

Abstract
Voltage-gated L-type Cav1.2 calcium channels couple membrane depolarization to transient increase in cytoplasmic free Ca2+ concentration that initiates a number of essential cellular functions including cardiac and vascular muscle contraction, gene expression, neuronal plasticity, and exocytosis. Inactivation or spontaneous termination of the calcium current through Cav1.2 is a critical step in regulation of these processes. The pathophysiological significance of this process is manifested in hypertension, heart failure, arrhythmia, and a number of other diseases where acceleration of the calcium current decay should present a benefit function. The central issue of this paper is the inactivation of the Cav1.2 calcium channel mediated by multiple determinants.
1. Introduction
The voltage-gated inward Ca2+ current (I Ca) is a common mechanism of transient increase in the cytoplasmic free Ca2+ concentration triggered by cell depolarization. This form of Ca2+ signaling activates essential cellular processes including cardiac contraction [1], regulation of a smooth muscle tone [2], gene expression [3], synaptic plasticity [4] and exocytosis [5]. Complete and rapid termination of Ca2+ influx is mediated by an intricate mechanism of spontaneous calcium channel inactivation, which is crucial for preventing Ca2+ overloading of the cell during action potentials and restoration of the resting sub-μM cytoplasmic free Ca2+ concentration [6]. This paper will focus on the molecular basis and multiple determinants of the Cav1.2 calcium channel inactivation.
2. Cav1.2: Challenges and Solutions
2.1. Molecular Complexity
The Cav1.2 calcium channel is an oligomeric complex composed of the α 1C, α 2 δ, and β subunits [7, 8]. The ion channel pore is formed by the α 1C peptide (Figure 1) that is encoded by the CACNA1C gene. The auxiliary β and α 2 δ subunits are essential for the functional expression and plasma membrane (PM) targeting of the channel [9, 10]. They exist in multiple genomic isoforms generated by four CACNB genes (CACNB1–4) and three CACNA2D genes (CACNA2D1–3). All three subunits are subject to alternative splicing. Adding to the complexity of the Cav1.2 molecular organization, β subunits tend to oligomerize [11]. All together, genomic variability, alternative splicing, and hetero-oligomerization generate a plethora of Cav1.2 splice variants that are expressed in cells in species-, tissue-, and developmental-dependent manner, while the change of their fine balance may have significant pathophysiological consequences [12, 13].
Figure 1.

Transmembrane topology of the α 1C subunit. To illustrate the sites of molecular diversity, the polypeptide sequence is schematically segmented according to the CACNA1C genomic map [14] and the corresponding invariant (black) and alternative (blue) exons are outlined by black bars and numbered (1–50). Four regions of homology (I–IV), each composed of 6 transmembrane segments (numbered), are believed to be folded around the central pore. α-interaction domain (AID) of a constitutive β-binding site is shown in green. LA and IQ motifs (red) constitute calmodulin-binding domain (CBD).
2.2. Challenges in the Selection of the Host Cell
Naturally occurring diversity of Cav1.2 complicates the interpretation of data obtained from native cells, let alone the single channel data. This underlies the importance of Cav1.2 research in recombinant expression systems where the molecular composition of the channel and the structure of its constituents are predefined. However, this experimental approach encountered the major problem of the selection of an appropriate host cell.
Most of the studies of calcium channels were carried out using HEK293 cells. These cells provide high expression efficiency of recombinant Ca2+ channels but, unfortunately, contain endogenous calcium channels exhibiting Ca2+ currents up to 3 pA/pF [15, 16]. Thus, HEK293 cells allow for the adequate study of recombinant Ca2+ channels only when the amplitude of the current is large enough to ignore the contribution of the endogenous channels. Correct assessment of the functional determinants of Ca2+ channels, however, requires the use of host cells that are completely free of endogenous Ca2+ channel subunits. COS1 or COS7 cells suit this requirement well because they generate no appreciable calcium current, do not contain endogenous Ca2+ channel subunits or their precursors, and show no induction of endogenous Cav1.2 subunits in response to the expression of the recombinant ones [17, 18]. Kinetics parameters and voltage dependence of activation and inactivation of the Cav1.2 channel currents measured in COS1 cells are consistent with data obtained in other expression systems [19]. An important advantage of COS cells is their relatively slow division rate that allows for better control over efficiency of expression and assembly of the Cav1.2 channel subunits of different size.
2.3. Problems of Fluorescent Labeling and Measurement
Fusion of GFP-like fluorophores to the N- and/or C-termini of the recombinant α 1C or to the N-terminus of β does not markedly change the electrophysiological properties of the expressed channels, enables the application of fluorescent and FRET (fluorescent resonance energy transfer) microscopy to the study of subcellular distribution and assembly of Cav1.2 as well as intricate aspects of molecular architecture and dynamics of the channel. The channel retains major electrophysiological characteristics unchanged when the α 1C C-terminal sequence encoded by distal exons 46–50 (Figure 1, residues 1833–2138 in α 1C,77) is replaced by ECFP. However, α 1C fused by its N- or/and C-termini to EYFP is highly sensitive to photobleaching that irreversibly inactivates it. Known as fluorophore-assisted light inactivation (FALI), this interesting property limits the applicability of acceptor photobleaching for the measurements of FRET in Cav1.2 because of uncertainty in the functional state of the channel [20]. However, the ratiometric analysis of corrected FRET between the fluorophores, fused to the tails of the α 1C and/or β subunits, reflects the reversible state-dependent structural rearrangements of the channel induced by the changes of transmembrane voltage under patch clamp [19, 21].
2.4. Recombinant Cav1.2: What Does It Need for Functional Expression and How Does It Appear?
Typical properties of a “wild-type” recombinant Cav1.2 are illustrated in Figure 2(A) using an example of the ubiquitous human α 1C,77 isoform (GenBank no. z34815). When the EYFP-labeled α 1C was expressed in COS1 cells alone, the fluorescent-tagged channel protein was diffusely distributed over the cytoplasm and did not generate measurable calcium current (Figure 2(A), panel a). The quantitative analysis of distribution of α 1C between PM and the cytoplasm (Figure 2(B)) [18] confirmed lack of significant PM targeting by α 1C independently on the presence of α 2 δ (bars a and b). Expression of Cav β in the absence of α 2 δ stimulated PM targeting of α 1C, but the channel remained silent (Figure 2(A), panel c) unless α 2 δ was coexpressed (panel d). Thus, β and α 2 δ subunits are sufficient for the functional channel; under these experimental conditions, β subunits stimulate PM targeting of the channel complex and, in the presence of α 2 δ, facilitate voltage gating of the Cav1.2 channel.
