

Abstract
In order to identify the most attractive starting points for drugs that can be used to prevent malaria, a diverse chemical space comprising tens of thousands to millions of small molecules may need to be examined. Achieving this throughput necessitates the development of efficient ultra-high-throughput screening methods. Here, we report the development and evaluation of a luciferase-based phenotypic screen of malaria exoerythrocytic-stage parasites optimized for a 1536-well format. This assay uses the exoerythrocytic stage of the rodent malaria parasite, Plasmodium berghei, and a human hepatoma cell line. We use this assay to evaluate several biased and unbiased compound libraries, including two small sets of molecules (400 and 89 compounds, respectively) with known activity against malaria erythrocytic-stage parasites and a set of 9886 diversity-oriented synthesis (DOS)-derived compounds. Of the compounds screened, we obtain hit rates of 12–13 and 0.6% in preselected and naïve libraries, respectively, and identify 52 compounds with exoerythrocytic-stage activity less than 1 μM and having minimal host cell toxicity. Our data demonstrate the ability of this method to identify compounds known to have causal prophylactic activity in both human and animal models of malaria, as well as novel compounds, including some exclusively active against parasite exoerythrocytic stages.
Keywords: malaria, drug discovery, high-throughput screening, exoerythrocytic-stage malaria, liver-stage malaria
Introduction
Despite being an ancient disease, malaria is still responsible for over a half million deaths and substantial morbidity, poverty, and suffering for hundreds of millions of people each year.1−3 It is a vector-borne disease caused by infection with Plasmodium parasites transmitted through the bite of Anopheles mosquitoes. While eradication campaigns have been successful in most of North America and Europe, malaria continues to devastate developing regions of Asia, Africa, and South America.4 The mortality rates are highest among African children, with an estimated one death per minute (WHO). The emergence of resistance to all of the current frontline antimalarial drugs warrants global concern.5 It is therefore critical that new drugs are developed that not only treat disease symptoms but also contribute toward the elimination and eradication of malaria infections. In order to achieve eradication, new drugs should inhibit multiple developmental stages of the parasite. Following the blood meal of an infected Anopheles mosquito, Plasmodium sporozoites travel through the bloodstream to reach the liver. The sporozoites traverse multiple cells within the liver before establishing productive invasion within hepatocytes, where they transform into exoerythrocytic-stage exoerythrocytic forms (EEFs).6 Depending on the species, these exoerythrocytic forms enter one of two developmental pathways: they can form mature exoerythrocytic-stage schizonts, or they can enter a dormant phase called hypnozoites. The determinant factors guiding exoerythrocytic-stage development toward hypnozoite formation in P. vivax and P. ovale are not understood. Hypnozoites can reinitiate development and give rise to malaria relapses weeks, months, or even years after the initial infection.7 Fully developed exoerythrocytic-stage merozoites within schizonts eventually egress from the liver and re-enter the bloodstream.6 The continuous replication of asexual blood stages within red blood cells (RBCs) causes RBC destruction and leads to the characteristic symptoms associated with malaria: anemia, fever, and chills.8 A small percentage of these asexual blood stage parasites will then differentiate into sexual erythrocytic-stage parasites as female and male gametocytes, and the transmission of the sexual blood stage back to the mosquito vector during a subsequent blood meal completes the life cycle.9
The majority of the current antimalarials only treat the symptom-causing erythrocytic stages of the parasite.10 A few classes, including cytochrome bc1 inhibitors (such as atovaquone) and antifolate drugs (such as pyrimethamine), are active against developing exoerythrocytic forms as well as erythrocytic forms and are able to prevent the establishment of infection (causal prophylactic activity) as well as relieve symptoms.10 Antibiotics, such as doxycycline, clindamycin, and azithromycin, are also an important class of antimalarial drugs. Doxycycline is commonly prescribed to travelers to endemic areas and is especially useful for its casual prophylaxis and slow acting blood schizontocidal activity.11 The 8-aminoquinolines (such as primaquine and tafenoquine) are a unique class in that they can eliminate hypnozoites as well and can provide a radical cure for P. vivax and P. ovale.7,10 Having new classes of drugs that could be used prophylactically and/or to provide a radical cure would be desirable. Resistance is developing to both napthoquinones and antifolates, and 8-aminoquinolines can be toxic to individuals with glucose-6-phosphate deficiency.10,12 Drugs targeting the exoerythrocytic stage only would also offer the reduced potential for drug resistance, as there are far fewer parasites at this “bottleneck” compared to the replicative erythrocytic stages.10 Accordingly, the development of an exoerythrocytic-stage-specific high-throughput screening assay is necessary in order to identify the next generation of antimalarial drugs.
