

Abstract
Eukaryotic genomes are regulated by epigenetic marks that act to modulate transcriptional control as well as to regulate DNA replication and repair. In Arabidopsis thaliana, mutation of the ATXR5 and ATXR6 histone methyltransferases causes reduction in histone H3 lysine 27 monomethylation, transcriptional upregulation of transposons, and a genome instability defect in which there is an accumulation of excess DNA corresponding to pericentromeric heterochromatin. We designed a forward genetic screen to identify suppressors of the atxr5/6 phenotype that uncovered loss-of-function mutations in two components of the TREX-2 complex (AtTHP1, AtSAC3B), a SUMO-interacting E3 ubiquitin ligase (AtSTUbL2) and a methyl-binding domain protein (AtMBD9). Additionally, using a reverse genetic approach, we show that a mutation in a plant homolog of the tumor suppressor gene BRCA1 enhances the atxr5/6 phenotype. Through characterization of these mutations, our results suggest models for the production atxr5 atxr6-induced extra DNA involving conflicts between the replicative and transcriptional processes in the cell, and suggest that the atxr5 atxr6 transcriptional defects may be the cause of the genome instability defects in the mutants. These findings highlight the critical intersection of transcriptional silencing and DNA replication in the maintenance of genome stability of heterochromatin.
Author Summary
In eukaryotic genomes cellular processes such as transcription and replication need to be tightly controlled in order to promote genomic stability and prevent deleterious mutations. In Arabidopsis thaliana, two redundant histone methyltransferases, ATXR5 and ATXR6, are responsible for the deposition of a silencing epigenetic mark, histone H3 lysine 27 monomethylation. Loss of ATXR5/6 results in transcriptional activation of transposable elements (TEs), upregulation of DNA damage response genes and a genomic instability defect characterized as an excess of DNA corresponding to heterochromatin regions. Using a genetic screen, we sought to find suppressors of the atxr5/6 phenotype, and interestingly, we identified multiple genes implicated in general transcriptional activity. Through genomic characterization of the mutants our data suggest a model where transcriptional silencing of heterochromatin during S-phase is required for proper replication and maintenance of genome stability. These findings emphasize the important relationship between chromatin, transcriptional control and replication in the maintenance of genome stability in a eukaryotic system and identify new players involved in these processes.
Introduction
The genome represents a biological entity that is necessarily static yet retains a level of plasticity. Cells must faithfully replicate their genomes to avoid deleterious mutations, but also must be responsive to external stimuli. Eukaryotes have evolved multiple layers of epigenetic regulation that allow the genome to respond to environmental and developmental changes as well as provide a level of genome defense against parasitic genetic elements such as transposons. While epigenetic and replication fidelity pathways have traditionally been thought to be independent, multiple lines of evidence have recently implicated epigenetic mechanisms in the regulation of DNA replication [1–4].
In Arabidopsis thaliana, we previously identified two redundant histone methyltransferases, ATXR5 and ATXR6 (referred to in the aggregate as ATXR5/6), that are responsible for monomethylating lysine 27 of histone H3 (H3K27me1) [5]. Loss of these methyltransferases in the atxr5/6 double mutant leads to a severe loss of transcriptional silencing at repetitive transposable elements (TEs) [5,6]. The atxr5/6 mutants also display an unusual phenotype, wherein heterochromatin regions of the Arabidopsis genome exhibit an aberrant gain in DNA copy number (here referred to simply as over-replication). The over-replication phenotype appears to be mainly in cells which have undergone endoreduplication, a form of cell cycle without mitosis frequently observed in terminally differentiated cells [7]. The regions producing extra DNA are highly repetitive and carry epigenetic marks characteristic of silent chromatin such as DNA methylation and H3K27me1, and largely overlap with the pericentromeric regions transcriptionally derepressed in the atxr5/6 mutant.
We previously showed that mutations that strongly reduce DNA methylation in an atxr5/6 mutant background suppress the over-replication phenotype of atxr5/6 [6], suggesting that the heterochromatic nature of these regions is necessary to engender the gain in DNA copy number phenotype of the atxr5/6 mutant. In addition, while the DNA methylation mutants suppressed the over-replication phenotype of atxr5/6 mutants, they actually enhanced the transcriptional derepression phenotype [6]. Thus, the extra DNA phenotype and the transcriptional silencing phenotypes were decoupled in these mutants, showing that the extra DNA production in atxr5/6 is not required for the aberrant transcriptional activation of transposable elements.
In order to better understand the relationship between the atxr5/6 silencing and DNA copy number phenotypes, we carried out an extensive analysis of the atxr5/6 transcriptome, and compared this with the transcriptome of plants undergoing DNA damage. We also identified a number of modulators of the atxr5/6 phenotype by forward and reverse genetics approaches. Most notably, we isolated mutations in several genes, including those encoding members of the TREX-2 complex, a methyl-binding domain protein, and a SUMO dependent E3 ligase that suppressed the transcriptional defects in atxr5/6 mutants together with the genomic instability defects. Furthermore, we found that mutation of a gene involved in DNA repair and replication fork stability, BRCA1, enhanced both the atxr5/6 transcriptional and over-replication phenotypes. These results suggest a very close relationship between the loss of transcriptional silencing in atxr5/6 mutants and over-replication, consistent with a model wherein inappropriate transcription in atxr5/6 conflicts with the normal replication of heterochromatin to cause genomic instability.
Results
Loss of transposon silencing in atxr5/6 mutants is tissue specific and correlates with extra DNA at pericentromeric heterochromatin
We previously observed that the atxr5/6 mutants produce excess DNA corresponding to heterochromatin regions, also referred to as over-replication, that was most obvious in nuclei that had undergone endoreduplication [7], a process that is systemic in many tissue types of Arabidopsis and roughly correlates with tissue age [8]. We sought to test whether the transcriptional silencing defect of atxr5/6 mutants is similarly confined to specific tissues. To do this we analyzed two tissue types with different levels of endoreduplication, immature floral tissue that shows very low levels of endoreduplication, and cotyledons (embryonic leaves) that are very highly endoreduplicated. These tissues were analyzed by flow cytometry (Fig 1A) and whole-genome re-sequencing to confirm the state of over-replication in atxr5/6 (Fig 1B). Consistent with previous profiling of nuclei of different ploidy levels [7], we found an increase in DNA copy number in atxr5/6 mutants in cotyledons (Fig 1A) that was localized to regions of pericentromeric heterochromatin (Fig 1B). The excess DNA was absent in floral tissue of atxr5/6 mutants by both flow cytometry and sequencing analysis (Fig 1A and 1B).
Fig 1. Transcriptional silencing defects in an atxr5/6 mutant are strongly correlated with over-replication defects and are not induced by DNA damage.

(A) Flow cytometry profiles of wild type plants (Col) and atxr5/6 mutants for flower and cotyledon tissue with ploidy levels labeled for Col profiles. Excess DNA production is seen as a broadening of the peaks in atxr5/6 cotyledons compared to wild type. (B) Chromosomal views of DNA sequencing read ratio of atxr5/6 mutants compared to Col for flower and cotyledon tissues with diagrammatic representations of the Arabidopsis chromosomes shown below with boxes identifying pericentromeric heterochromatin. Gaps in the plot represent areas of low coverage. (C) Boxplot of RNA-seq RPKM values for atxr5/6-induced TEs from cotyledon tissue in floral and cotyledon tissue. (D) Venn diagrams describing the relationship between genes de novo identified as up-regulated in the irradiation and atxr5/6 mutant transcriptomes. (E) Boxplots showing the behavior of atxr5/6- irradiation-induced protein coding genes (top) and TEs (bottom) for various radiation dosages, time points, and genotypes.
