

Abstract
We present an algorithm, COMPEL, (COnstant Molecular Pressure with Ewald sum for Long range forces) to conduct simulations in the NPT ensemble. The algorithm combines novel features recently proposed in the literature to obtain a highly efficient and accurate numerical integrator. COMPEL exploits the concepts of molecular pressure, rapid stochastic relaxation to equilibrium, exact calculation of the contribution to the pressure of long range non-bonded forces with Ewald summation, and the use of Trotter expansion to generate a robust, highly stable, symmetric and accurate algorithm. Explicit implementation in the MOIL program and illustrative numerical examples are discussed.
Graphical Abstract
Introduction
Molecular dynamics (MD) is a powerful and flexible tool for the study of complex systems at the molecular and atomic level. The models and algorithms that we use today are the product of some fifty years of continuous development. A major direction for study throughout the years has been the design of algorithms that allow the sampling of different statistical mechanical ensembles.
The macroscopic (thermodynamic) state of a statistical mechanical system can be characterized by the quantities that are fixed at defined values.1,2 Experiments carried out in laboratory conditions are typically defined by constant pressure, temperature and number of particles; these conditions correspond to the isobaric-isothermal ensemble, also known as the NPT ensemble. While all ensembles are equivalent in the thermodynamic limit, switching between ensembles for finite size systems can be challenging. For example, using the NVT ensemble to mimic NPT requires a specified choice of density. There is no simple way to estimate the density of non-homogeneous systems such as solvated proteins, membranes or micelles. Even in the case of liquid mixtures, only the densities of pure liquids are usually known. It is therefore essential to carry out equilibration simulations in which the volume can be adjusted such that the pressure matches a target value. Further complications arise when the system under study is subject to driving forces which alter its thermodynamical state; in such a case it is therefore necessary to perform molecular dynamics simulations in which the volume is automatically adjusted so that the pressure remains constant.
Many algorithms, including some quite recent proposals, have been suggested to sample from the NPT ensemble (e.g.3–6). In this paper we present a stochastic algorithm for the isobaric-isothermal ensemble that combines the best features previously introduced in different contexts to obtain a comprehensive, accurate and stable algorithm. Our approach exploits and combines state-of-the-art simulation techniques in two distinct areas: (i) algorithms for the integration of the equations of motion based on certain splittings of stochastic differential equations7–11 and (ii) algorithms for the calculation of the forces based on Ewald summation.12–15 We have implemented and evaluated our method in the molecular dynamics software package MOIL.16
In microcanonical molecular dynamics, Newton’s equations of motion are solved to advance the system in time, thus generating a trajectory. One typically assumes ergodicity of the dynamics and hence that the trajectory samples all accessible configurations of the micro-canonical ensemble (or NVE). To sample the isobaric-isothermal ensemble, the simulation needs to incorporate additional mechanisms in order to control temperature (thermostat) and pressure (barostat). The most common ways to impose a temperature control are velocity rescaling,17 weak coupling method,18 stochastic methods that mimic the behavior of a thermal bath such as the Langevin equation19 and Nosé-Hoover dynamics.20 Nosé introduced the idea that a thermal bath can be modelled using an additional degree of freedom over which one may integrate to provide the correct sampling of physical quantities. The approach has been referred to as pseudomicrocanonical simulation, as the integration of the extended system is essentially conservative, while averages of quantities which are functions of position and momenta only correspond to the NVT ensemble. Nosé’s seminal idea has led to a variety of different extended system methods.
All of the methods mentioned above are in principle able to reproduce correct thermodynamics, i.e. the correct microstate distribution of the corresponding ensemble. However, any thermostating algorithm alters the dynamics of the system with respect to Newton’s equations of motion. These thermostat-dependent artifacts in the dynamics can hinder the quality of measurement of transport properties and kinetics. Of particular importance is the interplay between ergodic sampling of the thermodynamic ensemble and the need to capture dynamical approximation properties of the underlying Hamiltonian system. With respect to other methods, the Nosé-Hoover thermostat is thought to better preserve dynamical properties since the perturbation to the system is acting through a single auxiliary degree of freedom. Evans21 pointed out that the perturbation of dynamics is O(1/ν) where ν is the number of degrees of freedom. Recently, Basconi and Shirts,22 in numerical experiments, have confirmed that the transport properties of several systems simulated using Nosé-Hoover are statistically indistinguishable from those of the same system simulated in the microcanonical ensemble. Leimkuhler and collaborators23 have studied the transport properties of stochastic thermostat methods, observing that for example the combination of Nosé-Hoover with degenerate noise in the auxiliary thermostat variable only (Nosé-Hoover-Langevin dynamics24) is capable of accurately representing dynamical properties.
For the control of pressure, most modern schemes build on the extended system approach of Andersen25 which maintains the pressure by allowing the volume of the simulation cell to fluctuate under the control of an additional degree of freedom, sometimes referred to as a “piston,” which is usually implemented in combination with some sort of thermostat. A drawback of the Andersen barostat is that the mass of the piston determines the way in which the system approaches to equilibrium. While the average volume and the magnitude of its fluctuations are correct there is a significant memory visible in volume fluctuations. The volume as a function of time contains in fact regular oscillations (“ringing”) inversely proportional to the mass of the piston.26 The ringing phenomenon leads to difficulties in the choice of parameters. A method for controlling the oscillations is the “Langevin piston” method due to Feller et al27 which couples an under-damped Langevin dynamics process to the piston equation. We incorporate a similar technique here so that, in addition of producing the correct dynamics and reasonable kinetics at the limit of long time, we also seek to control the mixing time (the necessary waiting time during which the system relaxes to the appropriate equilibrium state) to be as short as possible.