Figure 2.
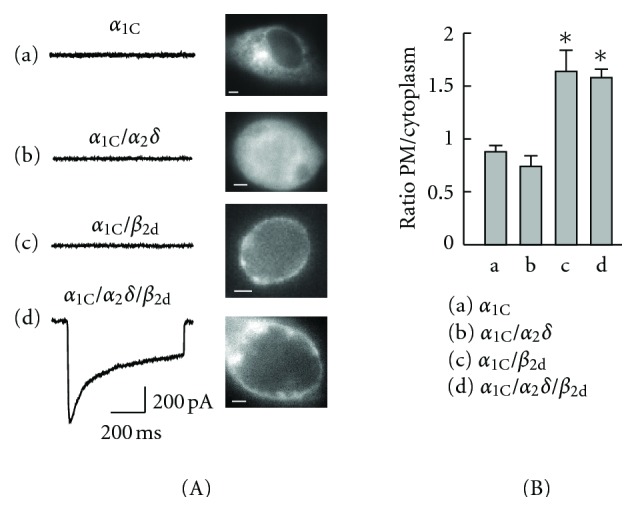
Role of the Cav1.2 auxiliary subunits. (A) Epifluorescent images of the expressing COS1 cells showing distribution of EYFPN-α 1C obtained with the YFP filter (scaling bars, 4 μm) and traces of the maximum calcium current recorded in response to 600 ms steps to +30 mV from the holding potential V h = −90 mV (left). (B) Relative distribution of EYFPN-α 1C in the plasma membrane (PM) over the cytoplasm in the absence (a) or presence of α 2 δ (b), β 2d (c), or α 2 δ + β 2d (d). The ratio of fluorescence intensity in PM over the area underneath PM was averaged after background subtraction in each cell. The ratio less than 1.0 indicates lack of significant PM targeting by α 1C. ∗P < 0.05 [18].
The shape and appearance of the peak calcium current shown in Figure 2(A) (panel d) is quite typical for the β 2-modulated Cav1.2 [22]. Its major features include the relatively slow rate of I Ca decay and a large fraction of the sustained I Ca remaining the end of the depolarizing pulse [18]. It is clear that during long-lasting action potentials such properties may lead to pathogenic calcium overload of the cell if it is not balanced by robust compensatory mechanisms. It was the ultimate role of Cav1.2 in defining the duration of the action potential in cardiac cells that triggered the research and development of calcium channel blockers, a class of drugs that by now has a billion dollar market. It is this role of Cav1.2 that stimulates the current interest to the identification of molecular determinants of Cav1.2 inactivation in hopes of finding more specific and more effective drugs.
2.5. Last but Not Least a Complication: Cav1.2 Clustering
A single ventricular myocyte contains ~300,000 Cav1.2 channels, but only ~3% of the channels are open at peak I Ca [23]. Contrary to the popular belief, Cav1.2 channels are not evenly distributed over the plasma membrane. In native neuronal [24–26] and cardiac muscle cells [27–29] they form large clusters. Single-molecule imaging of the functional recombinant EYFPN-α 1C/β 2a/α 2 δ channels expressed in HEK293 cells revealed clusters composed of ~40 channels that were mobile in the plasma membrane [30]. Both the fluorescence correlation spectroscopy and fluorescence recovery after photobleaching experiments yielded a lateral diffusion constant of D lat ≈ 0.1 μm2/s. The functional significance of the Cav1.2 clusters mobility is not clear. It is believed that in cardiac muscle cells such mobility may be restrained by interactions with other proteins, for example, ryanodine receptors [27]. The size of Cav1.2 clusters and their specific density in the plasma membrane depend on the type of β subunit expressed [31]. The distance between the termini of neighbor α 1C subunits varies from 67 Å with neuronal/cardiac β 1b to 79 Å with vascular β 3. The highest density of Cav1.2 clusters in the plasma membrane and the smallest cluster size were observed with β 1b present. Insight into molecular mechanisms defining the architecture and properties of Cav1.2 clusters is important for better understanding of pathophysiology of the coupling between the Cav1.2 activity and the induced responses in Ca2+ signal transduction.
3. Voltage- and Ca2+-Dependent Inactivation of the Cav1.2 Calcium Channel
In the case of Cav1.2 calcium channels, two different mechanisms are in control of Ca2+ current inactivation. One mechanism is driven by Ca2+ ions on the cytoplasmic side of the plasma membrane, whereas the other depends on transmembrane voltage. Experimentally, replacement of Ca2+ for Ba2+ as the charge carrier eliminates Ca2+-dependent inactivation (CDI) [32] so that the Ba2+-conducting calcium channels inactivate in a voltage-dependent manner by fast (FI) and slow (SI) mechanisms [33]. These three mechanisms of inactivation, FI, SI and CDI, and their major determinants are illustrated on Figure 3.
Figure 3.

Molecular determinants of Cav1.2 inactivation. Comparison of the wild-type Cav1.2 (A) with the same channel deprived of CDI (B) and SI (C) determinants. The five horizontal panels show (a) arrangement of critical determinants of inactivation. ADSI is composed of conserved hydrophobic amino acids in a -2 position of S6 segments in repeats II, III, and IV (yellow circles: Ala, Val, and Ile, resp.) as well as Ser residue in -1 position of IS6 (cyan circle). The CaM-binding domain (CBD) of the α 1C C-tail is shown by a red rounded rectangle. A β subunit (green) binds to the α-interaction domain in the linker between repeats I and II, and, in a Ca2+-dependent manner, to the IQ-region of the α 1C subunit C-tail ([45], not shown). The distal structure of β 2 (β 2CED, blue ball) binds to the CBD [46]. (b) Evidence of coimmunoprecipitation of the indicated subunits. (c) Normalized traces of I Ba (black) and I Ca (red), and (d) voltage dependence of I Ca (red) and time constant of FI (τ f, black) are presented to illustrate CDI in (A) and lack of CDI in (B) and (C). (e) Link between CDI and differential β-subunit modulation (DβM) of Cav1.2. (A) Differential modulation of the I Ba inactivation by β 1a (black trace) and β 2a (green trace) in the WT Cav1.2. Disruption of CBD (α 1C,86) eliminates CDI and SI targeted by CDI and DβM (B). Mutation of ADSI (α 1C,IS-IV) removed CDI and fully inhibited SI so that the channel remains conducting for the duration of the depolarization stimulus (C).