We next performed RNA sequencing (RNA-seq) of flower and cotyledon tissue. We observed a reduction of the atxr5/6 transcriptional silencing defect in flowers (Fig 1C) that paralleled the lack of the extra DNA phenotype. While we were able to identify 487 TEs up-regulated in atxr5/6 cotyledon tissue relative to wild type (S1 Table), we found only 5 TEs up-regulated in floral tissue (S1 Table) and the TEs identified in cotyledon tissue showed greatly reduced transcription in atxr5/6 flowers (Fig 1C). Together these results suggest that the co-occurrence of the transcriptional defect and the over-replication defect in the atxr5/6 mutant may be connected and specific to endoreduplicated tissues.
The atxr5/6 transcriptome resembles a genome undergoing constitutive, low-level DNA damage
We previously observed that, consistent with the over-replication phenotype in the atxr5/6 mutants that is likely to cause genomic instability, several genes involved in the homologous recombination (HR) DNA repair pathway were up-regulated in atxr5/6 [6]. To assess the extent of DNA damage pathway activation in the mutants, we generated RNA-seq data from wild type seedlings that had undergone gamma-irradiation, which is known to generate robust activation of DNA repair pathways. From this analysis, we identified 230 protein-coding genes that were activated 90 minutes post-irradiation (S2 Table). This identified gene set significantly overlapped (S1 Fig) with a previously published set of gamma-irradiation responsive genes identified by microarray analysis [9], though the RNA-seq method identified a larger set of genes than the microarray-based approach. Using this gene set we were able to conclude that the majority of the protein-coding genes up-regulated in atxr5/6 seedlings belong to genes that are upregulated upon irradiation (Fig 1D), implying that the atxr5/6 protein-coding gene expression changes mainly reflect a response to DNA damage. Furthermore, upon irradiation of atxr5/6 mutants, we observed a robust upregulation of the same DNA damage genes that were up-regulated upon irradiation in wild type plants (Fig 1E), indicating that DNA damage response signaling is intact in atxr5/6 mutants. Therefore, we concluded that the excessive DNA phenotype in atxr5/6 mutants is not due to a generalized failure to induce DNA damage pathways.
Using the seedling RNA-seq datasets, we also defined TEs up-regulated upon irradiation (Col 100Gy compared to Col 0Gy) as well as due to the atxr5/6 mutations (atxr5/6 0Gy compared to Col 0Gy) (Fig 1E). We identified fewer TEs up in the atxr5/6 seedling data (n = 69) than the cotyledon datasets (n = 487), likely due to the heterogeneous nature of tissues from whole seedlings that have fewer endoreduplicated nuclei than do cotyledons. Importantly, we failed to observe a large increase in transposon expression post-irradiation (Fig 1E). This was true at the 90 minute time point as well as at 24 hours post-irradiation (Fig 1E). These results indicate that the transposon silencing defect we observe in atxr5/6 mutants is most likely not simply a consequence of the DNA damage induced in those mutants.
Mutation of the Arabidopsis BRCA1 homolog (AtBRCA1) enhances the silencing and over-replication defects of atxr5/6 mutants
Given our observation that atxr5/6 mutants show a generalized activation of DNA damage response pathways (especially HR genes) we sought to assess the effect of loss of DNA damage response gene in atxr5/6 mutants. To do this we generated atbrca1 atxr5/6 triple mutants. In mammals BRCA1 is a well characterized HR pathway protein important in maintaining genome stability with additional functions in cell-cycle check point and transcriptional regulation [10–13]. In plants AtBRCA1 has been shown to be necessary for efficient DNA repair [14] and is among the most highly up-regulated genes in an atxr5/6 mutant [6]. Interestingly, we found that the atbrca1 atxr5/6 mutants exhibited an enhancement of atxr5/6-induced extra DNA phenotype by flow cytometry and whole-genome sequencing of sorted 16C nuclei (Fig 2A and 2B).
Fig 2. Loss of the Arabidopsis BRCA1 homolog enhances the atxr5/6 extra DNA and transcriptional silencing phenotypes.

(A) Flow cytometry of cotyledon tissue for Col, atxr5/6, and atbrca1-1 atxr5/6 lines. Col and atxr5/6 data is same as shown in Fig 1A and is shown here for comparison. (B) Chromosomal views as in Fig 1B comparing atbrca1-1 atxr5/6 sorted 16C DNA-seq to sorted 16C reads from atxr5/6. (C) Boxplot of RNA-seq RPKM values for atxr5/6-induced TEs in cotyledon tissue for genotypes derived from the listed genotype. (D) Venn diagram of TEs de novo identified from atxr5/6 and atbrca1-1 atxr5/6.
We used RNA-seq of cotyledons to compare wild type, atbrca1, atxr5/6, and atbrca1 atxr5/6 plants. The atbrca1 atxr5/6 mutants showed a marked increase in expression of TEs identified as being overexpressed in atxr5/6 (Fig 2C). In addition, de novo identification of up-regulated TEs identified a greater number of reactivated TEs in atbrca1 atxr5/6 plants than atxr5/6 mutants (Fig 2D). Together these results suggest that wild type BRCA1 acts to restrict the atxr5/6 phenotype.
Establishment of a forward genetic screen to identify factors that influence the genomic instability phenotype of atxr5/6 mutants
In order to learn more about the biological mechanisms underlying the apparent link between the over-replication and transcription phenotypes of atxr5/6, we established a forward genetic screen to identify suppressors of the atxr5/6 phenotype. We fused a GFP reporter to the promoter of the RAD51 DNA damage response gene that is highly over-expressed in atxr5/6 mutant. Transgenic lines carrying the RAD51 promoter-GFP fusion construct showed strong GFP fluorescence in cotyledons when in the presence of the atxr5/6 mutations but not when crossed to wild type plants (S2A Fig). Thus, GFP fluorescence correlated with endoreduplicated tissues showing upregulated expression of transposons and extra DNA in atxr5/6 mutants, and therefore appeared to be a suitable visual readout of the atxr5/6 phenotype. We carried out an EMS mutagenesis of atxr5/6 RAD51pro::GFP seed (referred to as RAD51pro::GFP from this point forward) and searched for suppressors of the atxr5/6 phenotype by screening for families segregating plants that had lost the cotyledon GFP expression (GFP-, S2B and S2C Fig).
Identification of mutations in two components of the TREX-2 complex as suppressors of the atxr5/6 phenotype
Utilizing two M2 lines, EMS_2_37 and EMS_2_300, segregating for mutations causing loss of GFP signal (ems_2_37 and ems_2_300, S2C Fig), we performed flow cytometry on the GFP- plants as well as GFP+ plants from the same M2 family. For both the EMS_2_37 and EMS_2_300 lines, we observed clear suppression of the atxr5/6 extra DNA defect for GFP- plants (Fig 3A and 3B). We backcrossed the GFP- ems_2_37 and ems_2_300 mutants to the atxr5/6 line to confirm the function of the GFP protein in the F1 generation and to confirm the resegregation of GFP- plants in the resultant F2 generation (S2B Fig). We performed RNA-seq on cotyledons from the GFP- plants from this F2 generation as well as the GFP+ segregants from the same families. We found a striking reduction of TE expression in the GFP- plants as compared to both the GFP+ siblings and the starting RAD51pro::GFP line (Fig 3C), revealing that the suppression of the extra DNA in atxr5/6 by these mutants was accompanied by suppression of the atxr5/6 transcriptional silencing defect. This result is markedly different from the previously characterized suppression of atxr5/6-induced over-replication by DNA methylation mutants [6], where the loss of DNA methylation caused an increase in TE expression while suppressing the generation of extra DNA in atxr5/6 mutants.
