that are overexpressed in CRPC.37 This reprogramming could in part reflect novel intracellular AR ligands or distinct activities of an unliganded AR, but increased H3K4 methylation at AR-binding sites in these genes suggest a role for epigenetic alterations. Significantly, previous studies have indicated that AR signaling, based on expression of genes that are normally stimulated by androgen, decreases in higher Gleason grade tumors,92 and in CRPC.10,79,92 This is consistent with lower androgen levels in CRPC, but may in addition reflect epigenetic reprogramming and narrowing of the spectrum of genes that are AR regulated with tumor progression.
Most recently, EZH2 has been identified as an AR coactivator that may contribute to altering AR function in CRPC.93 EZH2 is a histone methyltransferase associated with the polycomb-repressive complex 2 that is upregulated in CRPC, and had been presumed to be functioning by progressive silencing of tumor suppressor genes though H3K27 trimethylation. However, this recent study found that EZH2 forms a complex with AR in CRPC cells that is recruited to the cis-regulatory elements of AR-target genes including CDK1 and UBE2C, and that it functions as an AR coactivator on these genes by a mechanism that is dependent on its methyltransferase activity, but independent of its ability to methylate H3K27.93 This AR interaction and coactivator function of EZH2 is mediated by AKT-dependent phosphorylation of EZH2 on serine 21, which also impairs its ability to methylate H3K27 and presumably directs it to one or more alternative substrates. Finally, recent data indicate that AR splice variants lacking the LBD, which are increased in CRPC (see below), may regulate a distinct set of genes that include genes driving cell cycle progression.94 These findings could reflect novel interactions between AR splice variants and EZH2, but further studies are needed to determine their molecular basis.95,96
AR REACTIVATION IN CRPC AND RESISTANCE TO CYP17A1 INHIBITION MEDIATED BY INTRATUMORAL ANDROGEN SYNTHESIS
The normal prostate has the enzymatic machinery to synthesize testosterone from serum precursors supplied by the adrenal glands, and this testosterone is normally converted to the higher affinity ligand dihydrotestosterone by 5α-reductases (SRD5A1 and SRD5A2), primarily the type 2 5α-reducatase (SRD5A2) in normal prostate (Figure 4). Intraprostatic synthesis may not be a major source of androgen in men with intact testes and normal circulating testosterone, but can become a substantial source after ADT.21 Indeed, several studies have shown that intraprostatic androgen levels do not decline as markedly as serum levels after ADT.97–101 Moreover, an analysis of AR-regulated gene expression in prostatectomy specimens in patients receiving neoadjuvant (treatment prior to prostatectomy) ADT found that while several androgen-regulated genes (NDRG1, FKBP5, and TMPRSS2) were reduced, many others were not suppressed.102 Taken together, these data demonstrated that intraprostatic androgen synthesis can be a significant source of androgen and may buffer primary PCa against the acute effects of ADT.
Figure 4.

Increased intratumoral androgen synthesis in CRPC and potential mechanisms of resistance to CYP17A1 inhibitors.
Early studies examining intraprostatic androgen levels in men with CRPC and intact prostates suggested that androgen synthesis may be further increased,103,104 and recent studies using more reliable mass spectrometry methods have now clearly established that intraprostatic androgen levels in men with CRPC are not substantially reduced relative to levels in eugonadal men.18,19 Moreover, a study in metastatic CRPC samples showed that androgen levels in many of the CRPC samples were actually increased relative to control tissues, indicating that the progression to CRPC was associated with increased intratumoral accumulation or synthesis of androgen.20 In parallel with these assessments of androgen levels, gene expression studies in CRPC bone marrow metastases identified increased expression of enzymes mediating androgen synthesis from weak adrenal androgens as a molecular mechanism mediating the restoration of androgen levels in CRPC, with the largest increases being in the levels of AKR1C3 mRNA, which reduce androstenedione to testosterone (approximately five-fold median increase, with marked increases in approximately one-third of cases).10 This study also found significant increases in type 1 SRD5A and in HSD3B2, which converts DHEA to androstenedione.10 Subsequent studies in CRPC clinical samples have similarly found increased expression of enzymes mediating androgen synthesis, including AKR1C3 and SRD5A1.20,105,106
CRPC cells also may have increased expression of enzymes mediating cholesterol and fatty acid synthesis, which may be driven by increased SREBP activity and is consistent with their enhanced lipid synthesis.9,107 However, while it now appears clear that PCa cells adapt to ADT by enhancing their ability to metabolize weak androgens produced by the adrenal glands, whether they can synthesize physiologically significant amounts of androgen de novo from cholesterol has been less clear. Studies conducted primarily in LNCaP cells and xenografts showed that enzymes required for de novo steroid synthesis (including CYP17A1, the enzyme that catalyzes 2 steps in androgen synthesis to generate DHEA, see Figure 4), were increased in castration-resistant sublines and could generate detectable levels of androgens.108–110 However, studies in CRPC clinical samples have not consistently found increases in these enzymes.10,20,105 More recent studies in CRPC cell line and xenograft models have now confirmed that PCa cells can generate androgens de novo from cholesterol at levels that are adequate to drive AR transcriptional activity and tumor growth.78,111 Significantly, AR activity and growth of these CRPC xenografts could be suppressed by the CYP17A1 inhibitor abiraterone, and CYP17A1 mRNA was increased in xenografts that progressed after initial responses. Moreover, CYP17A1 mRNA also appeared to be increased in a small set of CRPC clinical samples from patients treated with a CYP17A1 inhibitor.78
Taken together, these findings suggest that CYP17A1-dependent de novo androgen synthesis may not be a major mechanism driving AR in tumors that relapse after standard ADT, where there are very high serum levels of precursor steroids that can be taken up by the tumor cells and converted to testosterone and dihydrotestosterone. However, this mechanism may become more important in patients treated with abiraterone or other CYP17A1 inhibitors that markedly decrease adrenal gland synthesis of precursor steroids. While abiraterone would presumably also be targeting CYP17A1 and androgen synthesis in tumor cells, relative resistance could emerge through mechanisms including CYP17A1 overexpression or mutations, increased synthesis of upstream substrates (possibly including novel substrates that may have higher affinity for CYP17A1 and be metabolized through alternative pathways), or increased drug metabolism or efflux (Figure 4). Alternatively, as abiraterone does not completely ablate serum precursor steroids (particularly DHEA-S, which is still present at substantial levels and can be transported and metabolized to DHEA by steroid sulfatase in PCa cells), tumor cells may adapt by further enhancing their ability to take up and metabolize steroid precursors downstream of CYP17A1. Further studies of clinical samples from CRPC patients with intrinsic and/or acquired resistance to CYP17A1 inhibitors will be critical to determine the role of these and other (see below) resistance mechanisms and to assess therapies targeting these mechanisms.