Motions along bonds in molecular systems are typically the fastest degrees of freedom. These set an upper bound on the timestep that can be used in integrating the equations on motion. For this reason, in MD it is often beneficial to impose holonomic constraints to speed up the simulation and to make it more stable by “freezing” the degrees of freedom with the fastest dynamics.28 The use of NPT algorithms in the presence of constraints is not entirely straightforward; the problem was solved by Kneller and Mulders29 and further investigated by Ciccotti et al.30 Their approach is demanding and, fortunately, not necessary; the use of the alternate approach of molecular pressure is a simpler and more efficient. In “molecular pressure” the pressure is calculated using positions and momenta of molecules instead of atoms; the equivalence between the molecular and the atomic formulations of the pressure was first proved Ciccotti and Ryckaert.31,32 This procedure avoids consideration of covalent bonding forces. The atomic and the molecular pressure are a special case of a generalized scheme in which the pressure is expressed by using the positions and momenta of subsets of atoms within molecules.4 The use of molecular pressure in MD recently gained new interest; see for example the recent implementation in the software Desmond of the Martyna-Tobias-Klein algorithm applied to molecular pressure.3
By the use of molecular pressure the NPT algorithm acts by rescaling molecular positions only; thus leaving bonds unaffected. Molecular pressure provides two advantages compared to pressure calculations based on atomic positions. First, to calculate the molecular pressure we only need to calculate the virial of the inter-molecular forces; i.e. only non-bonding interactions contribute to the pressure. Second, calculating the molecular virial greatly reduces fluctuations of pressure; once again, this is due to the fact that the virials of the rapidly changing forces due the covalent potential are not included in the calculation.33
To build a time-stepping algorithm from continuous equation of motion a discretization scheme is required. In MD it is common to use a symmetric Trotter expansion of the Liouville operator. This type of approach dates to the seminal paper of Ruth on symplectic integration34 and it has been used to build efficient integrators for a wide range of applications, such as multiple timestepping (e.g. RESPA).35 In our case we will use an under-damped Langevin equation for the motion of the piston. This equation can be decomposed into Hamiltonian components and Ornstein-Uhlenbeck equations. The integrator for the Langevin equation can then be constructed by composing Hamiltonian flows with the exact distributional solution of the Ornstein-Uhlenbeck (linear stochastic) system. While very natural, this type of method was introduced and studied relatively recently.7–11
The second key ingredient of molecular simulations is the model that we use to describe the system under consideration. The functional form of MD force fields remained essentially the same for decades; however, the way interactions are calculated has evolved. The interacting potential between atoms consists of bonding and non-bonding terms. The bonding interactions are modeled by two-, three- and four-body energy terms while the non-bonded interactions consist in fixed-point charge electrostatic forces and a Lennard-Jones (LJ) potential that models hard-core and dispersive forces.
The calculation of non-bonding interaction is computationally expensive and it tends to dominate the total runtime of MD simulations. In the early days, to save computer time, all the interactions were set to zero at a given cutoff distance. Eventually, the analysis of artifacts due to truncation led to the use of Ewald summation for electrostatic interaction. Particle Mesh Ewald12 is now a de facto standard in molecular simulations. Recently, the growing heterogeneity of simulated systems together with the availability more computational power support applying a similar approach to Lennard Jones (LJ) interactions.13 LJ interactions decay with distance as r−6, a decay rate that is much more rapid than electrostatic interactions (r−1), which explains the earlier focus on electrostatic terms. For example, Wennberg et al36 have shown that the use of a cutoff of 10Å for LJ interactions introduces deviations up to 5% in the order parameters of lipid bilayers with respect to simulations with no cutoff, i.e. using Ewald summation.
Truncation of dispersive interactions is highly relevant in measuring the pressure. The neglected interactions are all attractive and add up. This leads to errors in the estimation of the pressure that, at the typical cutoff of 10Å, are of the order of hundreds of Atm. Errors of this kind can be estimated in case of isotropic, homogeneous liquids.37 Lague et al38 have calculated the long-range correction for anisotropic systems such as membranes and proteins by measuring the pressure with a very long distance cutoff once every few timesteps. The correction is then applied statically until a new long cutoff calculation is executed and a new estimate for the correction is available.
We propose an algorithm for constant molecular pressure in which all the long-range interactions are calculated using Ewald summation. The algorithm consists of a Nosé-Hoover thermostat coupled with a Langevin piston. The stochastic equations of motion are discretized using a reversible multiple timestep Trotter expansion that takes advantage of the splitting method for the Langevin equation. We illustrate that the algorithm is highly accurate and stable and that it converges rapidly to the desired values. It is implemented in the simulation program MOIL.
The paper is organized as follow. The first section introduces the equations of motion. The second section is dedicated to the model and the representation of the potentials by Ewald summations. The third section is about molecular pressure and how it is measured. The fourth section outlines the discretization procedure and the explicit algorithm. The last section provides numerical examples.
Equations of Motion for the Isobaric-Isothermal Ensemble
The system consists of N molecules. A molecule is defined as a group of atoms that are covalently linked. The use of molecular pressure implies the study of gas and liquid phases. It does not make sense (for example) to investigate covalent solids such as diamond using molecular pressure. We will indicate the position vector of the center of mass of a molecule by , the momentum by and the mass by Mμ. The total number of atoms in the system is n with the number of atoms of molecule μ being nμ. We indicate the coordinates of atom i in the coordinate system of molecule μ by , the momentum by and the mass by mi. So we have:
| (1) |
We use the shorthand i ∈ μ to indicate that atom i is in molecule μ, thus the expression for the mass of molecule μ could be written Mμ = Σi∈μ mi
The atoms are subject to K holonomic constraints
| (2) |
The number of degrees of freedom of the system is g = 3n − K. The force induced by the constraints on atom i that belongs to molecule μ is . The atoms interact through the potential U(), with being the force acting on atom i and representing the vector of all internal forces acting on molecule μ.