3.1. Ca2+-Dependent Inactivation and Calmodulin-Binding Domain of α 1C
There are several different determinants of CDI, but it was not until 1997 that the Ca2+-sensing site of CDI had been narrowed down to a stretch of the 80-amino-acid C-terminal sequence of α 1C encoded by exons 40–42 [34] (Figure 2) marked by red block in Figure 3(A) (panel a). A naturally occurring splice variation in this region in α 1C,86 (Figure 3(B)) completely inhibited CDI as it is evident from the lack of deceleration of the current with Ba2+ as the charge carrier (panel c, black trace) as compared with I Ca (red trace). Another characteristic feature of the inhibited CDI was lack of the current size dependence of I Ca on voltage (Figure 3(B), panel d, open symbols) that stays in contrast to the U-shape dependence of the time constant of fast inactivation (τ f) on membrane potential in the wild-type Cav1.2 (see Figure 3(A), panel d). Two distinct sequences, L and K, were identified within this 80-amino-acid stretch whose α 1C,86-like mutations in the wild-type α 1C conform to the same characteristic features [35], suggesting the existence of two adjacent CDI sensors. One of them was outlined in the K region as the calmodulin- (CaM-) binding IQ motif [36] and, later on, the link of the IQ motif to CDI as the functional Ca2+-CaM binding site was confirmed in three independent studies [37–39] by the use of CaM mutants lacking affinity to Ca2+. Correspondingly, the LA motif was linked to CDI as apo-CaM binding site [40–42] endowed by the resting state of the channel. A single CaM molecule tethered to this Ca2+-dependent CaM-binding domain (CBD) of α 1C is the major Ca2+ sensor of the channel [43, 44].
Splice variation of α 1C in CBD region of α 1C,86 not only completely inhibits CDI, but also removes SI (Figure 3(B), panel c) and deprives the channel of differential sensitivity to β-subunit modulation (Figure 3(B), panel e) in spite of the fact that β remains associated with α 1C,86 (Figure 3(B), panel b). This indicates that all three properties of the channel—CDI, SI, and β-subunit modulation—are linked together [48, 49].
3.2. Slow Inactivation
A number of evidences have been presented that amino acids confined to the distal part of S6 segments in α 1C play important role in SI [50–52]. Systematic study of this region [53] outlined the “annual determinant of slow inactivation” (ADSI) as a structure composed of four highly conserved amino acids of four transmembrane segments S6, constituting the cytoplasmic end of the pore (Figure 3(A), panel a). Their simultaneous mutation (S405I in IS6, A752T in IIS6, V1165T in IIIS6, and I1475T in IVS6) generates the α 1C,IS-IV channel. Analysis of the current kinetics of the α 1C,IS-IV channel showed tremendous acceleration of the rapidly inactivating component (τ f ≤ 10 ms) that comprises about 50% of the total I Ba (or I Ca) amplitude. Slow voltage-dependent inactivation of α 1C,IS-IV is fully inhibited, and the channel remains conducting for the duration of depolarization. Replacement of Ca2+ for Ba2+ as the charge carrier (panel c) did not change significantly this pattern of inactivation, while the analysis of voltage dependence of τ f for the inactivating component of I Ca through the α 1C,IS-IV channel (panel d) confirmed lack of CDI. The replacement of β 1a for β 2a (panel e) did not change inactivation of the α 1C,IS-IV channel current suggesting lack of differential β-subunit modulation, while the co-immunoprecipitation analysis (panel b) provided direct evidence of association between α 1C,IS-IV and β.
Taken together, results presented in Figure 3 suggest that there is a cross-talk between ADSI, CBD and β, supported by direct interactions between them and/or specific conformational folding of the constituents of the polypeptide bundle underlying the pore. Indeed, both the interaction of β with CBD and the importance of functional conformation were directly demonstrated in live cells expressing recombinant Cav1.2.
3.3. Role of the α 1C C-tail Folding
Quantitative voltage-dependent FRET microscopy combined with patch clamp in the live cell showed that the α 1C subunit C-terminal tail is subject to reversible voltage-gated conformational rearrangements [21, 47]. The anchoring of the α 1C C-tail to the inner leaflet of the plasma membrane via the pleckstrin homology (PH) domain fused to the C-terminus of α 1C (α 1C-PHC) abolished this conformational rearrangement and inhibited both SI and CDI (Figure 4(A)) in a manner very similar to that observed with α 1C,IS-IV (Figure 3(C)). This modification limiting the mobility of the α 1C carboxyl terminus had major implication on Ca2+ signal transduction. CREB-dependent transcriptional activation associated with the activity of Cav1.2 was completely suppressed in spite of robust I Ca generated by the “C-anchored” channel in response to depolarization. Release of the α 1C C-tail by activation of PIP2 hydrolysis upon activation of phospholipase C fully restores all these deficient functions, including SI, CDI, and the effective coupling of I Ca to the CREB-dependent transcription [21]. Thus, it is specific functional folding of the α 1C C-terminal tail that is crucial for inactivation. It is crucial for signal transduction because it is designed to cage the permeating Ca2+ in CaM attached to CBD and to effectively move this caged Ca2+ to downstream signaling targets associated with CREB-dependent transcription or cardiac muscle contraction [54]. Above all, this function occurs in tight coordination with extracellular stimuli activating the channel. In terms of signal transduction, SI is a lock on the inside of the channel that is released by the permeating Ca2+ to accelerate its closure and initiate the movement of the C-terminal tail [49].
Figure 4.