Fig 3. Mutations in Arabidopsis TREX-2 complex proteins suppress the transcriptional silencing and extra-DNA phenotypes of atxr5/6 mutants.

(A) Flow cytometry of M2 atxr5/6 plants containing RAD51pro::GFP from the EMS_2_37 and (B) EMS_2_300 lines for both GFP+ and GFP- plants. (C) Heatmap of RNA-seq RPKM values over cotyledon atxr5/6-induced TEs for F2 EMS_2_37 or EMS_2_300 GFP+/- as well as control Col and atxr5/6 GFP+ plants. All lines except for Col contain RAD51pro::GFP and are in atxr5/6 background. (D) Boxplot of RNA-seq RPKM values from cotyledon tissue for identified atxr5/6-induced TEs (S1 Table) and (E) irradiation-induced genes (S2 Table) in TREX-2 insertional mutants.
We utilized EMS-induced mutations identified in RNA-seq datasets to map the ems_2_37 and ems_2_300 mutations (S3A and S3B Fig; S1 Text). The ems_2_37 mutation mapped to a splice site mutation at AtSAC3B (At3g06290, S3C–S3F Fig), a homolog of the yeast Sac3 protein [15]. The effect of the ems_2_37 mutation on AtSAC3B transcript splicing could be verified in the RNA-seq data, which showed clear intron retention relative to the control lines (S3C Fig). The ems_2_300 mutation was found to map to a nonsense mutation in AtTHP1 (At2g19560, S3F Fig), the homolog of yeast Thp1. AtSAC3B and AtTHP1 have been found to interact, analogously to their yeast homologs, in a complex termed TREX-2 [15]. The TREX-2 complex has been characterized in multiple systems as facilitating gene expression and RNA export from the nucleus via nuclear pore complexes (NPCs) [16–19]. Furthermore, the TREX-2 complex has also been found to act with transcribing RNA polymerase complexes to prevent the formation of deleterious transcriptional intermediates such as R-loops [20–22].
The molecular identities of ems_2_37 and ems_2_300 were confirmed by whole genome resequencing of the EMS lines to confirm the mapping of the EMS alleles (S3D and S3E Fig), as well as introgression of T-DNA-based insertional mutants for both AtSAC3B (atsac3b-3) and AtTHP1 (atthp1-1) into an atxr5/6 background to verify suppression of atxr5/6 extra DNA (S4A and S4B Fig) as well as atxr5/6-induced TE gene expression (Fig 3D). We also observed a reduction in expression of irradiation-induced genes in the trex-2 atxr5/6 triple mutants relative to atxr5/6, consistent with the suppression of the extra DNA and its relationship to the DNA damage response (Fig 3E). The RNA-seq analysis of atsac3b-3 and atthp1-1 single mutant cotyledon tissue revealed no gain in transcription for atxr5/6-induced TEs (Fig 3D) and de novo calling of up-regulated TEs in the single mutants identified only 2 and 4 TEs respectively. Finally, we verified the identities of the genes by performing complementation tests between the insertional mutants and EMS alleles (S4C Fig). We therefore renamed ems_2_37 and ems_2_300 as atsac3b-4 and atthp1-5 respectively. Subsequently we identified another EMS line, EMS_2_209, carrying a nonsense mutation in AtSAC3B (S3F Fig) by whole-genome re-sequencing and confirmed the identity of this mutation (renamed atsac3b-5) via non-complementation with the atsac3b-4 EMS line (S4C Fig).
Mutation of MBD9 suppresses the atxr5/6 phenotype
We isolated another suppressor of the extra DNA in atxr5/6 mutants, ems_2_129, which we mapped via whole-genome re-sequencing to a nonsense mutation at mbd9 (At3g01460; Fig 4A–4D). MBD9 is a protein with a methyl CpG binding domain previously identified as a regulator of flowering time with potential roles in histone H4 acetylation [23,24]. We confirmed the identity of ems_2_129 via introgression of the mbd9-3 insertional allele into an atxr5/6 background, and by performing complementation analysis (Fig 4E). RNA-seq analysis of the mbd9-3 atxr5/6 triple mutant revealed that suppression of the atxr5/6 extra DNA was accompanied by suppression of transcription at atxr5/6-induced TEs and irradiation-induced genes (Fig 4F).
Fig 4. The ems_2_129 mutation which suppresses the transcriptional silencing and extra-DNA phenotypes of atxr5/6 mutants maps to the MBD9 gene.

(A) Pie chart and (B) chromosomal view as shown in S3A and S3B Fig showing the distribution of significantly enriched mutations in EMS_2_129 (GFP-) plants identified in DNA-seq data. (C) Gene structure of MBD9 showing the newly identified point mutation from EMS mutagenesis as well the insertional mutant (triangles) used for complementation and downstream analysis. Black boxes represent exons. (D) Flow cytometry showing that the mbd9-3 insertional allele suppresses the atxr5/6 extra-DNA phenotype and (E) fails to complement the ems_2_129 EMS allele. (F) Boxplots showing the mbd9-3 allele suppresses the atxr5/6-induced expression of TEs and irradiation-responsive genes.
Mutation of At-STUbL2 suppresses the atxr5/6 phenotype
A fifth mutation, ems_2_325, like the TREX-2 and MBD9 mutants, showed suppression of both the atxr5/6 extra DNA and transposon over-expression phenotypes (Fig 5A and 5B). Mapping of the EMS-induced lesion by RNA-seq and subsequent whole-genome resequencing revealed a nonsense mutation in the coding region of At1g67180 (At-STUbL2), a relatively uncharacterized protein with a predicted N-terminal BRCT domain and C-terminal RING domain (S5A–S5C Fig). Complementation analysis was done using an insertional mutant (S5D–S5I Fig), the ems_2_325 allele was renamed stubl2-1 and the insertional FLAG_430E03 allele was renamed stubl2-2. At-STUbL2 was previously identified in a yeast two-hybrid screen for proteins that bind non-covalently to SUMO, and was shown to encode a SUMO-targeted ubiquitin E3 ligase capable of complementing the growth defects of Schizosaccharomyces pombe rfp1/rfp2 mutants [25]. In Arabidopsis, SUMO interacting proteins are highly enriched for those involved in chromatin regulation including histone and DNA methyltransferases [25]. Interestingly, At-STUbL2 was the only identified Arabidopsis STUbL protein containing the BRCT domain, which is a domain often found in proteins such as BRCA1 involved in DNA damage repair and cell cycle checkpoint control. Although the precise molecular function of At-STUbL2 is unknown, the At-STUbL2 RNA is co-expressed with ATXR6 as well as DNA repair and DNA replication genes, and the MET1 and CMT3 DNA methylation proteins that are known to function during DNA replication [26–28] (S3 Table).
Fig 5. Loss of AtSTUbL2 causes suppression of the atxr5/6 transcriptional silencing and extra-DNA phenotypes.