The molecules are contained in a volume V and the system is kept at equilibrium with a thermal bath at constant temperature Text and at constant pressure Πext. The internal kinetic temperature of the system is the average of T* given by
| (3) |
and the internal molecular pressure is obtained by averaging:32
| (4) |
We explain how to calculate the molecular pressure for periodic boundary conditions in a dedicated section, below.
In order to reproduce the sampling of the isobaric-isothermal ensemble, the equations of motion are not derived directly from a Hamiltonian, but instead include additional degrees of freedom. In addition to the set of coordinates already defined, we introduce the variables V, η and ξ (η and ξ are dimensionless). The volume of the system, V, is viewed as a dynamical variable associated to the barostat, with PV representing the associated barostat “momentum” and MV the “mass”. Note that the units of MV are mass × length−4. η is the coordinate associated with the thermostat; and Pη and Mη, respectively, are the momentum and mass (units mass×length2) associated with the coordinate η. ξ is associated with the coupling between the thermostat and the barostat and has momentum Pξ and mass Mξ (units mass×length2), respectively.
Our formulation is based on the following equations of motion:
| (5) |
Where W = W(t) is a one-dimensional Wiener process, γ is the friction constant for the volume control variable, with units of reciprocal time, and ; likewise γη and ση are friction and stochastic amplitude for the stochastic process attached to the thermostat variable.
The equations of motion above are similar to those of Marry and Ciccotti5 (see also the earlier paper of Hunenberger4), the main difference to be found in the equations for the momentum of the piston and thermostat which are here treated using a stochastic approach. In experiments, we find that the use of stochastic perturbation in the barostat significantly reduced the problem of “ringing,” as observed by Feller et al.27 in relation to the Andersen barostat.
In our simulations, the stochastic modification of the thermostat did not yield additional benefits as compared to the stochastic perturbation of the pressure variable alone, so we simply took γη = 0 in experiments presented here. However, the incorporation of this term is useful since it ensures ergodicity;39 its inclusion may be valuable in simulations of certain types of system with stiff harmonic internal coupling.
For γ = γη = 0 the equations of Marry and Ciccotti are recovered. In this case the equations of motion above preserve the following quantity that we will refer to as the extended energy in the sequel:
| (6) |
where H(, , , ) is the sum of the kinetic and potential energies of the physical system expressed in molecular coordinates. The extended energy is not relevant in the stochastic setting; it is however particularly useful during the coding process to provide evidence for the correctness of the implementation.
Derivation of the Partition Function
In this section we illustrate that the equations of motion we discussed above reproduce the probability density distribution of the isobaric-isothermal ensemble which, in the unconstrained case, is given by
| (7) |
The complete derivation of the partition function for the case of zero friction and in the presence of constraints may be found in Kalibaeva et al.6 The partition function for the deterministic equations of motions is found to be:
| (8) |
where represents the collection of all physical and artificial variables,
| (9) |
∫ d represents the usual integral with respect to all variables, and
| (10) |
This is easily seen to correspond to the desired NPT distribution following integration over the auxiliary variables PV, η, Pη, ξ, Pξ.
To demonstrate that ρ̃ is preserved under the evolution of the deterministic equations, one may write out the equations (5), for γ = γη = 0, in the compact form
| (11) |
The continuity equation corresponding to the evolution of the probability density of this system takes the form
| (12) |
where
| (13) |
The dagger indicates that this the adjoint of the Lie derivative associated to the system defined by Eq. 5 which acts on a function ϕ of the phase variables as
| (14) |
Using the equations Eq. 5 (with γ = γη = 0) and the definition of ρ̃ given above, it can be shown that5
| (15) |
meaning that ρ̃ is a steady state of the equations.
For completeness, and for later discussion in the setting of discretization, we write out the operator ℒ below. This can be obtained directly by substitution of ṽ (the vector field on the right hand side of Eq. 5, taking γ = γη = 0) into Eq. 14, we have
| (16) |
The above discussion pertains to the deterministic case, where no stochastic perturbation is incorporated. Unfortunately it is not possible to prove that the deterministic system is ergodic for the desired distribution. Following the usual approach taken in the physics literature, one typically assumes that, given sufficient internal complexity of the dynamical system, the ergodic property will hold in some practical sense, meaning simply that sufficiently long averages taken along dynamic paths will correspond to equilibrium averages.
When γ ≠ 0 or γη ≠ 0, the additional stochastic terms in Eq. 5 lead to a Fokker-Planck equation for the evolution of the probability density which may be written
| (17) |
where ℒdet is as described above, and
| (18) |
ρ̃ is stationary with respect to the δℒ† operator, as well, and thus with respect to the combined operator. Moreover, because the system is now subject to stochastic perturbations it is possible to prove that the system is ergodic,39 at least for the case γη ≠ 0 and for a harmonic underlying system. The proposed equations of motion therefore reproduce the NPT ensemble.
The Force Field
The atoms in the system are subject to a force field that reflects the atom-atom interactions both within the simulation cell and between atoms of different cells. The potential energy may be written
| (19) |
where Ucb represents terms involving covalently bound atoms and Unb represent non-bonded interactions. The covalent terms comprise bonds, angles, dihedrals and improper dihedrals potentials:
| (20) |
The covalent terms above, and the constraint forces do not contribute to the molecular pressure; therefore from now on only non-covalent terms are taken into account.
We define the set ℳ as the set of atom pairs (i, j) that are separated by less than three bonds. Non-bonding, non-imaged interactions for pairs of the ℳ set are either not calculated (separated by one or two bonds) or, if separated by three bonds (1–4 interactions), are calculated using special terms.