STRUCTURAL ALTERATIONS IN AR-MEDIATING RESISTANCE TO CASTRATION AND CYP17A1 INHIBITION
Increased AR expression as a result of AR gene amplification,11 or other mechanisms that increase AR gene transcription in CRPC,70,112 can clearly contribute to increasing AR responses to low levels of androgen. Mutations in the AR LBD can alter its structure so that it is activated by alternative ligands including progesterone, hydrocortisone, estradiol and certain AR antagonists.8,12,113–115 These latter mutations are found at increased frequency in patients treated with the AR antagonists flutamide (codons 874 and 877) and bicalutamide (codon 741), but are uncommon in patients treated with medical or surgical castration.12,116 However, as serum levels of progesterone and other upstream steroids are increased in patients treated with CYP17A1 inhibitors, mutant ARs that are activated by these upstream steroids may emerge as an important resistance mechanism.78
AR splice variants that have lost the LBD, and are therefore constitutively active in the absence of ligand, also have been identified in CRPC.117–123 In the majority of these variants, exon 3 (encoding the 3′ end of the DNA-binding domain) is spliced to a cryptic exon in the intron between exons 3 and 4, while exon 4 is spliced out of frame to exon 8 in another major variant. Based on mRNA levels, these variants generally appear to be expressed at low levels relative to the full length AR, and the extent to which they contribute to AR activity in CRPC (either as homodimers or as heterodimers with full length AR) is not yet clear. However, they can be expressed at high levels and drive androgen-independent growth in cells with deletions in the AR locus that impair expression of the full length AR.96,124 Therefore, it seems likely that genetic or epigenetic mechanisms mediating expression of AR variants at levels that can contribute substantially to AR-pathway reactivation will emerge as a resistance mechanism in response to agents that markedly deplete androgens such as CYP17A1 inhibitors or to novel AR antagonists targeting the LBD (see below).96,111 AR antagonists that target the NTD are currently being developed and may be active against these variants.125
Studies from many groups have identified post-translational modifications (serine/threonine phosphorylation, tyrosine phosphorylation, acetylation, methylation, ubiquitylation and sumoylation) of AR that can enhance AR activity in response to low levels of androgens, and may thereby contribute to AR reactivation in CRPC and to intrinsic and/or acquired resistance to CYP17A1 inhibitors.35 Proteins mediating these modifications that may have increased activity in CRPC include CDK1, AKT, SRC, ACK1, p38, JNK, RNF6 and SET9. SRC and ACK1 have been reported to tyrosine phosphorylate AR at distinct sites on the NTD and enhance AR transcriptional activity at low-androgen levels.126–128 CDK1 activated during M-phase phosphorylates serine 81 in the AR NTD, a site that is phosphorylated by CDK9 during interphase and enhances AR transcriptional activity.129–131 AKT can phosphorylate serine 213, but the major kinase targeting this site may be PIM1.132–135 Phosphorylation of this site can mediate MDM2 binding and AR degradation, but this appears to be context dependent and this site may also increase AR levels and modulate AR nuclear localization.132,134,136–138 Recent reports in PTEN-defficient mice indicate that PI3 kinase-pathway activation decreases AR expression, but the role of AR phosphorylation by AKT in this model remains to be defined.139,140 Phosphorylation of serine 650 in the AR hinge region by p38 and JNK stimulates AR nuclear export, and dephosphorylation of this site by PP1 can enhance AR nuclear retention.141,142 The ubiquitin ligase RNF6 is increased in CRPC and can enhance AR activity through non-lysine 48-linked polyubiquitylation, which presumably modulates its interaction with other chromatin-associated proteins.143 Finally, methylation of a site in the AR hinge region by SET9 can enhance the N–C terminal interaction and may increase AR activity in CRPC.144,145
Increased expression or activity of transcriptional coactivator proteins (including SRC2/TIF2, which is amplified in a subset of PCa), or decreases in corepressor proteins, also may enhance AR activity.146–150 Further proteins that associate with AR and may modulate its activity in CRPC include EZH2 and LSD1 (see above), and additional histone methytransferases and demethylases.69,151 Interestingly, exome sequencing indicates that multiple chromatin-modifying enzymes are mutated in advanced CRPC,152,153 suggesting that AR activity in general, or on subsets of genes (such as M-phase genes, see above), may be enhanced through changes in chromatin structure.