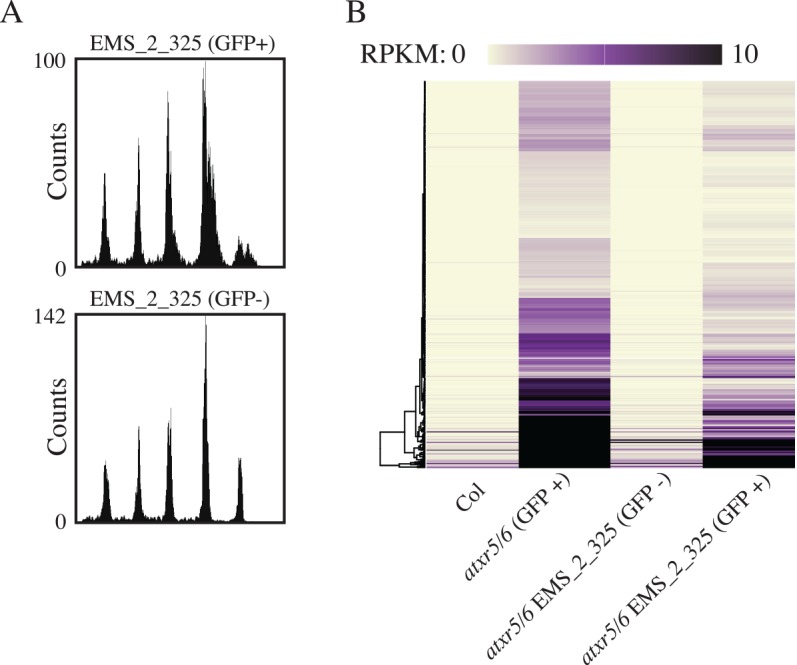
(A) Flow cytometry of M2 atxr5/6 plants containing RAD51pro::GFP from the EMS_2_325 line for both GFP+ and GFP- plants. (B) Heatmap for cotyledon RNA-seq, as in Fig 3C for EMS_2_325 F2 material with the same Col and atxr5/6 GFP+ data as in Fig 3C.
Identified suppressors of atxr5/6 show little to no alteration in DNA methylation patterns
Given our previous identification of DNA methylation mutants as suppressors of the atxr5/6 extra DNA phenotype, we questioned whether any of the newly identified suppressors of atxr5/6 from our forward genetic screen may have an effect on DNA methylation. To address this question, we performed whole-genome bisulfite sequencing on insertional mutants for each of the newly identified suppressors as well as wild type and atxr5/6 controls and performed differential methylated region (DMR) discovery for the mutants as well as on a previously published methyltransferase 1 (met1) dataset as a control [29]. MET1, a DNMT1 homolog, is a maintenance methyltransferase responsible for maintaining CG methylation as well as some of the non-CG cytosine methylation in the genome [29,30] and met1 mutants suppress the atxr5/6 extra-DNA phenotype while enhancing the TE silencing defect of atxr5/6 mutants [6].
Consistent with the lack of a strong TE silencing phenotype for any of the new suppressors of atxr5/6 (S6 Fig), we observed very limited alterations in DNA methylation in the mutants (Fig 6A). The one exception was the at-stubl2 mutant (Sample 5, Fig 6A), which showed a relatively large number of DMRs. We attribute this to the ecotype differences between the at-stubl2-2 Ws background and the control Col ecotype, since Arabidopsis ecotypes are known to contain differentially methylated regions [31]. In agreement with this interpretation, at-stubl2-2 mutants showed little to no alteration of DNA methylation patterns at the chromosomal level (Fig 6B). mbd9 mutants were previously reported to show global hypermethylation [24], however, while we observed a relatively higher number of CG-context hypermethylated DMRs for mbd9-3 mutants (Sample 2, Fig 6A) as compared to the other mutants, analysis of the overall genome levels of DNA methylation suggests this effect on DNA methylation is minor (Fig 6B).
Fig 6. Newly identified suppressors of atxr5/6 phenotypes do not have strong effects on DNA methylation.

(A) Barplot giving the number of DMRs identified in new suppressors of atxr5/6 with met1 used as positive control for DMR identification (Sample 1 = atxr5/6, 2 = mbd9-3, 3 = atsac3b-3, 4 = atthp1-1, 5 = atstubl2-2, 6 = met1-3). (B) Chromosomal views of log2 ratio of % cytosine methylation in mutants compared to a Col control.
Consistent with lack of strong methylation defects, the mutants isolated from our screen also did not exhibit dramatic morphological defects. This is also consistent with previously published data showing that a triple mutant of all three SAC3b related genes in Arabidopsis was reported to have no morphological defects [15] and that the mbd9 mutant displays only subtle flowering time and branching defects [23].
Discussion
ATXR5/6 represents a novel link between epigenetic gene regulation and genomic instability because the atxr5/6 mutant exhibits both de-repression of transposons in pericentromeric heterochromatin, as well as an over-replication defect manifested as the production of excessive DNA from these pericentromeric regions [7]. An open question has been the causal relationship between these two phenomena. A priori we can pose three different models of this relationship: 1) atxr5/6 mutations cause replication defects that indirectly cause transcriptional defects, 2) atxr5/6 mutations cause transcriptional defects that indirectly cause replication defects, or 3) atxr5/6 mutations affect replication and transcription independently. Based on the work presented here, we favor model 2 to the exclusion of model 1, but we cannot rule out model 3. Model 1, the model wherein transcriptional defects in atxr5/6 are due to defects in DNA replication, seems very unlikely based on our previous work characterizing the impact of the loss of DNA methylation pathways on the atxr5/6 phenotype. This work indicated that the transcriptional silencing phenotype of atxr5/6 mutants was not dependent on the over-replication phenotype, since DNA methyltransferase mutants suppressed the over-replication phenotype but actually enhanced the transcriptional derepression phenotype of atxr5/6 [6]. In addition, in the current study we found that irradiation-induced DNA damage caused an upregulation of DNA damage response genes resembling that found in the atxr5/6 mutant, but did not cause a transposable element silencing defect. In contrast, the possibility that transcriptional defects in the atxr5/6 mutant are the cause of the genomic instability defects (model 2) are consistent with the data in the current study. First, by comparing immature flower tissue with cotyledon tissue we observed a co-occurrence of the transcriptional silencing defect and the extra DNA phenotype in cotyledons, both of which were absent in immature flower tissue. Furthermore, the brca1 mutation acted as an enhancer of both the transposon derepression phenotype and the extra DNA phenotype of atxr5/6. Lastly, we performed a screen for mutants that suppress the atxr5/6 phenotype, and found that every suppressor reduced both the transposon derepression phenotype and the extra DNA phenotype of atxr5/6. Thus the transcriptional silencing defects of atxr5/6 were inseparable from the abnormal pericentromeric DNA content in these mutants, suggesting that the transcriptional misregulation may be the cause of the genome instability phenotype.
If our preferred model to explain the phenotype of atxr5/6 is correct, there remain many open questions. For instance, it is not clear why ATXR5/6 appears to be exceptional among transcriptional silencers, such as DNA methylation proteins, with regards to a link to aberrant DNA replication. Because ATXR6 is expressed at the G1/S transition of the cell cycle, showing close co-expression with DNA replication licensing factors such as CDT1 and ORC2, we previously speculated that ATXR5/6 and H3K27 monomethylation may act to limit DNA replication initiation, and that extra DNA in the atxr5/6 mutant was due to inappropriate multiple firing of origins of replication (re-replication), creating “onion skins” [32] of excessive DNA near origins [7]. Although this is still a possibility, because of the tight linkage between transposon upregulation and extra DNA production in atxr5/6 as well as in our newly identified suppressors, we favor a model in which aberrant G1/S phase transcription in atxr5/6 leads to replication-transcription conflicts, which ultimately lead the production of excessive DNA in heterochromatin. As part of the normal cell cycle, there must be coordination of DNA replication with transcription, and in both prokaryotic and eukaryotic systems, failure to coordinate DNA transcription and replication results in genome instability [33–35]. This instability can be caused by direct collision between DNA and RNA polymerase complexes as well as by indirect conflicts between the complexes as is the case with R-loop formation by RNA polymerases which can act as a barrier to DNA replication fork progression [21,36]. In both the direct and indirect cases of replication-transcription conflict, the result can be replication fork stalling and collapse [36–38] which in turn leads to the formation of single-stranded DNA and recombinagenic structures that can lead to mutagenic outcomes for the genome [39]. It is thus possible that atxr5/6 mutations generate genomic instability via the release of transcriptional silencing at a critical point during the cell cycle such as S-phase, which then creates replication-transcription conflicts and hyper-recombinagenic structures that result in the amplification of repetitive DNA in pericentromeric regions. In this way, the timing of transcriptional derepression may differentiate atxr5/6 mutants from other mutants such as DNA methylation mutants that exhibit loss of TE silencing, but no effect on DNA replication.