For the non-bonding term we need to take into account the fact that we will use periodic boundary conditions. If is the vector that shifts each periodic copy of the system, then the non-bonding terms are of the form
| (21) |
where
| (22) |
with qi representing the charge on atom i, and the * appearing in the summation indicating that one should omit terms for which both = 0 and (i, j) ∈ ℳ. Similarly
| (23) |
These terms together describe the Lennard-Jones potential for modelling the van der Waals interaction. Aij and Bij are parameters associated to the repulsive and attractive interactions between atoms, respectively. We assume a product combination rule, Aij = AiAj, Bij = BiBj, which is ideal for the implementation of Ewald summation. Other combination rules are possible but in this case the calculation of reciprocal summations are considerably more expensive.
In computing the fast decaying term U12, we assume a distance-dependent cutoff, i.e. the interaction is taken to be zero for distances greater than some fixed value. Due to the rapid decay of this term, the error is neglible beyond say 10Å.
The terms U6 and UC are calculated without cutoff using the technique of Ewald summation 1. We define to be the reciprocal lattice vector; the structure factor is therefore
| (24) |
Up to an arbitrary constant β, the electrostatic potential may be written as the sum of four terms:14
| (25) |
In a similar way, the U6 potential may decomposed as the sum of the following four terms:15
| (26) |
where the structure factor here is
| (27) |
For the calculation of the reciprocal part of both U6 and UC, the algorithm used is Particle Mesh Ewald (PME).12
Molecular Pressure
The atomic pressure is related to the trace of the internal stress tensor:
| (28) |
where the stress tensor for a system of atoms in periodic boundary conditions is:26,40,41
| (29) |
Where indicates the force that copy of atom j exercises on atom i. The symbol ⊗ indicates the tensor product. From this we recover the well known expression for the pressure:
| (30) |
The averaged quantity of the second term in the above expression is the atomic virial Watm. In a similar way the molecular pressure of a system with periodic boundary conditions can be written as:5,26,32
| (31) |
The averaged quantity in the second term of the above expression is the molecular virial Wmol. On average the molecular pressure and the atomic pressure are equal as proven by Ciccotti and Ryckaert42 for closed systems and by Akkermans and Ciccotti32 for open systems with periodic boundary conditions.
The positions of atoms are identified by their molecular coordinates plus their relative coordinates; therefore the molecular and the atomic virials are related as follows:
| (32) |
where we have used and where is the total force applied to atom i. We will use this relation to calculate the molecular virial Wmol by computing the atomic virial and post-processing it by subtracting the correction Σα αΣi∈α · as shown above.
This approach results in a cleaner implementation. First, the molecular positions do not need to be communicated to the routines that perform the force calculation. Second, and more importantly it solves a problem that arises when a molecule extends across the boundary of the simulation box (see Figure 1). In this case, atoms belonging to the same molecule actually interact through two different periodic copies, so in the implementation we would have two different molecular coordinates associated to the same molecule. The implementation of the atomic virial does not have the same problem.
Figure 1.

When a molecule extends across the periodic boundary atoms that belong to the same molecule interact through different periodic copies. When computing the molecular virial, atoms i1, j1, k1 are associated with the molecule centered in 1, while the atoms l2, m2, n2 are associated with a copy that is positioned at 2 = 1 + . Therefore, in implementing the molecular virial, it is necessary to allocate multiple molecular coordinates to a single molecule.
It is clear from the definition that the molecular virial of intramolecular forces is zero; therefore from now on we will deal exclusively with the intermolecular potentials, i.e. we will not calculate the virial of bonds, angles, torsions, improper torsions, 1–3, 1–4 interactions and constraints. This is a major advantage of using the molecular pressure.
The expression we have derived so far for the atomic virial is not suitable to be utilized in conjunction with Ewald summation because the individual atomic forces are lost. Following previous work in the literature,14,26,40 let us redefine the positions as:
| (33) |
where h is the 3 × 3 matrix defined by
| (34) |
where the three vectors are the edges of the unit cell of the periodic box, the components of the vector lie in [0, 1], and the vector has integer components (representing the translation of the cell in each direction). If the simulation box is a cuboid then h is given by
| (35) |
We can now derive an alternative expression for the atomic virial in the case of a uniform deformation. In a uniform deformation we keep constant and we change the size of the box by stretching or compressing the edges of the box Lγ. Thus
| (36) |
For a generic pair potential it is straightforward to show that the atomic virial can be expressed in terms of the deformation variables by40,41
| (37) |
Now we can calculate the molecular pressure (in terms of the molecular forces) as:
| (38) |
We next give the corresponding terms of the virial in the context of Ewald summation. The atomic virial of the electrostatic potential is given in terms of the Ewald components (direct, reciprocal and self) as14
| (39) |
| (40) |
| (41) |
We omit the virial of the correction term because this potential does not contribute to the molecular pressure being intramolecular in nature.
With regard to the Lennard-Jones attractive component, the atomic virial has a similar decomposition
| (42) |
| (43) |
| (44) |
| (45) |
As before, we do not calculate the virial of the correction term since, being an intramolecular term, it does not contribute to the molecular virial.
When LJ interactions are calculated using a distance cutoff a truncation error is introduced; calculating the pressure by the use of the virial of the Ewald summation avoids any truncation error. Using a cutoff to truncate the LJ interaction is a common solution in MD; the error introduced in the evaluation of the forces is indeed negligible. Unfortunately, the bias introduced in the calculation of the pressure is significant; the neglected interactions are all attractive, due to the long negative tail of the LJ potential. The resulting error in the value of the measured pressure is quite large at the typical cutoff distances of 10–12Å. For a homogenous isotropic liquid of density it is possible to compute the pressure truncation error:
| (46) |
For example, for TIP3P water the truncation error is ~200 Atm at 10Å and is still ~60 Atm at 15Å. The correction term is much more difficult to calculate for inhomogeneous and/or anisotropic liquids like mixtures of solvents or solutions, solvated proteins and membranes. To address this issue some heuristic methods have been proposed. Price et al43 have proposed a method in which the truncation correction is calculated using a very long Lennard Jones cutoff (every k steps); this approximation is valid under the assumption that the error is a quantity that varies adiabatically (i.e. slowly) in time.