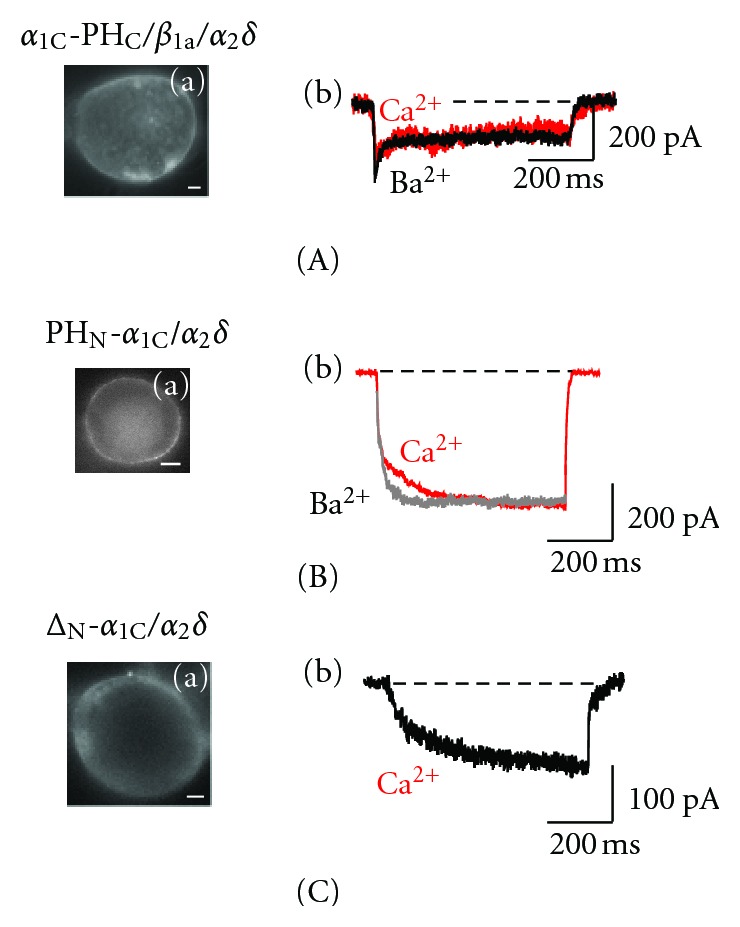
Differential role of the carboxyl- and amino-terminal tails of α 1C in Cav1.2 inactivation. Shown are (a) epifluorescent images illustrating the plasma membrane targeting of the EYFP-labeled α 1C (scaling bars, 4 μm) and (b) superimposed traces of the maximum I Ba (black) and I Ca (red) scaled to the same amplitude for α 1C-PHC/β 1a/α 2 δ [21, 47], (A) PHN-α 1C/α 2 δ (B) [47], and ΔN-α 1C/α 2 δ (C) [47].
3.4. Role of the α 1C N-Terminus
All the functions mentioned above depend also on the integrity of the α 1C N-terminus. Inactivation properties of the recombinant α 1C/β/α 2 δ channel are not greatly altered by structural changes of the proximal part of the α 1C N-tail, for example, by the fusion of a fluorescent protein [19, 21], by PH domain [47], or by alternative splicing of exons 1/1A generating the long isoform of α 1C [55]. The very first functional analysis of the effect of partial deletion of the α 1C N-terminus showed [56] that it is involved in inactivation while β prevents inhibition of the channel by the N-tail. Using FRET microscopy combined with patch clamp, we found that inactivation causes strong mutual reorientation of the α 1C and β 1a NH2-termini, but their distance vis-à-vis the plasma membrane is not appreciably changed [19]. This relative lack of mobility is conferred by β in a manner that facilitates the channel response to voltage gating. Experiments on uncoupling of the α 1C subunit N-terminal tail from the regulation of the channel were carried out in the absence of β. Anchoring of the α 1C N-tail in the inner leaflet of the plasma membrane via attached PH domain created conditions when PHN-α 1C and α 2 δ were sufficient to generate a robust inward current (Figure 4(B)). This channel, however, is deprived of CDI and any voltage-dependent inactivation. Indeed, neither Ba2+ nor Ca2+ current has shown appreciable decay (see overlapped traces). Release of the α 1C N-tail upon PIP2 hydrolysis by activation of phospholipase C completely inhibited the β-deficient channel [47]. Similar properties, except a much slower activation of the current, were observed on deletion of the entire (but 4 amino acids) N-terminal tail of α 1C (Figure 4(C)). With either type of uncoupling of the α 1C N-terminal tail—whether through a deletion or by PM anchoring,—a delay in the activation of the whole-cell current appears to be associated with prolongation of the first latency. Single channel recordings revealed that deletion of the N-tail essentially stabilized the open state of the ΔN-α 1C/α 2 δ channel, which showed longer openings during long-lasting depolarization [47].
Thus, CDI is mediated by CBD determinants of the α 1C C-tail, by the ADSI in the cytoplasmic pore region, and by the folding of the α 1C C- and N-termini. Calmodulin integrates these determinants, providing a Ca2+-dependent switch that terminates slow inactivation, releases the α 1C C-tail, and shuttles the associated Ca2+/calmodulin acting as an activating stimulus of the Ca2+ signal transduction [49].
3.5. Expression and Inactivation of Cav1.2 in the Absence of the β and α 2 δ
Are the β and α 2 δ subunits essential for the functional expression of the Cav1.2 channel? The analysis of the effects of exogenous CaM (CaMex) on the expression and properties of Cav1.2 in the absence of either β [18] or α 2 δ [57] clearly demonstrated that neither β nor α 2 δ is essential. Overexpression of CaMex only slightly modifies the voltage gating of the α 1C/β 2d/α 2 δ channel by shifting the voltage dependence of activation and inactivation towards more negative potentials, facilitating (but not accelerating) inactivation, and increasing the density of I Ca approximately 2-fold [18]. CDI is retained, as it is evident from the effect of the replacement of Ca2+ for Ba2+ as the charge carrier that significantly increased the time course of inactivation of the current (Figure 5(A)). New understanding of the roles of β and α 2 δ comes with the finding that CaMex renders expression and activity of the α 1C channel in the absence of β (Figure 5(B)) or α 2 δ (Figure 5(C)), but not both of these auxiliary subunits. Although CaMex is structurally unrelated to β and α 2 δ, it supports trafficking, CDI, and channel gating. Quantitative analysis showed that CaMex did not stimulate redistribution of α 1C in PM over the cytoplasm, but significantly enhanced plasma membrane targeting of α 1C/α 2 δ channels. On the other hand, CaMex did not enhance the relative distribution of the α 1C/β 2d and α 1C/β 2d/α 2 δ channels in the plasma membrane over the cytoplasm. Thus, depending on the auxiliary subunit present, CaMex-supported channel activity of α 1C/β 2d and α 1C/α 2 δ is under control of different mechanisms. In spite of that, the CaMex-facilitated, single-auxiliary-subunit channels exhibit quite similar properties including significantly slower inactivation kinetics of the calcium current and a strong shift of the voltage dependence of activation and inactivation towards more negative potentials. Similar to the conventional α 1C/β 2d/α 2 δ channels, these channels retain CDI and high sensitivity to dihydropyridine calcium channel blockers [18]. However, only the α 1C/β/CaMex channel shows facilitation of the calcium current by strong depolarization prepulse [57] (data not shown).