A replication-transcription conflict model would be in line with studies of the human [40] and yeast [41,42] genomes where it has been proposed that replication stress can lead to the generation of copy number variants at repetitive DNA. In support of the notion of such a replication-specific silencing function, ATXR5/6 have been characterized as cell-cycle regulated proteins which act with the PCNA proteins normally found at replication forks [43], suggesting that these proteins function during S phase. Furthermore, ATXR5/6 have been implicated in genetic and epigenetic control of normal rDNA repeat behavior [44], and the ribosomal repeats are known sources of replication-transcription conflict in yeast [45]. The reason for the specificity of the genome instability defect for heterochromatin regions is not known, but it seems possible that the resolution of transcription and replication fork collisions may be more difficult to complete in heterochromatin regions due to the more inaccessible nature of heterochromatin. This could also help explain why DNA methylation mutants suppress the genome instability defect of atxr5/6 mutants, since severe reduction of DNA methylation would render these regions much less like heterochromatin and more like euchromatin, for instance through reduced levels of the H3.1 histone variant recently shown to be required for over-replication in atxr5/6 [46].
Our identification of the TREX-2 complex as being necessary for the genomic instability defect in atxr5/6 mutants also supports the hypothesis of replication-transcription conflict driving the atxr5/6 genome instability, because components of this complex were isolated in a yeast genetic screen for factors affecting the viability of a strain genetically predisposed to accumulate aberrant replication intermediates [47]. TREX-2 mutants were found to rescue the viability of a replication-deficient strain where replication forks were destabilized in a manner that is phenomenologically similar to our observations of the trex-2 atxr5/6 triple mutants. In yeast, genetic rescue by the TREX-2 mutants was proposed to act via the loss of topological strain created by the normal gene gating facilitated by TREX-2/THO. Interestingly, this defect depended on transcription but not R-loop formation. In addition, the TREX-2 complex is also required for transcriptional efficiency [17], and TREX-2 was shown to promote RNA Pol II transcription through its interaction with the Mediator complex [16]. This is consistent with our finding that TREX-2 mutants reduce the inappropriate transcription of heterochromatin seen in atxr5/6 mutants, and suggests that loss of TREX-2 may alleviate replication stress present in an atxr5/6 genome, which is otherwise undergoing heterochromatic transcription. Similarly, MBD9, which is identified here and which has been characterized as a transcriptional activator of the flowering gene FLC [24], likely acts to promote transcription, such that loss of MBD9 would alleviate transcription-induced replication blocks in a manner similar to TREX-2 mutants. Finally, although the function of At-STUbL2 is not known, since this mutant also reduces the transposon over-expression phenotype of atxr5/6, we propose that STUbL2 acts via similar mechanisms as the TREX-2 and MBD9 mutants, and may encode a transcriptional regulator.
An alternative model to explain the correlation between the transcriptional defects and over-replication defects in atxr5/6 is that R-loops generated by inappropriately transcribing transposons directly cause mutagenic events leading to excessive DNA production, even in the absence of replication-transcription conflicts [48]. R-loops are formed during the process of transcription where the RNA strand pairs with the complementary DNA strand, leaving the other DNA strand free, and exposing the cell to potentially mutagenic single stranded DNA[21]. If atxr5/6 mutants fail to properly resolve R-loops in heterochromatin, this could explain the heterochromatin specificity of the excessive DNA phenotype, and also explain why mutants that suppress the transcriptional upregulation of atxr5/6 also suppress the over-replication defect. Consistent with this model, BRCA1 is known to play a role in the prevention of DNA damage due to transcription associated R-loops [13], and brca1 mutants enhanced the excessive DNA damage phenotype of atxr5/6 mutants. Although TREX-2 is also known to be involved in resolving R-loop structures[49], the trex-2 mutants from our screen dramatically reduced the transposon de-repression defect of atxr5/6 mutants, and therefore would also dramatically reduce the abundance of R-loops. One observation that does not fit well with the R-loop model is that brca1 mutant enhanced both the magnitude of the transposon over-expression phenotype and the over-replication defect, and it is difficult to understand how failure to resolve R-loop-induced DNA damage would lead to an increase in transcription. Clearly, the mode of action of Arabidopsis BRCA1, TREX-2, and the other suppressors identified here, will be an important question for future studies.
Given the emerging importance of the interaction between epigenome and genome stability for models of disease such as cancer [2,50,51], it will be important to further test the replication-transcription conflict and other models of the atxr5/6 phenotypes since further understanding of this phenomena may inform other models and systems where loss of transcriptional control leads to genomic instability.
Materials and Methods
Genetic strains
All strains used in this study, unless otherwise indicated, were in a Columbia (Col) ecotype background. Details regarding the strain information as well as the generation of the RAD51pro::GFP line can be found in the Supplemental Experimental Procedures (S1 Text). In addition, S4 Table details the genotypes of lines used in high-throughput sequencing experiments.
Irradiation
10-day old seedlings were irradiated on plates via exposure to a Cs-137 source following the general experimental design previously described [9].
EMS mutagenesis
EMS mutagenesis of ~2000 atxr5/6 seeds carrying the RAD51pro::GFP transgene was carried out as previously described [52].
Flow cytometry and FACS
All flow cytometry analysis and FACS was performed as previously described [6]. For cotyledon tissue, cotyledons from at least 20 plants were pooled, whereas for leaf or floral tissue 3 plants were typically pooled.
Sequencing library generation
The DNA-seq libraries presented in Figs 1 and 2 were generated as previously described [6,7]. The DNA-seq libraries in S3, S5 Figs and Fig 4 were similarly prepared regarding DNA extraction and Covaris shearing, but the libraries were prepared using either the Illumina DNA TruSeq or Nugen Ultralow Ovation kits (see GEO accession GSE77735 for details).
All RNA-seq libraries were prepared using a standard Trizol (Life Technologies) RNA extraction followed by library generation with the Illumina RNA TruSeq kit. All RNA was derived from the cotyledon tissue of >20 plants unless otherwise indicated. For all libraries two biological replicates were performed unless otherwise indicated.
For whole genome bisulfite sequencing libraries, libraries were generated from 3-week-old adult leaf material using the NuGen Ovation Ultralow Methyl-Seq kit before being bisulfite converted with the Qiagen Epitect bisulfite kit using the FFPE protocol. All libraries were sequenced on an Illumina HiSeq instrument.
Data analysis
Base calls were performed using the standard Illumina pipeline and all reads were aligned to the TAIR10 genome (www.arabidopsis.org). For DNA-seq libraries reads were aligned using the Bowtie aligner [53], for RNA-seq Tophat2 [54]was used, and for whole-genome bisulfite data the BSmap aligner was used [55]. Protein-coding genes were defined as described in the TAIR10 annotation (www.arabidopsis.org) and transposable elements were defined using a previously described list [56] that had been updated to the TAIR10 assembly. All statistical analysis was performed in an R environment. Details of bioinformatics data analysis can be found in the Supplemental Experimental Procedures (S1 Text).
Data deposition
The sequencing data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE77735.
Supporting Information
Venn diagram detailing the significant overlap (P<2.2e-16, Fisher’s Exact Test) of genes identified as up-regulated in the RNA-seq dataset as compared to the previously published microarray study.