Discretization of the Equations of Motion
The phase space probability density ρ(t) evolves in a step of size Δt according to
| (47) |
Functions ϕ of the phase variables evolve under the adjoint of this operator, i.e. if (t + Δt; , t) represents the solution at time t + Δt, given that (t) = , then we have, for any scalar valued function ϕ of the phase variables,
| (48) |
The operator etℒ is referred to as the evolution operator. ℒ can be decomposed as
| (49) |
where ℒdet is the Lie-derivative of the deterministic part of the system which is given in Eq. 16 and δℒ is the Fokker-Planck operator corresponding to the linear stochastic differential equations (SDEs) combining dissipation and random forcing, see Eq. 18.
The natural method for integrating the equations, which is inspired by prior work in symplectic integrators for Hamiltonian systems, is to split the deterministic part into pieces which are directly and exactly integrable and to additionally treat the stochastic Ornstein-Uhlenbeck terms by exact distributional integration; the flows of each of the pieces are then composed to define the numerical method.7–11
A variety of splitting methods are possible. Based on previous experience we employ a decomposition of ℒ into simple terms as follows:
| (50) |
where the various components are as follows:
|
|
|
||
|
|
|
||
|
|
|
||
|
|
|
||
|
|
|
||
|
|
|
||
|
|
|
||
|
|
|
The operators ℒ1, …, ℒ6, ℒ11 and δℒ1, δℒ2 are responsible for updating the extended variables, while the remaining operators drive the evolution of atomic and molecular positions and momenta.
Operators ℒ1, ℒ7, ℒ9, and ℒ12 give rise to multiplication by an exponential factor:
| (51) |
while the other deterministic operators result in translations
| (52) |
The stochastic terms are evolved using an exact weak (distributionally correct) solution of exp(tδℒ) which is easily written down. For example, for the SDE , an exact weak solution is
| (53) |
where R(t) is a standard Gaussian random variable with mean zero and unit variance.
Rescaling the simulation box is an expensive task since the non-bonded interaction lists must be recomputed, thus there is an advantage to delaying this task for several timesteps. This leads us to the use of a symmetric multiple timestepping procedure in which a longer timestep is used between changes to the box size parameter V than is used for updating the other variables. Specifically, we employ a symmetric Trotter factorization, based on performing ν basic timesteps between changes of the box size, of the following form:
| (54) |
In our case
| (55) |
Each factor is approximated by a further decomposition in to the enumerated components in such a way that the resulting discrete approximation is symmetric.
The constraint forces are treated using the SHAKE28 and RATTLE44 algorithms, with the resulting explicit integrator reported in the appendix.
Numerical Examples
We use three systems to test the algorithm described above.
We first test the deterministic algorithm (i.e. we set γ= γη = 0) in order to verify the correctness of the implementation. We then compare the behavior of the stochastic algorithm for two different friction values with the behavior of the deterministic algorithm. In the simulation the short time step was set to 1fs and the long time step (the one associated with the pressure control) was 8fs.
The first system is the peptide LKKLGKKLLKKLLKKGLKKL solvated in 6588 TIP3P45 water molecules also containing 12 sodium ions and 22 chloride ions; the system has no net charge. The peptide was simulated with periodic boundary conditions in a cube with an edge of 59Å. Particle Mesh Ewald algorithm was used for both electrostatic and dispersive interaction. Short-range interactions were calculated up to the distance of 9.8Å; long-range interactions were calculated using a grid of 64×64×64 points.
We used the following coupling constants:
The force field utilized for the peptide and the ions was OPLS462. The peptide was compressed from standard conditions to 1200 Atm at constant temperature of 300K. The total simulation time was 100ps. The system was compressed from a volume of ~ 206000Å3 to a volume of ~ 192000Å3. The results of this test are shown in Figure 2. The compression phase lasted about 80ps. There is no significant drift of the conserved extended energy or the temperature; the fluctuations are about 1Kcal/mol.
Figure 2.

The peptide system is compressed to a target pressure of 1200 Atm (see main text for details). In Figure 2A the extended energy is shown as a function of time. In Figure 2B the pressure is shown in red and the black horizontal line marks the target pressure of 1200 Atm. Figures 2C and 2D show volume and temperature also as functions of time.
The second system used was a membrane composed of 128 DOPC molecules surrounded by 6097 TIP3P water molecules. The force field used for the DOPC molecules was the united-atom Berger force field47 for the acyl chain atoms and OPLS46 for the head groups; this set up was the same as used in.48
In this case the size of the periodic box was initially set to 66.026Å×66.026Å×74.854Å. Ewald summation was used as before but with a grid 64×64×76. The membrane was expanded from ~ 1500 Atm to the standard conditions of 1 Atm at constant temperature of 300K. The total time of simulation was 500 ps. The relaxation to standard conditions resulted into an expansion from a volume of ~ 323000Å3 to a volume of ~ 345000Å3. The results of this test are shown in Figure 3.
Figure 3.

The membrane system is expanded to a target pressure of 1 Atm (see main text for details). In Figure 3A the extended energy is shown as a function of time. In Figure 3B the pressure is shown in red and the black horizontal line marks the target pressure. Figures 3C and 3D show volume and temperature also as function of time. After the initial expansion periodic ringing is evident in both figures 3C and 3D.