MECHANSIMS OF ACTION AND RESISTANCE TO AR ANTAGONISTS
Nonsteroidal AR antagonists including bicalutamide and hydroxyflutamide (active metabolite of flutamide) bind to the steroid-binding pocket in the AR LBD and presumably mediate an alternative conformational change that interferes with formation of the coactivator-binding site (crystal structures of the wild-type AR LBD bound to antagonists have not yet been obtained). Mutations in codons 874 or 877 alter the steroid-binding pocket and allow hydroxyflutamide to generate an agonist confirmation and function as a strong agonist, while a mutation in codon 741 similarly allows bicalutamide to function as a potent AR agonist.26,154,155 Importantly, ChIP and FRAP studies have shown that bicalutamide can still stimulate nuclear accumulation and binding of the wild-type AR to chromatin (although more weak and transient binding compared to androgen), and that its antagonist activity reflects ineffective recruitment of coactivator proteins.32,156,157 In contrast, enzalutamide stimulates less nuclear accumulation of AR, and reporter gene and related studies show that the enzalutamide liganded AR does not bind to chromatin (Figure 1).158 A sensitive fluorescence resonance energy transfer assay indicates that this may reflect more effective impairment of the AR–N–C terminal interaction, and possibly AR interaction with other proteins. However, the precise basis for this loss of chromatin binding, which likely contributes to the improved efficacy of enzalutamide versus bicalutamade in CRPC, remains to be determined.158 Another AR antagonist related to enzalutamide that is currently in clinical trials (ARN-509),159 as well as a series of recently reported unrelated AR antagonists, similarly inhibit AR chromatin binding.160
The agonist-liganded AR can also interact weakly with transcriptional corepressor proteins including NCoR and SMRT, and RNA interference targeting NCoR and SMRT can enhance androgen-stimulated AR transcriptional activity (see above). This AR interaction with NCoR and SMRT appears to be mediated primarily by the AR NTD, as the agonist-liganded coactivator-binding site in the AR LBD cannot accommodate the extended CoRNR boxes in NCoR and SMRT that mediate binding to other unliganded nuclear receptors. ChIP studies indicate that AR recruitment of NCoR and SMRT are increased by the AR antagonist bicalutamide, which may reflect an alternative non-agonist conformation of the AR LBD that can better accommodate CoRNR box binding.36,161,162 The interaction between AR and NCoR/SMRT also can be further enhanced by mifepristone.64,65 However, the extent to which NCoR/SMRT recruitment contributes to the antagonist activity of bicalutamide is not clear, as bicalutamide still functions as an AR antagonist in cells treated with NCoR and SMRT small interfering RNA.163
One study found that bicalutamide could function as an AR agonist in the setting of inflammation due to NCoR/SMRT translocation out of the nucleus, but the extent of this agonist activity and whether factors in addition to decreased nuclear NCoR/SMRT mediate this activity are not clear.164 Bicalutamide also has detectable partial agonist activity in cells that are transiently or stably overexpressing AR.14 However, this activity is very weak compared to the potent activity mediated by true agonists or by bicalutamide-stimulated Y741-mutant ARs, and bicalutamide still functions as an antagonist of androgen-stimulated AR activity in cells that overexpress AR. Nonetheless, it is reasonable to suggest that a number of factors, including altered expression or post-translational modification of AR, coactivators or corepressors, may enhance the very weak partial agonist activity of bicalutamide adequately to achieve a threshold level of AR activity required to support tumor growth. However, it remains to be determined whether bicalutamide resistance in CRPC reflects bicalutamide function as an agonist for wild-type AR that can drive the substantial AR reactivation in CRPC, versus lack of potency in competing for increased intratumoral androgens, or other mechanisms. Mechanisms mediating intrinsic or acquired AR-pathway resistance to enzalutamide remain to be determined, but similarly to bicalutamide may include AR mutations, constitutively active AR splice variants lacking the LBD (see above), or increased intratumoral androgens. Moreover, further novel mechanisms may emerge that can drive chromatin binding by the enzalutamide liganded wild-type AR.
CONCLUSIONS
It is now apparent that increased intratumoral androgen synthesis and subsequent partial restoration of AR transcriptional activity make a significant contribution to CRPC. To build on the clinical success of abiraterone and enzalutamide, it will clearly be necessary to identify and target mechanisms of resistance to these and related agents (and/or to employ these agents earlier in the course of the disease). Multiple mechanisms may contribute to restoration of AR activity after treatment with CYP17A1 inhibitors, including further increases in intratumoral androgen synthesis (de novo or from serum DHEA-S) and alterations in AR or associated proteins that can enhance AR responses to very low levels of ligand. The former mechanism may be addressed by targeting additional steps in androgen synthesis or possibly by addition of an AR antagonist, while the latter may be addressed by combination therapies with agents blocking kinase or other pathways that enhance AR activity. Resistance to enzalutamide or other AR antagonists may be mediated by similar mechanisms, or by mechanisms that stimulate partial agonist activity, and there is clear interest in the development of additional antagonists that may circumvent these mechanisms by enhancing AR degradation. As treatment with flutamide or bicalutamide selects mutant ARs that are activated by these agents, it also is reasonable to assume that mutant ARs will emerge that have decreased affinity or are stimulated by enzalutamide. The increased levels of pregnanes in patients treated with CYP17A1 inhibitors may similarly select for AR mutations (including those in codons 874 and 877) that enhance AR activation by progesterone or other steroids upstream of CYP17A1. Increased expression of AR variants lacking the LBD also may emerge as a significant mechanism of resistance to inhibitors of steroid synthesis and to antagonists targeting the LBD, which should spur the further development of agents targeting the AR NTD or DNA-binding domain. Finally, one can anticipate that more effective targeting of AR will lead to the increased emergence of tumors that are no longer dependent on AR, and that will require alternative therapeutic strategies.