(EPS)
(A) GFP fluorescence of RAD51pro::GFP in atxr5/6 cotyledons is lost upon crossing to a Col control. (B) Diagrammatic representation of the mutagenesis, screening, and mapping schema. GFP positive cotyledons are colored green and GFP negative tissues are colored red (due to the autofluoresence of chlorophylls). The RAD51pro::GFP transgene was maintained at each step by growing plants on selective media (hygromycin). (C) Identification of the ems_2_37 (atsac3b-4) mutant in an M2 family using UV without a band-pass filter and with a band-pass filter (removes chlorophyll autofluoresence). Mutant plants segregated for GFP- cotyledons.
(TIF)
(A) Pie charts showing the distribution of significantly enriched mutations in the EMS_2_37 (chromosome 3) or EMS_2_300 (chromosome 2) GFP- RNA-seq libraries across the 5 Arabidopsis chromosomes. The “Genome” pie chart gives the distribution of all bp in the Arabidopsis genome. (B) Chromosomal view of the position of significantly enriched mutations in the EMS_2_37/EMS_2_300 GFP- RNA-seq libraries as well as the percent mismatch for each mutation. (C) Screen shot of aligned RNA-seq reads showing intron retention at the AtSAC3B gene in the ems_2_37 (atsac3b-4) library. The atsacb3-4 lesion can be seen as a red mismatch in the sequencing reads at the left intron-exon boundary. (D) Pie charts showing the distribution of significantly enriched mutations for the five Arabidopsis chromosomes from DNA-seq libraries of the ems_2_37 and ems_2_300 mutants. (E) Chromosomal distribution and mismatch frequency of significant mutations derived from DNA-seq data of the ems_2_37 and ems_2_300 mutants. (F) Gene structure of AtSAC3B and AtTHP1 showing newly identified point mutations from EMS mutagenesis as well as insertional mutants (triangles) used for complementation and downstream analysis. Exons are represented by black boxes.
(EPS)
(A) Flow cytometry of nuclei counts as in Fig 1A showing that the atsac3b-3 insertional allele suppresses the atxr5/6 extra-DNA phenotype similar to the mapped point mutations. (B) The insertional atthp1-1 allele suppresses the atxr5/6 extra-DNA phenotype. (C) Complementation analysis showing the TREX-2 insertional alleles fail to complement the EMS alleles because the atxr5/6 extra-DNA phenotype is suppressed. Also shown is the non-complementation of the ems_2_209 line crossed to ems_2_37, confirming the identity of ems_2_209 as atsac3b-5. Also shown is a control cross of ems_2_37 crossed to atxr5/6 grown in parallel to show the re-emergence of the atxr5/6 extra-DNA phenotype in the F1.
(EPS)
(A) Pie charts and chromosomal views as shown in S3A and S3B Fig showing the distribution of significantly enriched mutations in ems_2_325 (GFP-) plants identified in RNA-seq and (B) DNA-seq data. (C) Gene (top) and protein (bottom) structure of At-STUbL2 showing the newly identified point mutation from EMS mutagenesis as well the insertional mutant (triangles) used for complementation and downstream analysis. For the gene structure the black boxes represent exons, and for the protein structure gray boxes represent pfam domains. (D) No insertional allele of At1g67180 exists in the Col ecotype used for all other lines in this study for complementation purposes, so we obtained an insertional mutant (FLAG_430E03) isolated in the Ws ecotype [57]. The hybrid nature of the genome resulting from complementation crosses between the Ws allele and our ems_2_325 line made direct comparison to the control atxr5/6 line difficult and we also noted that the atxr5/6 extra-DNA defect was severely reduced in the 50% Ws atxr5/6 plants regardless of the At1g67180 genotype. The graph shows flow cytometry showing that the atstubl2-2 insertional allele suppresses the atxr5/6 extra-DNA phenotype as well as partial suppression of the extra-DNA phenotype in the control 50% Ws line. (E) Complementation analysis comparing crosses between ems_2_325 line crossed to the atstubl2-2 atxr5/6 triple mutant (showing strong non-complementation) with a control cross to atxr5/6. (F) Quantitation of the 16C peak, shown in S5E. Coefficient of variation (CV) of the F1 complementation material shows a slight reduction in extra-DNA (small CV value) for the atstubl2-2 atxr5/6 x ems_2_325 plants as compared to control atstubl2-2 atxr5/6 x atxr5/6 plants. (G) To overcome the confounding factor of the genetic background, we performed RNA-seq on F1 plants resulting from a cross of a FLAG_430E03 atxr5/6 triple mutant (50% Col; 50% Ws ecotype) with pollen from either an atxr5/6 mutant or the ems_2_325 line. The resultant progeny were all 75% Col, 25% Ws in genome composition and the RNA-seq results showed clear suppression of atxr5/6 transposon expression and irradiation-induced genes in those plants carrying both the insertional FLAG_430E03 allele and the ems_2_325 allele relative to plants heterozygous for a functional copy of At1g67180. The box plots show RNA-seq RPKM values for atxr5/6-induced TEs and (H) irradiation-induced genes for ems_2_325 complementation material showing non-complementation (atxr5/6 suppression) by atstubl2 alleles. (I) Chromosomal views of the log2 ratio of normalized RNA-seq reads between the non-complementing atstubl2-2 atxr5/6 x ems_2_325 F1 material compared to a control atstubl2-2 atxr5/6 x atxr5/6 control F1 cross.
(EPS)
Aggregation of data shown throughout this study for comparative purposes showing cotyledon RNA-seq RPKM values for atxr5/6-induced TEs and irradiation-induced genes for the newly identified atxr5/6 suppressors.
(EPS)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(DOCX)
Acknowledgments
We thank M. Akhavan and S. Feng for Illumina sequencing. Sequencing was performed at the University of California, Los Angeles (UCLA) Broad Stem Cell Research Center BioSequencing Core Facility. Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center Flow Cytometry Core Facility.
Data Availability
The sequencing data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE77735. All other relevant data are within the paper and its Supporting Information files.