Figure 5.
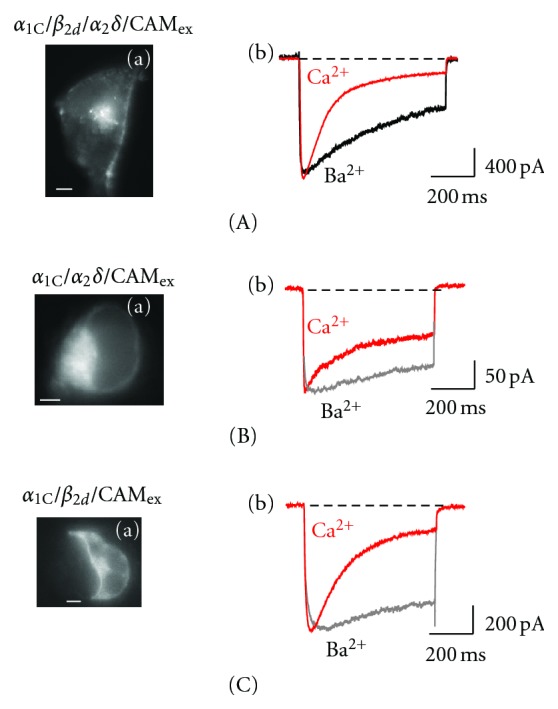
Activity of Cav1.2 expressed in the absence of β or α 2 δ subunits. Shown are (a) epifluorescent images illustrating the predominant PM localization of EYFPN-α 1C (scaling bars, 4 μm) in COS1 cells and (b) superimposed traces of the maximum I Ba (black) and I Ca (red) scaled to the same amplitude for α 1C/β 2d/α 2 δ/CaMex (A), β-free α 1C/α 2 δ/CaMex [18] (B) and α 2 δ-free α 1C/β 2d/CaMex channel [57] (C).
Because CaM associated with CBD is involved in CDI, it is clear that the effect of CaMex is mediated by different CaM-binding site(s). One of the potential candidates of such a site is present in the distal part of the α 1C N-terminal tail [58, 59]. It remains to be seen whether this site indeed plays an integrating role in the regulatory bundle of several molecular determinants supporting Cav1.2 inactivation. Another possibility confines the role of CaMex to the activation of silent Cav1.2 within the large clusters, where limited local availability of CaM may be the reason of the low fractional activity described in Section 2.5. Whatever the mechanisms associated with regulation of Cav1.2 by CaM are, they seems to have little practical implication for use in medicine at this time exactly because CaM is a ubiquitous and multifunctional peptide that regulates many other cellular functions, while its presence in Cav1.2 is vital for CDI.
4. β-Subunit Modulation of Cav1.2
Remarkable molecular variability of β subunits, reflected in altered inactivation properties of the differentially modulated Cav1.2 [12, 60], exemplified in Figure 3(A) (panel e) presents a new opportunity for the development of innovative approaches to the treatment of the diseases associated with Ca2+ mishandling. Several recent observations provide a foundation for such an optimistic view. First, β subunits exhibit a tendency to form homo- and hetero-oligomers [11, 61] that was directly demonstrated by a variety of biochemical techniques in both native cells and in recombinant expression system. While an augmentation of β homooligomerization significantly increases the density of I Ca, heterooligomerization of β 2 splice variants with other β subunits may also change the voltage-dependence and inactivation kinetics of Cav1.2 [11]. The β-oligomerization is mediated by several molecular determinants and thus needs multiple interventions to be managed, for example, in case of pathogenic overexpression of β 2. However, it seems to be more feasible to target β 2 itself; molecular determinant of β 2-specific slow and incomplete inactivation (see Figure 2(A), panel d) was identified [46] as the 40-amino-acid C-terminal determinant (β 2CED) present in all 7 known naturally occurring β 2 splice variants. Uncoupling of its Ca2+- and CaM-independent interaction with CBD (Figure 3(A), panel a) recovers the inactivation properties characteristic for β 1b/β 3-modulated Cav1.2 exhibiting rapid and complete inactivation of I Ca, as it was shown in deletion experiments. In my view, such selective uncoupling of β 2CED from binding to its receptor in CBD is a new attractive strategy to manage Ca2+ overload because other β subunits are not to be affected. Moreover, a cross-talk between Cav1.2 and the nearest target Ca2+/CaM-dependent protein kinase II [62, 63] will be preserved.
5. Conclusions
This paper has demonstrated that we know how to accelerate inactivation of Cav1.2 to τ f less than 10 ms (Figure 3(C)), to deprive it from inactivation completely (Figures 4(B) and 4(C)), or to eliminate dependence of its expression from β or α 2 δ without significant consequences for inactivation. We outlined the ultimate roles of the α 1C termini and CaM for inactivation, and yet none of these studies have brought us any closer to the ultimate goal of managing calcium mishandling associated with Cav1.2 except of old and, unfortunately, not too selective calcium channel blockers. The only new feasible target is pathogenic β 2 modulation of Cav1.2, where effector-receptor interaction is established.