ACKNOWLEDGEMENTS
Work from the authors cited in this review has been supported by awards from the National Institutes of Health, Department of Defense Prostate Cancer Research Program and the Prostate Cancer Foundation. We apologize to the many colleagues whose work we were unable to discuss or cite.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Mahoney EM, Harrison JH. Bilateral adrenalectomy for palliative treatment of prostatic cancer. J Urol. 1972;108:936–938. doi: 10.1016/s0022-5347(17)60911-x. [DOI] [PubMed] [Google Scholar]
- 2.Small EJ, Halabi S, Dawson NA, Stadler WM, Rini BI, Picus J, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583) J Clin Oncol. 2004;22:1025–1033. doi: 10.1200/JCO.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 3.Yap TA, Carden CP, Attard G, de Bono JS. Targeting CYP17: established and novel approaches in prostate cancer. Curr Opin Pharmacol. 2008;8:449–457. doi: 10.1016/j.coph.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Taplin ME, Regan MM, Ko YJ, Bubley GJ, Duggan SE, Werner L, et al. Phase II study of androgen synthesis inhibition with ketoconazole, hydrocortisone, and dutasteride in asymptomatic castration-resistant prostate cancer. Clin Cancer Res. 2009;15:7099–7105. doi: 10.1158/1078-0432.CCR-09-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenberger MA, Blumenstein BA, Crawford ED, Miller G, McLeod DG, Loehrer PJ, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–1042. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- 6.Samson DJ, Seidenfeld J, Schmitt B, Hasselblad V, Albertsen PC, Bennett CL, et al. Systematic review and meta-analysis of monotherapy compared with combined androgen blockade for patients with advanced prostate carcinoma. Cancer. 2002;95:361–376. doi: 10.1002/cncr.10647. [DOI] [PubMed] [Google Scholar]
- 7.Ruizeveld de Winter JA, Janssen PJ, Sleddens HM, Verleun-Mooijman MC, Trapman J, Brinkmann AO, et al. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am J Pathol. 1994;144:735–746. [PMC free article] [PubMed] [Google Scholar]
- 8.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 9.Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 11.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 12.Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–2515. [PubMed] [Google Scholar]
- 13.Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, et al. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58:5718–5724. [PubMed] [Google Scholar]
- 14.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 15.Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, et al. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994;54:2577–2581. [PubMed] [Google Scholar]
- 16.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008–1013. [PubMed] [Google Scholar]
- 17.Yuan X, Li T, Wang H, Zhang T, Barua M, Borgesi RA, et al. Androgen receptor remains critical for cell-cycle progression in androgen-independent CWR22 prostate cancer cells. Am J Pathol. 2006;169:682–696. doi: 10.2353/ajpath.2006.051047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohler JL, Gregory CW, Ford OH, 3rd, Kim D, Weaver CM, Petrusz P, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 19.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–4657. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 20.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer. 2011;18:R175–R182. doi: 10.1530/ERC-10-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 24.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem. 2000;275:26164–26171. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- 26.Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, An Y, et al. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc Natl Acad Sci USA. 2001;98:4904–4909. doi: 10.1073/pnas.081565498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He B, Gampe RT, Jr., Kole AJ, Hnat AT, Stanley TB, An G, et al. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol Cell. 2004;16:425–438. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 28.Hur E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, Fletterick RJ. Recognition and accommodation at the androgen receptor coactivator binding interface. PLoS Biol. 2004;2:E274. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaufele F, Carbonell X, Guerbadot M, Borngraeber S, Chapman MS, Ma AA, et al. The structural basis of androgen receptor activation: intramolecular and intermolecular amino-carboxy interactions. Proc Natl Acad Sci USA. 2005;102:9802–9807. doi: 10.1073/pnas.0408819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Royen ME, Cunha SM, Brink MC, Mattern KA, Nigg AL, Dubbink HJ, et al. Compartmentalization of androgen receptor protein-protein interactions in living cells. J Cell Biol. 2007;177:63–72. doi: 10.1083/jcb.200609178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Royen ME, van Cappellen WA, de Vos C, Houtsmuller AB, Trapman J. Step-wise androgen receptor dimerization. J Cell Sci. 2012;125:1970–1979. doi: 10.1242/jcs.096792. [DOI] [PubMed] [Google Scholar]