Funding Statement
CJH and MEP were supported by a Damon Runyon Cancer Research Postdoctoral Fellowship. Research in SDM lab was supported by a grant from the National Institutes of Health (GM075060). Work in the Jacobsen lab was supported by NSF grant 1121245. SEJ is an Investigator of the Howard Hughes Medical Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ding Q, MacAlpine DM. Defining the replication program through the chromatin landscape. Crit Rev Biochem Mol Biol. 2011;46: 165–79. 10.3109/10409238.2011.560139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alabert C, Groth A. Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2012;13: 153–67. 10.1038/nrm3288 [DOI] [PubMed] [Google Scholar]
- 3.Yoshida K, Poveda A, Pasero P. Time to be versatile: regulation of the replication timing program in budding yeast. J Mol Biol. 2013;425: 4696–705. 10.1016/j.jmb.2013.09.020 [DOI] [PubMed] [Google Scholar]
- 4.Brustel J, Tardat M, Kirsh O, Grimaud C, Julien E. Coupling mitosis to DNA replication: the emerging role of the histone H4-lysine 20 methyltransferase PR-Set7. Trends Cell Biol. 2011;21: 452–60. 10.1016/j.tcb.2011.04.006 [DOI] [PubMed] [Google Scholar]
- 5.Jacob Y, Feng S, LeBlanc C a, Bernatavichute Y V, Stroud H, Cokus S, et al. ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat Struct Mol Biol. 2009;16: 763–8. 10.1038/nsmb.1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stroud H, Hale CJ, Feng S, Caro E, Jacob Y, Michaels SD, et al. DNA methyltransferases are required to induce heterochromatic re-replication in Arabidopsis. PLoS Genet. 2012;8: e1002808 10.1371/journal.pgen.1002808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacob Y, Stroud H, Leblanc C, Feng S, Zhuo L, Caro E, et al. Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature. 2010;466: 987–91. 10.1038/nature09290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galbraith DW, Harkins KR, Knapp S. Systemic Endopolyploidy in Arabidopsis thaliana. Plant Physiol. 1991;96: 985–9. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1080875&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Culligan KM, Robertson CE, Foreman J, Doerner P, Britt AB. ATR and ATM play both distinct and additive roles in response to ionizing radiation. Plant J. 2006;48: 947–61. [DOI] [PubMed] [Google Scholar]
- 10.Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7: a016600 10.1101/cshperspect.a016600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Q, Greenberg RA. Deciphering the BRCA1 Tumor Suppressor Network. J Biol Chem. 2015;290: 17724–32. 10.1074/jbc.R115.667931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savage KI, Harkin DP. BRCA1, a “complex” protein involved in the maintenance of genomic stability. FEBS J. 2015;282: 630–46. 10.1111/febs.13150 [DOI] [PubMed] [Google Scholar]
- 13.Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell. 2015;57: 636–647. 10.1016/j.molcel.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Block-Schmidt AS, Dukowic-Schulze S, Wanieck K, Reidt W, Puchta H. BRCC36A is epistatic to BRCA1 in DNA crosslink repair and homologous recombination in Arabidopsis thaliana. Nucleic Acids Res. 2011;39: 146–54. 10.1093/nar/gkq722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Q, Tang X, Tian G, Wang F, Liu K, Nguyen V, et al. Arabidopsis homolog of the yeast TREX-2 mRNA export complex: components and anchoring nucleoporin. Plant J. 2010;61: 259–70. 10.1111/j.1365-313X.2009.04048.x [DOI] [PubMed] [Google Scholar]
- 16.Schneider M, Hellerschmied D, Schubert T, Amlacher S, Vinayachandran V, Reja R, et al. The Nuclear Pore-Associated TREX-2 Complex Employs Mediator to Regulate Gene Expression. Cell. Elsevier; 2015;162: 1016–28. 10.1016/j.cell.2015.07.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rondón AG, Jimeno S, Aguilera A. The interface between transcription and mRNP export: from THO to THSC/TREX-2. Biochim Biophys Acta. 2010;1799: 533–8. 10.1016/j.bbagrm.2010.06.002 [DOI] [PubMed] [Google Scholar]
- 18.García-Oliver E, García-Molinero V, Rodríguez-Navarro S. mRNA export and gene expression: the SAGA-TREX-2 connection. Biochim Biophys Acta. 2012;1819: 555–65. 10.1016/j.bbagrm.2011.11.011 [DOI] [PubMed] [Google Scholar]
- 19.Blobel G. Gene gating: a hypothesis. Proc Natl Acad Sci. 1985;82: 8527–8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poveda AM, Le Clech M, Pasero P. Transcription and replication: breaking the rules of the road causes genomic instability. Transcription. Landes Bioscience; 2010;1: 99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aguilera A, García-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46: 115–24. 10.1016/j.molcel.2012.04.009 [DOI] [PubMed] [Google Scholar]
- 22.Aguilera A, Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. Nature Publishing Group; 2008;9: 204–17. 10.1038/nrg2268 [DOI] [PubMed] [Google Scholar]
- 23.Peng M, Cui Y, Bi Y-M, Rothstein SJ. AtMBD9: a protein with a methyl-CpG-binding domain regulates flowering time and shoot branching in Arabidopsis. Plant J. 2006;46: 282–96. [DOI] [PubMed] [Google Scholar]
- 24.Yaish MWF, Peng M, Rothstein SJ. AtMBD9 modulates Arabidopsis development through the dual epigenetic pathways of DNA methylation and histone acetylation. Plant J. 2009;59: 123–35. 10.1111/j.1365-313X.2009.03860.x [DOI] [PubMed] [Google Scholar]
- 25.Elrouby N, Bonequi MV, Porri A, Coupland G. Identification of Arabidopsis SUMO-interacting proteins that regulate chromatin activity and developmental transitions. Proc Natl Acad Sci U S A. 2013;110: 19956–61. 10.1073/pnas.1319985110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du J, Zhong X, Bernatavichute Y V, Stroud H, Feng S, Caro E, et al. Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell. Elsevier; 2012;151: 167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kankel MW, Ramsey DE, Stokes TL, Flowers SK, Haag JR, Jeddeloh JA, et al. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics. Genetics; 2003;163: 1109–22. Available: http://genetics.org/content/163/3/1109.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obayashi T, Okamura Y, Ito S, Tadaka S, Aoki Y, Shirota M, et al. ATTED-II in 2014: evaluation of gene coexpression in agriculturally important plants. Plant Cell Physiol. 2014;55: e6 10.1093/pcp/pct178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stroud H, Greenberg MVC, Feng S, Bernatavichute Y V, Jacobsen SE. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell. 2013;152: 352–64. 10.1016/j.cell.2012.10.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson LM, Du J, Hale CJ, Bischof S, Feng S, Chodavarapu RK, et al. SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2014;507: 124–8. 10.1038/nature12931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, et al. Patterns of population epigenomic diversity. Nature. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2013;495: 193–8. 10.1038/nature11968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Botchan M, Topp W, Sambrook J. Studies on Simian Virus 40 Excision from Cellular Chromosomes. Cold Spring Harb Symp Quant Biol. 1979;43: 709–719. [DOI] [PubMed] [Google Scholar]
- 33.Merrikh H, Zhang Y, Grossman AD, Wang JD. Replication-transcription conflicts in bacteria. Nat Rev Microbiol. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2012;10: 449–58. 10.1038/nrmicro2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mirkin E V, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71: 13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bermejo R, Lai MS, Foiani M. Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell. 2012;45: 710–8. 10.1016/j.molcel.2012.03.001 [DOI] [PubMed] [Google Scholar]
- 36.Hamperl S, Cimprich KA. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst). 2014;19: 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim N, Jinks-Robertson S. Transcription as a source of genome instability. Nat Rev Genet. 2012;13: 204–14. 10.1038/nrg3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan YA, Hieter P, Stirling PC. Mechanisms of genome instability induced by RNA-processing defects. Trends Genet. 2014;30: 245–253. 10.1016/j.tig.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16: 2–9. 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arlt MF, Wilson TE, Glover TW. Replication stress and mechanisms of CNV formation. Curr Opin Genet Dev. 2012;22: 204–10. 10.1016/j.gde.2012.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finn KJ, Li JJ. Single-stranded annealing induced by re-initiation of replication origins provides a novel and efficient mechanism for generating copy number expansion via non-allelic homologous recombination. PLoS Genet. Public Library of Science; 2013;9: e1003192 10.1371/journal.pgen.1003192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green BM, Finn KJ, Li JJ. Loss of DNA replication control is a potent inducer of gene amplification. Science. 2010;329: 943–6. 10.1126/science.1190966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sozzani R, Glab N. Two cell-cycle regulated SET-domain proteins interact with proliferating cell nuclear antigen (PCNA) in Arabidopsis. Plant J. 2006;47: 395–407. [DOI] [PubMed] [Google Scholar]
- 44.Pontvianne F, Blevins T, Chandrasekhara C, Feng W, Stroud H, Jacobsen SE, et al. Histone methyltransferases regulating rRNA gene dose and dosage control in Arabidopsis. Genes Dev. 2012;26: 945–57. 10.1101/gad.182865.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsang E, Carr AM. Replication fork arrest, recombination and the maintenance of ribosomal DNA stability. DNA Repair (Amst). 2008;7: 1613–23. [DOI] [PubMed] [Google Scholar]
- 46.Jacob Y, Bergamin E, Donoghue MTA, Mongeon V, LeBlanc C, Voigt P, et al. Selective methylation of histone H3 variant H3.1 regulates heterochromatin replication. Science. 2014;343: 1249–53. 10.1126/science.1248357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bermejo R, Capra T, Jossen R, Colosio A, Frattini C, Carotenuto W, et al. The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell. 2011;146: 233–46. 10.1016/j.cell.2011.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamperl S, Cimprich KA. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst). 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.González-Aguilera C, Tous C, Gómez-González B, Huertas P, Luna R, Aguilera A. The THP1-SAC3-SUS1-CDC31 complex works in transcription elongation-mRNA export preventing RNA-mediated genome instability. Mol Biol Cell. 2008;19: 4310–8. 10.1091/mbc.E08-04-0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evertts AG, Coller HA. Back to the origin: reconsidering replication, transcription, epigenetics, and cell cycle control. Genes Cancer. 2012;3: 678–96. 10.1177/1947601912474891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Putiri EL, Robertson KD. Epigenetic mechanisms and genome stability. Clin Epigenetics. 2011;2: 299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moissiard G, Cokus SJ, Cary J, Feng S, Billi AC, Stroud H, et al. MORC family ATPases required for heterochromatin condensation and gene silencing. Science. 2012;336: 1448–51. 10.1126/science.1221472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2009;10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14: R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10: 232 10.1186/1471-2105-10-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buisine N, Quesneville H, Colot V. Improved detection and annotation of transposable elements in sequenced genomes using multiple reference sequence sets. Genomics. 2008;91: 467–75. 10.1016/j.ygeno.2008.01.005 [DOI] [PubMed] [Google Scholar]
- 57.Samson F, Brunaud V, Balzergue S, Dubreucq B, Lepiniec L, Pelletier G, et al. FLAGdb/FST: a database of mapped flanking insertion sites (FSTs) of Arabidopsis thaliana T-DNA transformants. Nucleic Acids Res. 2002;30: 94–7. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=99145&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Venn diagram detailing the significant overlap (P<2.2e-16, Fisher’s Exact Test) of genes identified as up-regulated in the RNA-seq dataset as compared to the previously published microarray study.