The time trace of the extended energy (Eq. 6) shows an initial relaxation resulting into a decrease of conserved quantity of about 1–2 Kcal/mol; after relaxation there is no measurable energy drift. The temperature shows no drift. During the initial 120ps the system undergoes the main expansion; then the membrane evolves at approximately constant volume with fluctuations of about 1000Å3, i.e. ~0.03%. Typical oscillations of the volume are present with period ~30ps; this is the phenomenon of ringing.
The last system we consider consists of 2033 TIP3P45 water molecules. For this system we analyze the behavior of both the deterministic and the stochastic algorithm. The system was let to equilibrate to a target pressure of 1 Atm from the initial condition of ~1000 Atm. The initial box size was set to 39.35Å×39.35Å×39.35Å. Short-range electrostatic and dispersive interactions were calculated up to the cutoff distance of 9.5Å while the remaining interactions were calculated in Fourier space using a grid 64×64×64. The following coupling constants were used:
The system was simulated using three different values for the volume friction coefficient γ = 0/ps, γ= 0.01/ps and γ = .1/ps. The total simulation time was 1ns for each set up.
The resulting evolution in time for the variable volume is shown in Figure 4. The expansion phase of the system lasts 20 to 70 ps depending on the value of the friction; the higher the friction the slower the relaxation to equilibrium. Associated to a higher friction one also introduces a proportionately larger noise term; the effects of the noise are especially evident with the highest friction. After the initial expansion we observe volume fluctuations near an average determined by the preset pressure. Periodic ringing is clearly present in the deterministic algorithm and is reduced with moderate friction and practically eliminated with the highest friction value. As shown in Figure 6 all the runs exhibit the same probability distribution for the variable volume as expected. The measured density of TIP3P water at the average volume is 0.984 g/cm3. This value is consistent with what was previously found Price et al43 when the TIP3P model is used in combination with PME. The time traces for pressure and temperature are reported in Figure 5.
Figure 4.

The volume as a function of time in constant pressure simulations. The results of the deterministic simulation are shown in green while the simulations with friction γ equal to 0.01 and 0.1 are shown in blue and red, respectively. In panel A we plot a nanosecond simulation while in panel B only the first 100 ps are shown. Ringing phenomenon is clearly present in the deterministic simulation and is reduced in the stochastic algorithm depending on the size of the friction coefficient. The relaxation time to the preset pressure also depends on the value of the friction coefficient.
Figure 6.
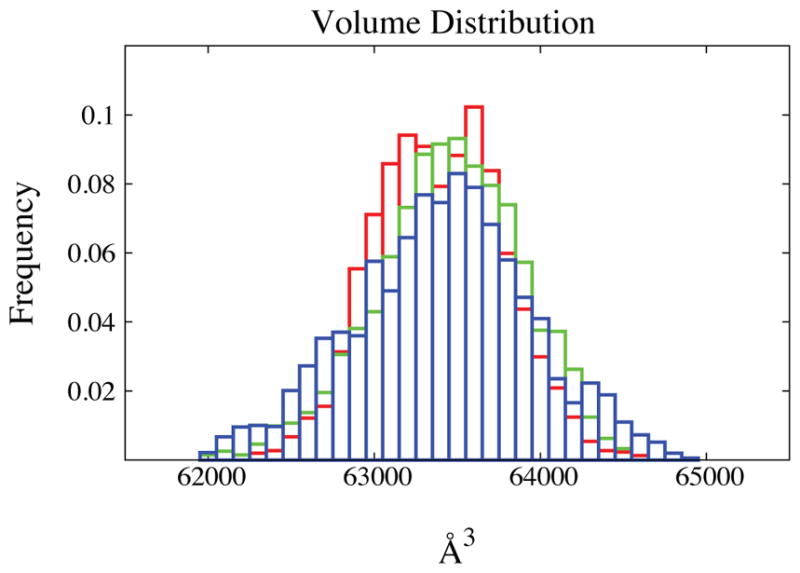
Volume distribution for the water box (see text for details). Histograms are reported histogram for the three different simulations: green bins are the results of the the deterministic simulations, blue and red are results of simulations with friction coefficients of 0.01 and 0.1 respectively. The three simulations with different level of stochasticity converge to the same average volume and share the similar distributions for the volume variable.
Figure 5.

Simulation of a water box under constant pressure conditions. Pressure and temperature are shown for 3 values of the friction constant 0, 0.01 and 0.1. Panels A, B and C show the instantaneous value of the pressure in red. The blue line indicates the value of the pressure averaged over 10 ps windows while the green line indicates the running average. The black horizontal line is the target pressure. Convergence to the target pressure is evident in all the runs. Panel D shows the temperature as a function of time for the three simulations: green correspond to friction coefficient γ equal to 0 and blue and red to 0.01 and 0.1 respectively. Once again, the horizontal black line indicates the target temperature.
Conclusions
We developed a new algorithm that reproduces the isobaric-isothermal sampling. The algorithm integrates several previous ideas into a single, efficient, and accurate algorithm.
The inclusion of a stochastic forcing term avoids non-ergodic behavior. By tuning the friction value is also possible to adjust the rate of decay to equilibrium, at least as measured by the decay of autocorrelation of the piston variable. Although we considered the use of stochastic noise in both thermostat and barostat variables, in our simulations, the ringing behavior was well controlled by just the single stochastic process coupled to the piston. For this reason, and in view of works such as,22,23 it is reasonable to expect that our algorithm will inherit the good characteristics of the deterministic algorithms in reproducing kinetic and transport properties; even though we have not specifically investigated those properties here. Retaining the option for a deterministic algorithm (obtained by setting γ= 0) provides a valuable tool for debugging in the form of the conserved effective energy.