- 32.Klokk TI, Kurys P, Elbi C, Nagaich AK, Hendarwanto A, Slagsvold T, et al. Ligand-specific dynamics of the androgen receptor at its response element in living cells. Mol Cell Biol. 2007;27:1823–1843. doi: 10.1128/MCB.01297-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, et al. Nucleosome dynamics define transcriptional enhancers. Nat Genet. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gioeli D, Paschal BM. Post-translational modification of the androgen receptor. Mol Cell Endocrinol. 2012;352:70–78. doi: 10.1016/j.mce.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 36.Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9:601–610. doi: 10.1016/s1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 37.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30:2719–2733. doi: 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783–7792. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- 40.Knudsen KE, Arden KC, Cavenee WK. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J Biol Chem. 1998;273:20213–20222. doi: 10.1074/jbc.273.32.20213. [DOI] [PubMed] [Google Scholar]
- 41.Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008;6:e001. doi: 10.1621/nrs.06001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu L, Schulz H, Wolf DA. The F-box protein SKP2 mediates androgen control of p27 stability in LNCaP human prostate cancer cells. BMC Cell Biol. 2002;3:22. doi: 10.1186/1471-2121-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang Z, Zhang T, Dizeyi N, Chen S, Wang H, Swanson KD, et al. Androgen receptor enhances p27 degradation in prostate cancer cells through rapid and selective TORC2 activation. J Biol Chem. 2012;287:2090–2098. doi: 10.1074/jbc.M111.323303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Xu Y, Fang Z, Chen S, Balk SP, Yuan X. Doxycycline regulated induction of AKT in murine prosate drives proliferation independently of p27 cyclin dependent kinase inhibitor downregulation. PLoS ONE. 2012;7:e41330. doi: 10.1371/journal.pone.0041330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu S, Liu M, Epner DE, Tsai SY, Tsai MJ. Androgen regulation of the cyclin-dependent kinase inhibitor p21 gene through an androgen response element in the proximal promoter. Mol Endocrinol. 1999;13:376–384. doi: 10.1210/mend.13.3.0254. [DOI] [PubMed] [Google Scholar]
- 46.Yan G, Fukabori Y, Nikolaropoulos S, Wang F, McKeehan WL. Heparin-binding keratinocyte growth factor is a candidate stromal-to-epithelial-cell andromedin. Mol Endocrinol. 1992;6:2123–2128. doi: 10.1210/mend.6.12.1491693. [DOI] [PubMed] [Google Scholar]
- 47.Curtin D, Jenkins S, Farmer N, Anderson AC, Haisenleder DJ, Rissman E, et al. Androgen suppression of GnRH-stimulated rat LHbeta gene transcription occurs through Sp1 sites in the distal GnRH-responsive promoter region. Mol Endocrinol. 2001;15:1906–1917. doi: 10.1210/mend.15.11.0723. [DOI] [PubMed] [Google Scholar]
- 48.Verras M, Lee J, Xue H, Li TH, Wang Y, Sun Z. The androgen receptor negatively regulates the expression of c-Met: implications for a novel mechanism of prostate cancer progression. Cancer Res. 2007;67:967–975. doi: 10.1158/0008-5472.CAN-06-3552. [DOI] [PubMed] [Google Scholar]
- 49.Grosse A, Bartsch S, Baniahmad A. Androgen receptor-mediated gene repression. Mol Cell Endocrinol. 2012;352:46–56. doi: 10.1016/j.mce.2011.06.032. [DOI] [PubMed] [Google Scholar]
- 50.Truica CI, Byers S, Gelmann EP. Beta-catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res. 2000;60:4709–4713. [PubMed] [Google Scholar]
- 51.Shah S, Hecht A, Pestell R, Byers SW. Trans-repression of beta-catenin activity by nuclear receptors. J Biol Chem. 2003;278:48137–48145. doi: 10.1074/jbc.M307154200. [DOI] [PubMed] [Google Scholar]
- 52.Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, et al. Linking beta-catenin to androgen-signaling pathway. J Biol Chem. 2002;277:11336–11344. doi: 10.1074/jbc.M111962200. [DOI] [PubMed] [Google Scholar]
- 53.Chesire DR, Isaacs WB. Ligand-dependent inhibition of beta-catenin/TCF signaling by androgen receptor. Oncogene. 2002;21:8453–8469. doi: 10.1038/sj.onc.1206049. [DOI] [PubMed] [Google Scholar]
- 54.Mulholland DJ, Read JT, Rennie PS, Cox ME, Nelson CC. Functional localization and competition between the androgen receptor and T-cell factor for nuclear beta-catenin: a means for inhibition of the Tcf signaling axis. Oncogene. 2003;22:5602–5613. doi: 10.1038/sj.onc.1206802. [DOI] [PubMed] [Google Scholar]
- 55.Chen SY, Wulf G, Zhou XZ, Rubin MA, Lu KP, Balk SP. Activation of beta-catenin signaling in prostate cancer by peptidyl-prolyl isomerase Pin1-mediated abrogation of the androgen receptor-beta-catenin interaction. Mol Cell Biol. 2006;26:929–939. doi: 10.1128/MCB.26.3.929-939.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuan X, Lu ML, Li T, Balk SP. SRY interacts with and negatively regulates androgen receptor transcriptional activity. J Biol Chem. 2001;276:46647–46654. doi: 10.1074/jbc.M108404200. [DOI] [PubMed] [Google Scholar]
- 57.Jouravel N, Sablin E, Arnold LA, Guy RK, Fletterick RJ. Interaction between the androgen receptor and a segment of its corepressor SHP. Acta Crystallogr D Biol Crystallogr. 2007;63:1198–1200. doi: 10.1107/S0907444907045702. [DOI] [PubMed] [Google Scholar]
- 58.Moehren U, Papaioannou M, Reeb CA, Hong W, Baniahmad A. Alien interacts with the human androgen receptor and inhibits prostate cancer cell growth. Mol Endocrinol. 2007;21:1039–1048. doi: 10.1210/me.2006-0468. [DOI] [PubMed] [Google Scholar]
- 59.Gamble SC, Chotai D, Odontiadis M, Dart DA, Brooke GN, Powell SM, et al. Prohibitin, a protein downregulated by androgens, represses androgen receptor activity. Oncogene. 2007;26:1757–1768. doi: 10.1038/sj.onc.1209967. [DOI] [PubMed] [Google Scholar]