(EPS)
(A) GFP fluorescence of RAD51pro::GFP in atxr5/6 cotyledons is lost upon crossing to a Col control. (B) Diagrammatic representation of the mutagenesis, screening, and mapping schema. GFP positive cotyledons are colored green and GFP negative tissues are colored red (due to the autofluoresence of chlorophylls). The RAD51pro::GFP transgene was maintained at each step by growing plants on selective media (hygromycin). (C) Identification of the ems_2_37 (atsac3b-4) mutant in an M2 family using UV without a band-pass filter and with a band-pass filter (removes chlorophyll autofluoresence). Mutant plants segregated for GFP- cotyledons.
(TIF)
(A) Pie charts showing the distribution of significantly enriched mutations in the EMS_2_37 (chromosome 3) or EMS_2_300 (chromosome 2) GFP- RNA-seq libraries across the 5 Arabidopsis chromosomes. The “Genome” pie chart gives the distribution of all bp in the Arabidopsis genome. (B) Chromosomal view of the position of significantly enriched mutations in the EMS_2_37/EMS_2_300 GFP- RNA-seq libraries as well as the percent mismatch for each mutation. (C) Screen shot of aligned RNA-seq reads showing intron retention at the AtSAC3B gene in the ems_2_37 (atsac3b-4) library. The atsacb3-4 lesion can be seen as a red mismatch in the sequencing reads at the left intron-exon boundary. (D) Pie charts showing the distribution of significantly enriched mutations for the five Arabidopsis chromosomes from DNA-seq libraries of the ems_2_37 and ems_2_300 mutants. (E) Chromosomal distribution and mismatch frequency of significant mutations derived from DNA-seq data of the ems_2_37 and ems_2_300 mutants. (F) Gene structure of AtSAC3B and AtTHP1 showing newly identified point mutations from EMS mutagenesis as well as insertional mutants (triangles) used for complementation and downstream analysis. Exons are represented by black boxes.
(EPS)
(A) Flow cytometry of nuclei counts as in Fig 1A showing that the atsac3b-3 insertional allele suppresses the atxr5/6 extra-DNA phenotype similar to the mapped point mutations. (B) The insertional atthp1-1 allele suppresses the atxr5/6 extra-DNA phenotype. (C) Complementation analysis showing the TREX-2 insertional alleles fail to complement the EMS alleles because the atxr5/6 extra-DNA phenotype is suppressed. Also shown is the non-complementation of the ems_2_209 line crossed to ems_2_37, confirming the identity of ems_2_209 as atsac3b-5. Also shown is a control cross of ems_2_37 crossed to atxr5/6 grown in parallel to show the re-emergence of the atxr5/6 extra-DNA phenotype in the F1.
(EPS)
(A) Pie charts and chromosomal views as shown in S3A and S3B Fig showing the distribution of significantly enriched mutations in ems_2_325 (GFP-) plants identified in RNA-seq and (B) DNA-seq data. (C) Gene (top) and protein (bottom) structure of At-STUbL2 showing the newly identified point mutation from EMS mutagenesis as well the insertional mutant (triangles) used for complementation and downstream analysis. For the gene structure the black boxes represent exons, and for the protein structure gray boxes represent pfam domains. (D) No insertional allele of At1g67180 exists in the Col ecotype used for all other lines in this study for complementation purposes, so we obtained an insertional mutant (FLAG_430E03) isolated in the Ws ecotype [57]. The hybrid nature of the genome resulting from complementation crosses between the Ws allele and our ems_2_325 line made direct comparison to the control atxr5/6 line difficult and we also noted that the atxr5/6 extra-DNA defect was severely reduced in the 50% Ws atxr5/6 plants regardless of the At1g67180 genotype. The graph shows flow cytometry showing that the atstubl2-2 insertional allele suppresses the atxr5/6 extra-DNA phenotype as well as partial suppression of the extra-DNA phenotype in the control 50% Ws line. (E) Complementation analysis comparing crosses between ems_2_325 line crossed to the atstubl2-2 atxr5/6 triple mutant (showing strong non-complementation) with a control cross to atxr5/6. (F) Quantitation of the 16C peak, shown in S5E. Coefficient of variation (CV) of the F1 complementation material shows a slight reduction in extra-DNA (small CV value) for the atstubl2-2 atxr5/6 x ems_2_325 plants as compared to control atstubl2-2 atxr5/6 x atxr5/6 plants. (G) To overcome the confounding factor of the genetic background, we performed RNA-seq on F1 plants resulting from a cross of a FLAG_430E03 atxr5/6 triple mutant (50% Col; 50% Ws ecotype) with pollen from either an atxr5/6 mutant or the ems_2_325 line. The resultant progeny were all 75% Col, 25% Ws in genome composition and the RNA-seq results showed clear suppression of atxr5/6 transposon expression and irradiation-induced genes in those plants carrying both the insertional FLAG_430E03 allele and the ems_2_325 allele relative to plants heterozygous for a functional copy of At1g67180. The box plots show RNA-seq RPKM values for atxr5/6-induced TEs and (H) irradiation-induced genes for ems_2_325 complementation material showing non-complementation (atxr5/6 suppression) by atstubl2 alleles. (I) Chromosomal views of the log2 ratio of normalized RNA-seq reads between the non-complementing atstubl2-2 atxr5/6 x ems_2_325 F1 material compared to a control atstubl2-2 atxr5/6 x atxr5/6 control F1 cross.
(EPS)
Aggregation of data shown throughout this study for comparative purposes showing cotyledon RNA-seq RPKM values for atxr5/6-induced TEs and irradiation-induced genes for the newly identified atxr5/6 suppressors.
(EPS)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(DOCX)
Data Availability Statement
The sequencing data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE77735. All other relevant data are within the paper and its Supporting Information files.