Although it is possible to create new chemical derivatives of existing drugs with improved therapeutic and resistance profiles, phenotypic screening offers the opportunity to find entirely new classes of small molecules that are active against exoerythrocytic stages of the life cycle.13 We have previously reported an immunofluorescence-based malaria exoerythrocytic-stage assay that we used to screen a library of >4000 commercially available compounds with erythrocytic-stage activity.14 While this platform led to the identification of 275 exoerythrocytic-stage active compounds, the assay is limited to a 384-well throughput and is therefore not suitable for the screening of larger chemical libraries, in part because of the high cost of sporozoites obtained by manual mosquito dissections ($1.00 per well screened). In addition, the requirement for a specialized high-content imaging device limits the accessibility of the assay. Other malaria exoerythrocytic-stage drug screens have utilized P. berghei sporozoites that express a luciferase reporter (Pb-Luc);15−17 however, these assays are also limited by a 384-well assay throughput. In this report, we describe the development of a high-throughput luciferase-based assay that can be used to screen chemical libraries in a 1536-well plate format. We demonstrate that the assay is highly sensitive, reproducible, and efficient. As a proof of concept, we use this assay to screen the Medicines for Malaria Venture (MMV) Malaria Box for compounds with exoerythrocytic-stage activity18 as well as a larger collection of chemical compounds from the Broad Diversity-Oriented Synthesis Library, a set that includes compounds with and without demonstrated erythrocytic-stage antimalarial activity.
Results and Discussion
Development of a Luciferase-Based High-Throughput Exoerythrocytic-Stage Assay
In order to develop a high-throughput exoerythrocytic-stage malaria assay capable of screening large libraries of chemical compounds, a number of tests were performed to optimize a 48 h in vitro PbGFP-Luc-SMCON19 infection of HepG2-A16-CD81EGFP hepatocytes20 (Figure S1). This rodent Plasmodium strain was previously generated through the integration of a GFP-Luc cassette into the c-rrna locus and selecting transgenic P. berghei by flow sorting GFP-expressing parasites. For simplicity, we will refer to this strain as Pb-Luc. For these tests, HepG2-A16-CD81EGFP cells were seeded in 1536-well plates 24 h prior to infection and luciferase bioluminescence measured 48 h postinfection to detect parasite viability. We found the ideal ratio of sporozoites to cells per well to be 1:3, respectively (1 × 103 sporozoites in 5 μL to 3 × 103 cells in 5 μL) (Figure 1a and Figure S1a). At these concentrations, the cells were ideally confluent, and the infection rate produced luciferase values that were significantly greater than background values at 48 h postinfection (Figure S1a). Furthermore, tests without hepatocytes showed that there was no residual luciferase activity from Pb-Luc sporozoites at 24 h postinfection at 37 °C (Figure S1b), eliminating the possibility that sporozoites, which had not invaded, contribute to the luciferase signal. We also tested different DMSO concentrations (added 18 h preinfection) to assess their impact on parasite viability and found that concentrations up to 0.88% DMSO had an insignificant effect on luciferase activity 48 h postinfection (Figure S1c). The final protocol was to add 50 nL of compound in DMSO (resulting in 50 μM compound and 0.5% DMSO concentration in the assay plates) 18 h preinfection in the optimized screening assay (Figure 1a). An example of the luciferase signal for two replicate plates seeded with a representative small molecule library is shown in Figure 1b. Z factor for these plates was between 0.7 and 0.9, an excellent value for a phenotypic screen.
Figure 1.

Luciferase-based high-throughput screening assay to identify malaria exoerythrocytic-stage inhibitors. (a) Assay workflow. Twenty-four hours prior to infection, 3 × 103 HepG2-A16-CD81EGFP cells in 5 μL media were added to wells in a 1536-well assay plate. Then 1–4 h later, 50 nL of compound dissolved in DMSO was added to the wells. At the time of infection, Pb-Luc sporozoites were freshly prepared from infected A. stephensi mosquitoes and diluted to a concentration of 1 × 103 in 5 μL media per well. After 48 h, Pb-Luc growth within hepatocytes was measured by bioluminescence. (b) As a proof of concept, we screened two plates containing 2816 natural compounds (GNF) in replicate. One set of replicates is shown here. The average Z factor for these plates was 0.82.
Assay Validation through Screening of Known Antimalarial Compounds
To validate this assay further, we next examined several compound collections. We first evaluated 50 established antimalarial clinical or tool compounds (Table S1) that have been tested in other antimalarial phenotypic assays.10 The most active compounds included the electron transport chain inhibitor atovaquone, antifolate pathway inhibitors P218·HCl, pyrimethamine and cycloguanil, the protein biosynthesis inhibitor cycloheximide, and the glutathione reductase inhibitor methylene blue. For these compounds, the exoerythrocytic-stage half-maximal inhibitory concentration (IC50) values were similar to the P. falciparum erythrocytic-stage IC50 values. Compounds that were less active relative to erythrocytic stages, on the other hand, included 4-aminoquinolines, amino alcohols, and endoperoxides, which presumably act primarily against hemoglobin degradation, a process that does not occur during hepatic stages. Overall, these results were highly consistent with our previously established high-content imaging (HCI) assay of the hepatic stages against the related rodent parasite, Plasmodium yoelii.10
For active compounds, we also compared the IC50 values for Pb-Luc generated by the luciferase-based enzymatic assay to values produced by screening Pb-Luc parasites using our previously established (HCI) assay that uses polyclonal Plasmodium HSP70 antibody staining at 48 h as an indicator of infection14 (Figure 2). We found that there was a strong correlation between compound activities in both assays, as indicated by an R2 of 0.83, but the high-throughput luciferase-based assay resulted in IC50s roughly 10× lower (Figure 2). Since luciferase is relatively unstable, with a half-life of less than 2 h,21 this may lead to a higher rate of reporter turnover and therefore increased sensitivity to compounds that inhibit parasite growth at later stages compared to that of parasite HSP70 (as measured in the HCI assay). Additionally, nonviable parasites may be stained using the HCI assay, whereas they may not produce luciferase.