- 60.Belandia B, Powell SM, Garcia-Pedrero JM, Walker MM, Bevan CL, Parker MG. Hey1, a mediator of notch signaling, is an androgen receptor corepressor. Mol Cell Biol. 2005;25:1425–1436. doi: 10.1128/MCB.25.4.1425-1436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu X, Li P, Roeder RG, Wang Z. Inhibition of androgen receptor-mediated transcription by amino-terminal enhancer of split. Mol Cell Biol. 2001;21:4614–4625. doi: 10.1128/MCB.21.14.4614-4625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoon HG, Wong J. The corepressors silencing mediator of retinoid and thyroid hormone receptor and nuclear receptor corepressor are involved in agonist- and antagonist-regulated transcription by androgen receptor. Mol Endocrinol. 2006;20:1048–1060. doi: 10.1210/me.2005-0324. [DOI] [PubMed] [Google Scholar]
- 63.Cheng S, Brzostek S, Lee SR, Hollenberg AN, Balk SP. Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol Endocrinol. 2002;16:1492–1501. doi: 10.1210/mend.16.7.0870. [DOI] [PubMed] [Google Scholar]
- 64.Hodgson MC, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E, et al. The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J Biol Chem. 2005;280:6511–6519. doi: 10.1074/jbc.M408972200. [DOI] [PubMed] [Google Scholar]
- 65.Song LN, Coghlan M, Gelmann EP. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol. 2004;18:70–85. doi: 10.1210/me.2003-0189. [DOI] [PubMed] [Google Scholar]
- 66.Zhao JC, Yu J, Runkle C, Wu L, Hu M, Wu D, et al. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012;22:322–331. doi: 10.1101/gr.131508.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chng KR, Chang CW, Tan SK, Yang C, Hong SZ, Sng NY, et al. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J. 2012;31:2810–2823. doi: 10.1038/emboj.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 69.Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9:347–353. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 70.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20:457–471. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 72.Garcia-Bassets I, Kwon YS, Telese F, Prefontaine GG, Hutt KR, Cheng CS, et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128:505–518. doi: 10.1016/j.cell.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu Q, Kwon YS, Nunez E, Cardamone MD, Hutt KR, Ohgi KA, et al. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci USA. 2008;105:19199–19204. doi: 10.1073/pnas.0810634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, et al. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- 75.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–887. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 76.Metzger E, Yin N, Wissmann M, Kunowska N, Fischer K, Friedrichs N, et al. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol. 2008;10:53–60. doi: 10.1038/ncb1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 2010;464:792–796. doi: 10.1038/nature08839. [DOI] [PubMed] [Google Scholar]
- 78.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–6513. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mendiratta P, Mostaghel E, Guinney J, Tewari AK, Porrello A, Barry WT, et al. Genomic strategy for targeting therapy in castration-resistant prostate cancer. J Clin Oncol. 2009;27:2022–2029. doi: 10.1200/JCO.2008.17.2882. [DOI] [PubMed] [Google Scholar]
- 80.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 81.Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang Z, Hurley PJ, Simons BW, Marchionni L, Berman DM, Ross AE, et al. Sox9 is required for prostate development and prostate cancer initiation. Oncotarget. 2012;3:651–663. doi: 10.18632/oncotarget.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schaeffer EM, Marchionni L, Huang Z, Simons B, Blackman A, Yu W, et al. Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene. 2008;27:7180–7191. doi: 10.1038/onc.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thomsen MK, Butler CM, Shen MM, Swain A. Sox9 is required for prostate development. Dev Biol. 2008;316:302–311. doi: 10.1016/j.ydbio.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 86.Wang H, Leav I, Ibaragi S, Wegner M, Hu GF, Lu ML, et al. SOX9 is expressed in human fetal prostate epithelium and enhances prostate cancer invasion. Cancer Res. 2008;68:1625–1630. doi: 10.1158/0008-5472.CAN-07-5915. [DOI] [PubMed] [Google Scholar]
- 87.Cai C, Wang H, He HH, Chen S, He L, Ma F, et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J Clin Invest. 2013;123:1109–1122. doi: 10.1172/JCI66666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thomsen MK, Ambroisine L, Wynn S, Cheah KS, Foster CS, Fisher G, et al. SOX9 elevation in the prostate promotes proliferation and cooperates with PTEN loss to drive tumor formation. Cancer Res. 2010;70:979–987. doi: 10.1158/0008-5472.CAN-09-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12:559–571. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 90.Wang H, McKnight NC, Zhang T, Lu ML, Balk SP, Yuan X. SOX9 is expressed in normal prostate basal cells and regulates androgen receptor expression in prostate cancer cells. Cancer Res. 2007;67:528–536. doi: 10.1158/0008-5472.CAN-06-1672. [DOI] [PubMed] [Google Scholar]
- 91.Bhatt RS, Werner L, Regan MM, Yannucci J, Ko Y-J, Wang H-Y, et al. Possible risk factors associated with relpase in patients treated with neoadjuvant chemohormonal therapy for high risk prostate cancer. Open Prostate Cancer J. 2011;4:1–8. [Google Scholar]
- 92.Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 93.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–3462. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287:19736–19749. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Geller J, Albert J. Effects of castration compared with total androgen blockade on tissue dihydrotestosterone (DHT) concentration in benign prostatic hyperplasia (BPH) Urol Res. 1987;15:151–153. doi: 10.1007/BF00254427. [DOI] [PubMed] [Google Scholar]