As already observed by Feller et al,27 the inclusion of a stochastic term also eliminates the periodic oscillations in both pressure and volume. By using the molecular definition of pressure we have reduced the number of operations necessary to calculate the pressure itself since intramolecular interactions can be neglected. In particular, no special treatment for holonomic constraints is required. We have also shown how the molecular pressure can be calculated by using the atomic virial of intermolecular interactions only. Finally, we calculated all the long-range interactions and their stress tensors with Ewald sum. In this way we have avoided any truncation error in calculating the pressure and thus we also have avoided the necessity to introduce any correction scheme. The resulting algorithm it is implemented in the software package MOIL and it has been tested on several complex systems.
Acknowledgments
We thank our collaborators and the developers of MOIL, which is the code used in the present manuscript, that assisted in this work. In particular, Arnold P. Ruymgaart who collaborated to develop code essential to the presented algorithm and Juan M. Bello-Rivas who assisted with the theory of the PME and developed the code of the Particle Mesh Ewald of dispersive forces for the calculations of energies, forces, and virials in MOIL. Thanks also to Alfredo Cardenas and Daniel Walker for helping with the set up of the systems and some simulations. M.DP. thanks Mauro Lorenzo Mugnai for the many useful discussions that were instrumental to the development of concepts and ideas in this manuscript. This work was support by NIH grant GM59796 and Welch grant F1783 to RE.
Appendix
In this section we report the explicit algorithm as it is implemented in MOIL. For the definition of symbols please refer to the main text, we only remind that here Δt is the short time step and ν is the ratio between long and short time steps. The implemented algorithm is:
| 1 |
|
|
||
| 2 |
|
|||
| 3 |
|
|
||
| 4 |
|
|
||
| 5 |
|
|
Repeat ν times:
| 6 |
|
|
||
| 7 |
|
|
||
| 8 |
|
|
||
| 9 |
|
|
||
| 10 |
|
|
||
| 11 |
|
|
||
| 12 |
|
|
||
| 13 | ← SHAKE COORDINATES | |||
| 14 |
|
|||
| 15 |
|
|
||
| 16 |
|
|
||
| 17 | eℒ13Δt |
|
||
| 18 | eℒ14Δt |
|
||
| 29 | , ← FORCES CALCULATION | |||
| 20 |
|
|
||
| 21 |
|
|
||
| 22 |
|
|
||
| 23 |
|
|
||
| 24 |
|
|
||
| 25 |
|
|
||
| 26 | ← SHAKE MOMENTA | |||
| 27 |
|
|
||
| 28 |
|
|
||
| 29 |
|
|
||
| 30 |
|
|
||
| 31 |
|
|||
| 32 |
|
|
||
| 33 |
|
|
||
| 34 |
|
|
Footnotes
In the calculation of Ewald summation we use the standard tinfoil infinite boundary term.
All the force field parameters utilized in this manuscript are available for download together with the molecular dynamics package MOIL http://clsb.ices.utexas.edu/web/moil.html
References
- 1.McQuarrie DA. Statistical Mechanics. Harper and Row; New York: 1976. [Google Scholar]
- 2.Chandler D. Introduction to Modern Statistical Mechanics. Oxford University Press; New York: 1987. [Google Scholar]
- 3.Lippert RA, Predescu C, Ierardi DJ, Mackenzie KM, Eastwood MP, Dror RO, Shaw DE. Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. J Chem Phys. 2013;139(16):164106. doi: 10.1063/1.4825247. [DOI] [PubMed] [Google Scholar]
- 4.Hunenberger PH. Calculation of the group-based pressure in molecular simulations. I. A general formulation including Ewald and particle-particle-particle-mesh electrostatics. J Chem Phys. 2002;116(16):6880. [Google Scholar]
- 5.Marry V, Ciccotti G. Trotter derived algorithms for molecular dynamics with constraints: Velocity Verlet revisited. J Comput Phys. 2007;222(1):428. [Google Scholar]
- 6.Kalibaeva G, Ferrario M, Ciccotti G. Constant pressure-constant temperature molecular dynamics: a correct constrained NPT ensemble using the molecular virial. Mol Phys. 2003;101(6):765. [Google Scholar]
- 7.Serrano M, De Fabritiis G, Español P, Coveney PV. A stochastic Trotter integration scheme for dissipative particle dynamics. Math Comput Simulat. 2006;72(2–6):190. [Google Scholar]
- 8.De Fabritiis G, Serrano M, EspañŰol P, Coveney P. Efficient numerical integrators for stochastic models. Phys A. 2006;361(2):429. [Google Scholar]
- 9.Bussi G, Parrinello M. Accurate sampling using Langevin dynamics. Phys Rev E. 2007;75(5 Pt 2):056707. doi: 10.1103/PhysRevE.75.056707. [DOI] [PubMed] [Google Scholar]
- 10.Leimkuhler B, Matthews C. Robust and efficient configurational molecular sampling via Langevin dynamics. J Chem Phys. 2013;138(17):174102. doi: 10.1063/1.4802990. [DOI] [PubMed] [Google Scholar]
- 11.Leimkuhler B, Margul D, Tuckerman M. Stochastic resonance-free multiple time-step algorithm for molecular dynamics with very large time steps. Mol Phys. 2013;111:3579. [Google Scholar]
- 12.Darden T, York D, Pedersen L. Particle Mesh Ewald: an N. log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089. [Google Scholar]
- 13.in t’ Veld P, Ismail A, Grest G. Application of Ewald summations to long-range dispersion forces. J Chem Phys. 2007;127:14471. doi: 10.1063/1.2770730. [DOI] [PubMed] [Google Scholar]
- 14.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577. [Google Scholar]