In terms of molecular biology, Cav1.2 is certainly among the most complicated regulatory systems known. Remarkable molecular diversity of each of the Cav1.2 constituents gives rise to multiple genetic/splice variants of the channel that are subject to segregation into large and diverse clusters and to continuous functional change through homo- and hetero-oligomerization of β and other signaling components, not to speak about species, tissue, and developmental variability. We are surprised by the redundancy of the properties of multiple Cav1.2 isoforms [64, 65] and are even more surprised when some of them, showing just “conventional” electrophysiological properties, turn out to be associated with a disease [13]. In looking for an explanation, our insight should not be intuitively focused just on the characteristics of the calcium current-voltage dependence, amplitude, and duration. The end response, such as spatial and temporal organization of CREB signaling events associated with specific Cav1.2 isoform [66], and its competition with other (e.g., cAMP dependent) signaling mechanisms, or other Cav1.2 isoforms present, may provide new ideas and open new frontiers for investigation of the roles of individual Cav1.2 splice variants in normal and diseased cells and tissues.
Abbreviations
- ADSI:
Annual determinant of slow inactivation
- CaM:
Calmodulin
- CBD:
Calmodulin-binding domain
- CDI:
Ca-dependent inactivation
- ECFP:
Enhanced cyan fluorescent protein
- EYFP:
Enhanced yellow fluorescent protein
- FALI:
Fluorophore-assisted light inactivation
- FI:
Fast inactivation
- FRET:
Fluorescent resonance energy transfer
- GFP:
Green fluorescent protein
- SI:
Slow voltage-dependent inactivation.
References
- 1.Bers D. M. Calcium cycling and signaling in cardiac myocytes. Annual Review of Physiology. 2008;70(1):23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 2.Nelson M. T., Standen N. B., Brayden J. E., Worley J. F. Noradrenaline contracts arteries by activating voltage-dependent calcium channels. Nature. 1988;336(6197):382–385. doi: 10.1038/336382a0. [DOI] [PubMed] [Google Scholar]
- 3.Bading H., Ginty D. D., Greenberg M. E. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260(5105):181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 4.Johnston D., Williams S., Jaffe D., Gray R. NMDA-receptor-independent long-term potentiation. Annual Review of Physiology. 1992;54:489–505. doi: 10.1146/annurev.ph.54.030192.002421. [DOI] [PubMed] [Google Scholar]
- 5.Artalejo C. R., Adams M. E., Fox A. P. Three types of Ca2+ channel trigger secretion with different efficacies in chromaffin cells. Nature. 1994;367(6458):72–76. doi: 10.1038/367072a0. [DOI] [PubMed] [Google Scholar]
- 6.Carafoli E. Intracellular calcium homeostasis. Annual Review of Biochemistry. 1987;56:395–433. doi: 10.1146/annurev.bi.56.070187.002143. [DOI] [PubMed] [Google Scholar]
- 7.Flockerzi V., Oeken H. J., Hofmann F. Purification of a functional receptor for calcium-channel blockers from rabbit skeletal-muscle microsomes. European Journal of Biochemistry. 1986;161(1):217–224. doi: 10.1111/j.1432-1033.1986.tb10145.x. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi M., Seagar M. J., Jones J. F., Reber B. F. X., Catterall W. A. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(15):5478–5482. doi: 10.1073/pnas.84.15.5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dolphin A. C. β Subunits of voltage-gated calcium channels. Journal of Bioenergetics and Biomembranes. 2003;35(6):599–620. doi: 10.1023/B:JOBB.0000008026.37790.5a. [DOI] [PubMed] [Google Scholar]
- 10.Klugbauer N., Marais E., Hofmann F. Calcium channel α 2 δ subunits: differential expression, function, and drug binding. Journal of Bioenergetics and Biomembranes. 2003;35(6):639–647. doi: 10.1023/B:JOBB.0000008028.41056.58. [DOI] [PubMed] [Google Scholar]
- 11.Lao Q. Z., Kobrinsky E., Liu Z., Soldatov N. M. Oligomerization of Cav β subunits is an essential correlate of Ca2+ channel activity. FASEB Journal. 2010;24(12):5013–5023. doi: 10.1096/fj.10-165381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colecraft H. M., Alseikhan B., Takahashi S. X., et al. Novel functional properties of Ca2+ channel β subunits revealed by their expression in adult rat heart cells. Journal of Physiology. 2002;541(2):435–452. doi: 10.1113/jphysiol.2002.018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiwari S., Zhang Y., Heller J., Abernethy D. R., Soldatov N. M. Artherosclerosis-related molecular alteration of the human Cav1.2 calcium channel α 1C subunit. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(45):17024–17029. doi: 10.1073/pnas.0606539103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soldatov N. M. Genomic structure of human L-type Ca2+ channel. Genomics. 1994;22(1):77–87. doi: 10.1006/geno.1994.1347. [DOI] [PubMed] [Google Scholar]
- 15.Berjukow S., Döring F., Froschmayr M., Grabner M., Glossmann H., Hering S. Endogenous calcium channels in human embryonic kidney (HEK293) cells. British Journal of Pharmacology. 1996;118(3):748–754. doi: 10.1111/j.1476-5381.1996.tb15463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurejová M., Uhrík B., Sulová Z., Sedláková B., Križanová O., Lacinová L. Changes in ultrastructure and endogenous ionic channels activity during culture of HEK 293 cell line. European Journal of Pharmacology. 2007;567(1-2):10–18. doi: 10.1016/j.ejphar.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Meir A., Bell D. C., Stephens G. J., Page K. M., Dolphin A. C. Calcium channel β subunit promotes voltage-dependent modulation of α1B by Gβγ . Biophysical Journal. 2000;79(2):731–746. doi: 10.1016/S0006-3495(00)76331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravindran A., Lao Q. Z., Harry J. B., Abrahimi P., Kobrinsky E., Soldatov N. M. Calmodulin-dependent gating of Cav1.2 calcium channels in the absence of Cav β subunits. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(23):8154–8159. doi: 10.1073/pnas.0711624105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobrinsky E., Kepplinger K. J. F., Yu A., et al. Voltage-gated rearrangements associated with differential β-subunit modulation of the L-type Ca2+ channel inactivation. Biophysical Journal. 2004;87(2):844–857. doi: 10.1529/biophysj.104.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobrinsky E., Lee J.-H., Soldatov N. M. Selective fluorophore-assisted light inactivation of voltage-gated calcium channels. Channels. 2012;6(3):154–156. doi: 10.4161/chan.20867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobrinsky E., Schwartz E., Abernethy D. R., Soldatov N. M. Voltage-gated mobility of the Ca2+ channel cytoplasmic tails and its regulatory role. Journal of Biological Chemistry. 2003;278(7):5021–5028. doi: 10.1074/jbc.M211254200. [DOI] [PubMed] [Google Scholar]