- 98.Belanger B, Belanger A, Labrie F, Dupont A, Cusan L, Monfette G. Comparison of residual C-19 steroids in plasma and prostatic tissue of human, rat and guinea pig after castration: unique importance of extratesticular androgens in men. J Steroid Biochem. 1989;32:695–698. doi: 10.1016/0022-4731(89)90514-1. [DOI] [PubMed] [Google Scholar]
- 99.Mizokami A, Koh E, Fujita H, Maeda Y, Egawa M, Koshida K, et al. The adrenal androgen androstenediol is present in prostate cancer tissue after androgen deprivation therapy and activates mutated androgen receptor. Cancer Res. 2004;64:765–771. doi: 10.1158/0008-5472.can-03-0130. [DOI] [PubMed] [Google Scholar]
- 100.Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clin Cancer Res. 2004;10:7121–7126. doi: 10.1158/1078-0432.CCR-04-0913. [DOI] [PubMed] [Google Scholar]
- 101.Page ST, Lin DW, Mostaghel EA, Hess DL, True LD, Amory JK, et al. Persistent intraprostatic androgen concentrations after medical castration in healthy men. J Clin Endocrinol Metab. 2006;91:3850–3856. doi: 10.1210/jc.2006-0968. [DOI] [PubMed] [Google Scholar]
- 102.Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67:5033–5041. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 103.Geller J, Albert JD, Nachtsheim DA, Loza D. Comparison of prostatic cancer tissue dihydrotestosterone levels at the time of relapse following orchiectomy or estrogen therapy. J Urol. 1984;132:693–696. doi: 10.1016/s0022-5347(17)49829-6. [DOI] [PubMed] [Google Scholar]
- 104.Geller J. Rationale for blockade of adrenal as well as testicular androgens in the treatment of advanced prostate cancer. Semin Oncol. 1985;12:28–35. [PubMed] [Google Scholar]
- 105.Hofland J, van Weerden WM, Dits NF, Steenbergen J, van Leenders GJ, Jenster G, et al. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010;70:1256–1264. doi: 10.1158/0008-5472.CAN-09-2092. [DOI] [PubMed] [Google Scholar]
- 106.Cai C, Wang H, Xu Y, Chen S, Balk SP. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res. 2009;69:6027–6032. doi: 10.1158/0008-5472.CAN-09-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. Dys-regulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004;64:2212–2221. doi: 10.1158/0008-5472.can-2148-2. [DOI] [PubMed] [Google Scholar]
- 108.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 109.Locke JA, Nelson CC, Adomat HH, Hendy SC, Gleave ME, Guns ES. Steroidogenesis inhibitors alter but do not eliminate androgen synthesis mechanisms during progression to castration-resistance in LNCaP prostate xenografts. J Steroid Biochem Mol Biol. 2009;115:126–136. doi: 10.1016/j.jsbmb.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 110.Dillard PR, Lin MF, Khan SA. Androgen-independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Mol Cell Endocrinol. 2008;295:115–120. doi: 10.1016/j.mce.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–5925. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, Thangavel C, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120:4478–4492. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Culig Z, Hobisch A, Cronauer MV, Cato AC, Hittmair A, Radmayr C, et al. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol Endocrinol. 1993;7:1541–1550. doi: 10.1210/mend.7.12.8145761. [DOI] [PubMed] [Google Scholar]
- 114.Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, et al. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 2000;6:703–706. doi: 10.1038/76287. [DOI] [PubMed] [Google Scholar]
- 115.Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534–540. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 116.Taplin ME, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J, et al. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol. 2003;21:2673–2678. doi: 10.1200/JCO.2003.11.102. [DOI] [PubMed] [Google Scholar]
- 117.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci USA. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18:R183–R196. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS ONE. 2011;6:e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31:4759–4767. doi: 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 126.Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, et al. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci USA. 2007;104:8438–8443. doi: 10.1073/pnas.0700420104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309–319. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 128.Kraus S, Gioeli D, Vomastek T, Gordon V, Weber MJ. Receptor for activated C kinase 1 (RACK1) and Src regulate the tyrosine phosphorylation and function of the androgen receptor. Cancer Res. 2006;66:11047–11054. doi: 10.1158/0008-5472.CAN-06-0596. [DOI] [PubMed] [Google Scholar]
- 129.Chen S, Gulla S, Cai C, Balk SP. Androgen receptor serine 81 phosphorylation mediates chromatin binding and transcriptional activation. J Biol Chem. 2012;287:8571–8583. doi: 10.1074/jbc.M111.325290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chen S, Xu Y, Yuan X, Bubley GJ, Balk SP. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc Natl Acad Sci USA. 2006;103:15969–15974. doi: 10.1073/pnas.0604193103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gordon V, Bhadel S, Wunderlich W, Zhang J, Ficarro SB, Mollah SA, et al. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Mol Endocrinol. 2010;24:2267–2280. doi: 10.1210/me.2010-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lin HK, Hu YC, Yang L, Altuwaijri S, Chen YT, Kang HY, et al. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer LNCaP cells with different passage numbers. J Biol Chem. 2003;278:50902–50907. doi: 10.1074/jbc.M300676200. [DOI] [PubMed] [Google Scholar]