- 15.Williams D. Accelerated convergence of crystal-lattice potential sums. Acta Crystallogr Sect A. 1971;27(5):452. [Google Scholar]
- 16.Ruymgaart AP, Cardenas AE, Elber R. MOIL-opt: Energy-Conserving Molecular Dynamics on a GPU/CPU System. J Chem Theory Comput. 2011;7(10):3072. doi: 10.1021/ct200360f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1):014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 18.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular Dynamics with Coupling to an External Bath. J Chem Phys. 1984;81:3684. [Google Scholar]
- 19.Frenkel D, Smit B. Understanding molecular simulation : from algorithms to applications. 2. Academic Press; San Diego: 2002. [Google Scholar]
- 20.Hoover WG. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A. 1985;31(3):1695. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 21.Evans D, Holian B. The Nose-Hoover thermostat. J Chem Phys. 1985;83:4069. [Google Scholar]
- 22.Basconi JE, Shirts MR. Effects of Temperature Control Algorithms on Transport Properties and Kinetics in Molecular Dynamics Simulations. J Chem Theory Comput. 2013;9(7):2887. doi: 10.1021/ct400109a. [DOI] [PubMed] [Google Scholar]
- 23.Leimkuhler B, Noorizadeh E, Penrose O. Comparing the efficiencies of stochastic isothermal molecular dynamics methods. J Stat Phys. 2011;143:921. [Google Scholar]
- 24.Samoletov AA, Dettmann CP, Chaplain MAJ. Thermostats for “slow” configurational modes. J Stat Phys. 2007;128:1321. [Google Scholar]
- 25.Andersen HC. Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys. 1980;72(4):2384. [Google Scholar]
- 26.Nose S, Klein ML. Constant Pressure Molecular-Dynamics for Molecular-Systems. Mol Phys. 1983;50(5):1055. [Google Scholar]
- 27.Feller SE, Zhang YH, Pastor RW, Brooks BR. Constant-Pressure Molecular-Dynamics Simulation - the Langevin Piston Method. J Chem Phys. 1995;103(11):4613. [Google Scholar]
- 28.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys. 1977;23(3):327. [Google Scholar]
- 29.Kneller GR, Mulders T. Nose-Andersen dynamics of partially rigid molecules: coupling all degrees of freedom to heat and pressure baths. Phys Rev E. 1996;54(6):6825. doi: 10.1103/physreve.54.6825. [DOI] [PubMed] [Google Scholar]
- 30.Ciccotti G, Martyna GJ, Melchionna S, Tuckerman ME. Constrained isothermal-isobaric molecular dynamics with full atomic virial. J Phys Chem B. 2001;105(28):6710. [Google Scholar]
- 31.Ryckaert JP, Ciccotti G. Introduction of Andersen Demon in the Molecular-Dynamics of Systems with Constraints. J Chem Phys. 1983;78(12):7368. [Google Scholar]
- 32.Akkermans RLC, Ciccotti G. On the equivalence of atomic and molecular pressure. J Phys Chem B. 2004;108(21):6866. [Google Scholar]
- 33.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-Dynamics with Coupling to an External Bath. J Chem Phys. 1984;81(8):3684. [Google Scholar]
- 34.Ruth Ronald D. A canonical integration technique. IEEE Trans Nucl Sci. 1983;30:2669. no. CERN-LEP-TH-83-14. [Google Scholar]
- 35.Tuckerman M, Berne BJ, Martyna GJ. Reversible Multiple Time Scale Molecular-Dynamics. J Chem Phys. 1992;97(3):1990. [Google Scholar]
- 36.Wennberg CL, Murtola T, Hess B, Lindahl E. Lennard-Jones lattice summation in bilayer simulations has critical effects on surface tension and lipid properties. J Chem Theory Comput. 2013;9(8):3527. doi: 10.1021/ct400140n. [DOI] [PubMed] [Google Scholar]
- 37.Allen MP, Tildesley DJ. Computer simulation of liquids. Oxford university press; New York: 1989. [Google Scholar]
- 38.Lague P, Pastor RW, Brooks BR. Pressure-based long-range correction for Lennard-Jones interactions in molecular dynamics simulations: Application to alkanes and interfaces. J Phys Chem B. 2004;108(1):363. [Google Scholar]
- 39.Leimkuhler B, Noorizadeh E, Theil F. A gentle stochastic thermostat for molecular dynamics. J Stat Phys. 2009;135:261. [Google Scholar]
- 40.Parrinello M, Rahman A. Crystal-structure and pair potentials - a molecular-dynamics study. Phys Rev Lett. 1980;45(14):1196. [Google Scholar]
- 41.Landau L, Lifshitz E. Elasticity theory. Pergamon Press Inc; New York: 1980. [Google Scholar]
- 42.Ciccotti G, Ryckaert JP. Molecular-dynamics simulation of rigid molecules. Comput Phys Rep. 1986;4(6):345. [Google Scholar]
- 43.Price DJ, Brooks CL. A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys. 2004;121(20):10096. doi: 10.1063/1.1808117. [DOI] [PubMed] [Google Scholar]
- 44.Andersen HC. Rattle - a velocity version of the Shake algorithm for molecular-dynamics calculations. J Comput Phys. 1983;52(1):24. [Google Scholar]
- 45.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926. [Google Scholar]
- 46.Jorgensen WL, Tiradorives J. The Opls potential functions for proteins–energy minimizations for crystals of cyclic-peptides and crambin. J Am Chem Soc. 1988;110(6):1657. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 47.Berger O, Edholm O, Jahnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 1997;72(5):2002. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardenas AE, Elber R. Modeling kinetics and equilibrium of membranes with fields: Milestoning analysis and implication to permeation. J Chem Phys. 2014;141(5):054101. doi: 10.1063/1.4891305. [DOI] [PMC free article] [PubMed] [Google Scholar]