- 22.Herzig S., Khan I. F. Y., Gründemann D., et al. Mechanism of Cav1.2 channel modulation by the amino terminus of cardiac β2-subunits. FASEB Journal. 2007;21(7):1527–1538. doi: 10.1096/fj.06-7377com. [DOI] [PubMed] [Google Scholar]
- 23.Lew W. Y. W., Hryshko L. V., Bers D. M. Dihydropyridine receptors are primarily functional L-type calcium channels in rabbit ventricular myocytes. Circulation Research. 1991;69(4):1139–1145. doi: 10.1161/01.res.69.4.1139. [DOI] [PubMed] [Google Scholar]
- 24.Hell J. W., Westenbroek R. E., Warner C., et al. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. Journal of Cell Biology. 1993;123(4):949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipscombe D., Madison D. V., Poenie M., Reuter H., Tsien R. Y., Tsien R. W. Spatial distribution of calcium channels and cytosolic calcium transients in growth cones and cell bodies of sympathetic neurons. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(7):2398–2402. doi: 10.1073/pnas.85.7.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westenbroek R. E., Ahlijanian M. K., Catterall W. A. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347(6290):281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 27.Franzini-Armstrong C., Protasi F., Tijskens P. The assembly of calcium release units in cardiac muscle. Annals of the New York Academy of Sciences. 2005;1047:76–85. doi: 10.1196/annals.1341.007. [DOI] [PubMed] [Google Scholar]
- 28.Gathercole D. V., Colling D. J., Skepper J. N., Takagishi Y., Levi A. J., Severs N. J. Immunogold-labeled L-type calcium channels are clustered in the surface plasma membrane overlying junctional sarcoplasmic reticulum in guinea-pig myocytes—implications for excitation-contraction coupling in cardiac muscle. Journal of Molecular and Cellular Cardiology. 2000;32(11):1981–1994. doi: 10.1006/jmcc.2000.1230. [DOI] [PubMed] [Google Scholar]
- 29.Takagishi Y., Yasui K., Severs N. J., Murata Y. Species-specific difference in distribution of voltage-gated L-type Ca2+ channels of cardiac myocytes. American Journal of Physiology. 2000;279(6):C1963–C1969. doi: 10.1152/ajpcell.2000.279.6.C1963. [DOI] [PubMed] [Google Scholar]
- 30.Harms G. S., Cognet L., Lommerse P. H. M., et al. Single-molecule imaging of L-type Ca2+ channels in live cells. Biophysical Journal. 2001;81(5):2639–2646. doi: 10.1016/S0006-3495(01)75907-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobrinsky E., Abrahimi P., Duong S. Q., et al. Effect of Cav β subunits on structural organization of Cav1.2 calcium channels. PLoS ONE. 2009;4(5) doi: 10.1371/journal.pone.0005587.e5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee K. S., Marban E., Tsien R. W. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. Journal of Physiology. 1985;364:395–411. doi: 10.1113/jphysiol.1985.sp015752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zong X., Hofmann F. Ca2+-dependent inactivation of the class C L-type Ca2+ channel is a property of the α1 subunit. FEBS Letters. 1996;378(2):121–125. doi: 10.1016/0014-5793(95)01434-9. [DOI] [PubMed] [Google Scholar]
- 34.Soldatov N. M., Zühlke R. D., Bouron A., Reuter H. Molecular structures involved in L-type calcium channel inactivation. Role of the carboxyl-terminal region encoded by exons 40–42 in α 1C subunit in the kinetics and Ca2+ dependence of inactivation. Journal of Biological Chemistry. 1997;272(6):3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- 35.Soldatov N. M., Oz M., O'Brien K. A., Abernethy D. R., Morad M. Molecular determinants of L-type Ca2+ channel inactivation: segment exchange analysis of the carboxyl-terminal cytoplasmic motif encoded by exons 40-42 of the human α 1C subunit gene. Journal of Biological Chemistry. 1998;273(2):957–963. doi: 10.1074/jbc.273.2.957. [DOI] [PubMed] [Google Scholar]
- 36.Zühlke R. D., Reuter H. Ca2+-sensitive inactivation of L-type Ca2+ channels depends on multiple cytoplasmic amino acid sequences of the α 1C subunit. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):3287–3294. doi: 10.1073/pnas.95.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson B. Z., DeMaria C. D., Adelman J. P., Yue D. T. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22(3):549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 38.Qin N., Olcese R., Bransby M., Lin T., Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(5):2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zühlke R. D., Pitt G. S., Deisseroth K., Tsien R. W., Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399(6732):159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 40.Erickson M. G., Alseikhan B. A., Peterson B. Z., Yue D. T. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31(6):973–985. doi: 10.1016/S0896-6273(01)00438-X. [DOI] [PubMed] [Google Scholar]
- 41.Pate P., Mochca-Morales J., Wu Y., et al. Determinants for calmodulin binding on voltage-dependent Ca2+ channels. Journal of Biological Chemistry. 2000;275(50):39786–39792. doi: 10.1074/jbc.M007158200. [DOI] [PubMed] [Google Scholar]
- 42.Romanin C., Gamsjaeger R., Kahr H., et al. Ca2+ sensors of L-type Ca2+ channel. FEBS Letters. 2000;487(2):301–306. doi: 10.1016/S0014-5793(00)02361-9. [DOI] [PubMed] [Google Scholar]