- 133.Lin HK, Yeh S, Kang HY, Chang C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc Natl Acad Sci USA. 2001;98:7200–7205. doi: 10.1073/pnas.121173298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Taneja SS, Ha S, Swenson NK, Huang HY, Lee P, Melamed J, et al. Cell-specific regulation of androgen receptor phosphorylation in vivo. J Biol Chem. 2005;280:40916–40924. doi: 10.1074/jbc.M508442200. [DOI] [PubMed] [Google Scholar]
- 135.Ha S, Iqbal NJ, Mita P, Ruoff R, Gerald WL, Lepor H, et al. Phosphorylation of the androgen receptor by PIM1 in hormone refractory prostate cancer. Oncogene. doi: 10.1038/onc.2012.412. e-pub ahead of print 17 September 2012; doi:10.1038/onc.2012.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gaughan L, Logan IR, Neal DE, Robson CN. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005;33:13–26. doi: 10.1093/nar/gki141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ha S, Ruoff R, Kahoud N, Logan SK, Franke TF. Androgen receptor levels are upregulated by Akt in prostate cancer. Endocr Relat Cancer. 2011;18:245–255. doi: 10.1530/ERC-10-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J. 2002;21:4037–4048. doi: 10.1093/emboj/cdf406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, et al. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol Endocrinol. 2006;20:503–515. doi: 10.1210/me.2005-0351. [DOI] [PubMed] [Google Scholar]
- 142.Chen S, Kesler CT, Paschal BM, Balk SP. Androgen receptor phosphorylation and activity are regulated by an association with protein phosphatase 1. J Biol Chem. 2009;284:25576–25584. doi: 10.1074/jbc.M109.043133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu K, Shimelis H, Linn DE, Jiang R, Yang X, Sun F, et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell. 2009;15:270–282. doi: 10.1016/j.ccr.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gaughan L, Stockley J, Wang N, McCracken SR, Treumann A, Armstrong K, et al. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011;39:1266–1279. doi: 10.1093/nar/gkq861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ko S, Ahn J, Song CS, Kim S, Knapczyk-Stwora K, Chatterjee B. Lysine methylation and functional modulation of androgen receptor by Set9 methyltransferase. Mol Endocrinol. 2011;25:433–444. doi: 10.1210/me.2010-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–4319. [PubMed] [Google Scholar]
- 147.Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, et al. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem. 2004;279:7119–7130. doi: 10.1074/jbc.M307649200. [DOI] [PubMed] [Google Scholar]
- 148.Agoulnik IU, Vaid A, Bingman WE, 3rd, Erdeme H, Frolov A, Smith CL, et al. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 2005;65:7959–7967. doi: 10.1158/0008-5472.CAN-04-3541. [DOI] [PubMed] [Google Scholar]
- 149.Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE, 3rd, Erdem H, et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 2006;66:10594–10602. doi: 10.1158/0008-5472.CAN-06-1023. [DOI] [PubMed] [Google Scholar]
- 150.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 152.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, Dumpit RF, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci USA. 2011;108:17087–17092. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci USA. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bohl CE, Wu Z, Miller DD, Bell CE, Dalton JT. Crystal structure of the T877A human androgen receptor ligand-binding domain complexed to cyproterone acetate provides insight for ligand-induced conformational changes and structure-based drug design. J Biol Chem. 2007;282:13648–13655. doi: 10.1074/jbc.M611711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J Biol Chem. 2002;277:26321–26326. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- 157.Farla P, Hersmus R, Trapman J, Houtsmuller AB. Antiandrogens prevent stable DNA-binding of the androgen receptor. J Cell Sci. 2005;118:4187–4198. doi: 10.1242/jcs.02546. [DOI] [PubMed] [Google Scholar]
- 158.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72:1494–1503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shen HC, Shanmugasundaram K, Simon NI, Cai C, Wang H, Chen S, et al. In silico discovery of androgen receptor antagonists with activity in castration resistant prostate cancer. Mol Endocrinol. 2012;26:1836–1846. doi: 10.1210/me.2012-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kang Z, Janne OA, Palvimo JJ. Coregulator recruitment and histone modifications in transcriptional regulation by the androgen receptor. Mol Endocrinol. 2004;18:2633–2648. doi: 10.1210/me.2004-0245. [DOI] [PubMed] [Google Scholar]
- 162.Hodgson MC, Shen HC, Hollenberg AN, Balk SP. Structural basis for nuclear receptor corepressor recruitment by antagonist-liganded androgen receptor. Mol Cancer Ther. 2008;7:3187–3194. doi: 10.1158/1535-7163.MCT-08-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hodgson MC, Astapova I, Hollenberg AN, Balk SP. Activity of androgen receptor antagonist bicalutamide in prostate cancer cells is independent of NCoR and SMRT corepressors. Cancer Res. 2007;67:8388–8395. doi: 10.1158/0008-5472.CAN-07-0617. [DOI] [PubMed] [Google Scholar]
- 164.Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]