- 43.Mori M. X., Erickson M. G., Yue D. T. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 2004;304(5669):432–435. doi: 10.1126/science.1093490. [DOI] [PubMed] [Google Scholar]
- 44.Xiong L., Kleerekoper Q. K., He R., Putkey J. A., Hamilton S. L. Sites on calmodulin that interact with the C-terminal tail of Cav1.2 channel. Journal of Biological Chemistry. 2005;280(8):7070–7079. doi: 10.1074/jbc.M410558200. [DOI] [PubMed] [Google Scholar]
- 45.Zhang R., Dzhura I., Grueter C. E., Thiel W., Colbran R. J., Anderson M. E. A dynamic α-β inter-subunit agonist signaling complex is a novel feedback mechanism for regulating L-type Ca2+ channel opening. FASEB Journal. 2005;19(11):1573–1575. doi: 10.1096/fj.04-3283fje. [DOI] [PubMed] [Google Scholar]
- 46.Lao Q. Z., Kobrinsky E., Harry J. B., Ravindran A., Soldatov N. M. New determinant for the Cav β 2 subunit modulation of the Cav1.2 calcium channel. Journal of Biological Chemistry. 2008;283(23):15577–15588. doi: 10.1074/jbc.M802035200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kobrinsky E., Tiwari S., Maltsev V. A., et al. Differential role of the α 1C subunit tails in regulation of the Cav1.2 channel by membrane potential, β subunits, and Ca2+ ions. Journal of Biological Chemistry. 2005;280(13):12474–12485. doi: 10.1074/jbc.M412140200. [DOI] [PubMed] [Google Scholar]
- 48.Hering S., Berjukow S., Aczél S., Timin E. N. Ca2+ channel block and inactivation: common molecular determinants. Trends in Pharmacological Sciences. 1998;19(11):439–443. doi: 10.1016/S0165-6147(98)01258-9. [DOI] [PubMed] [Google Scholar]
- 49.Soldatov N. M. Ca2+ channel moving tail: link between Ca2+-induced inactivation and Ca2+ signal transduction. Trends in Pharmacological Sciences. 2003;24(4):167–171. doi: 10.1016/S0165-6147(03)00065-8. [DOI] [PubMed] [Google Scholar]
- 50.Hering S., Berjukow S., Sokolov S., et al. Molecular determinants of inactivation in voltage-gated Ca2+ channels. Journal of Physiology. 2000;528(2):237–249. doi: 10.1111/j.1469-7793.2000.t01-1-00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soldatov N. M., Zhenochin S., AlBanna B., Abernethy D. R., Morad M. New molecular determinant for inactivation of the human L-type α 1C Ca2+ channel. Journal of Membrane Biology. 2000;177(2):129–135. doi: 10.1007/s002320001106. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J. F., Ellinor P. T., Aldrich R. W., Tsien R. W. Molecular determinants of voltage-dependent inactivation in calcium channels. Nature. 1994;372(6501):97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]
- 53.Shi C., Soldatov N. M. Molecular determinants of voltage-dependent slow inactivation of the Ca2+ channel. Journal of Biological Chemistry. 2002;277(9):6813–6821. doi: 10.1074/jbc.M110524200. [DOI] [PubMed] [Google Scholar]
- 54.Morad M., Soldatov N. Calcium channel inactivation: possible role in signal transduction and Ca2+ signaling. Cell Calcium. 2005;38(3-4):223–231. doi: 10.1016/j.ceca.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 55.Blumenstein Y., Kanevsky N., Sahar G., Barzilai R., Ivanina T., Dascal N. A novel long N-terminal isoform of human L-type Ca2+ channel is up-regulated by protein kinase C. Journal of Biological Chemistry. 2002;277(5):3419–3423. doi: 10.1074/jbc.C100642200. [DOI] [PubMed] [Google Scholar]
- 56.Shistik E., Ivanina T., Blumenstein Y., Dascal N. Crucial role of N terminus in function of cardiac L-type Ca2+ channel and its modulation by protein kinase C. Journal of Biological Chemistry. 1998;273(28):17901–17909. doi: 10.1074/jbc.273.28.17901. [DOI] [PubMed] [Google Scholar]
- 57.Ravindran A., Kobrinsky E., Lao Q. Z., Soldatov N. M. Functional properties of the Cav1.2 calcium channel activated by calmodulin in the absence of α 2 δ subunits. Channels. 2009;3(1):25–31. doi: 10.4161/chan.3.1.7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benmocha A., Almagor L., Oz S., Hirsch J. A., Dascal N. Characterization of the calmodulin-binding site in the N terminus of Cav1.2 . Channels. 2009;3(5):337–342. doi: 10.4161/chan.3.5.9686. [DOI] [PubMed] [Google Scholar]
- 59.Ivanina T., Blumenstein Y., Shistik E., Barzilai R., Dascal N. Modulation of L-type Ca2+ channels by Gβγ and calmodulin via interactions with N and C termini of α 1C . Journal of Biological Chemistry. 2000;275(51):39846–39854. doi: 10.1074/jbc.M005881200. [DOI] [PubMed] [Google Scholar]
- 60.Buraei Z., Yang J. The β subunit of voltage-gated Ca2+ channels. Physiological Reviews. 2010;90(4):1461–1506. doi: 10.1152/physrev.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miranda-Laferte E., Gonzalez-Gutierrez G., Schmidt S., et al. Homodimerization of the Src homology 3 domain of the calcium channel β-subunit drives dynamin-dependent endocytosis. Journal of Biological Chemistry. 2011;286(25):22203–22210. doi: 10.1074/jbc.M110.201871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grueter C. E., Abiria S. A., Wu Y., Anderson M. E., Colbran R. J. Differential regulated interactions of calcium/calmodulin-dependent protein kinase II with isoforms of voltage-gated calcium channel β subunits. Biochemistry. 2008;47(6):1760–1767. doi: 10.1021/bi701755q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koval O. M., Guan X., Wu Y., et al. Cav1.2β-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(11):4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao P., Yu D., Lu S., et al. Smooth muscle-selective alternatively spliced exon generates functional variation in Cav1.2 calcium channels. Journal of Biological Chemistry. 2004;279(48):50329–50335. doi: 10.1074/jbc.M409436200. [DOI] [PubMed] [Google Scholar]
- 65.Liao P., Yu D., Li G., et al. A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. Journal of Biological Chemistry. 2007;282(48):35133–35142. doi: 10.1074/jbc.M705478200. [DOI] [PubMed] [Google Scholar]
- 66.Kobrinsky E., Duong S. Q., Sheydina A., Soldatov N. M. Microdomain organization and frequency-dependence of CREB-dependent transcriptional signaling in heart cells. FASEB Journal. 2011;25(5):1544–1555. doi: 10.1096/fj.10-176198. [DOI] [PMC free article] [PubMed] [Google Scholar]