Figure 2.
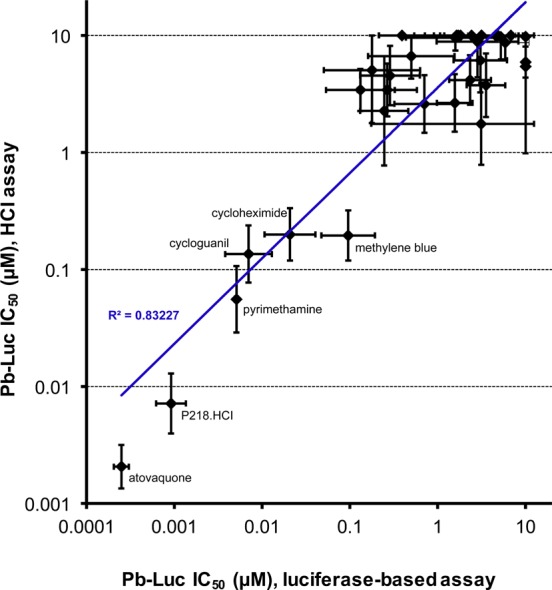
A 1536-well luciferase-based screening assay is higher-throughput and more sensitive than former 384-well HCI assay. Pb-Luc IC50 values for the MMV validation set of antimalarials screened by the luciferase-based 1536-well assay and by the 384-well high-content imaging (HCI) assay are compared. Each data point represents a single antimalarial compound. The most active compounds in both assays are labeled. The assays generate Pb-Luc IC50 values that correlate very well with each other, as demonstrated by an R2 value of 0.83; however, the luciferase-based assay resulted in IC50 values roughly 10× lower than that in the HCI assay.
Screening the MMV Malaria Box for Exoerythrocytic-Stage Inhibitors
After establishing the high-throughput luciferase-based assay quality, reproducibility, and sensitivity, we sought to test its validity as a platform to screen diverse chemical libraries by screening the MMV Malaria Box.18 This open-access set consists of 400 compounds that were selected from a group of ∼20 000 antimalarial hits generated from a large-scale erythrocytic-stage screening of >4 000 000 compounds by St. Jude Children’s Hospital, Novartis, and GSK (Figure S2). The library contains 200 “probe-like” and 200 “drug-like” compounds, selected based on their chemical diversity, erythrocytic-stage antimalarial activity, and commercial availability. The Malaria Box compounds have erythrocytic-stage activity ranging from an IC50 of 30 nM to 4 μM.18 Compounds originating in the Novartis collection had been previously tested in the P. yoelii assay,14 but others had not, providing valuable internal controls.
After a first-round screening of the MMV Malaria Box at compound concentrations of 50 μM in duplicate plates, 48 compounds were selected for reconfirmation based on Pb-Luc inhibition of more than 90% and HepG2 cytotoxicity of less than 25% in both assay plates, a hit rate of 12%. These were tested in a 12-point serial dilution dose response beginning at 10 μM. Of those retested, 36 had a Pb-Luc IC50 < 10 μM, and counter-screening for HepG2 cytotoxicity and luciferase inhibition produced IC50 values greater than 10 μM (Figure 3; see additional file 1 for full screening results), leading to a confirmation rate of 75%. Furthermore, more than half of these compounds were very active against Pb-Luc exoerythrocytic stages with IC50 values of less than 1 μM (Figure 3). Six of the compounds which had been previously tested and confirmed14 were reconfirmed here, but there were 18 compounds that were considered exoerythrocytic-stage active hits in the HCI assay and not in the luciferase-based assay. This is likely due to the higher initial screening concentrations used in the luciferase-based assay (50 μM compared to 10 μM in the HCI assay), leading to increased HepG2 cytotoxicity, as all of these compounds inhibited Pb-Luc activity but were toxic at 50 μM. It should be noted that due to the sensitivity of the luciferase assay and to minimize cytotoxicity, we advise starting with lower initial screening concentrations for future high-throughput screening.
Figure 3.

MMV Malaria Box compounds are identified as potent malaria exoerythrocytic-stage inhibitors: 36 exoerythrocytic-stage active MMV Malaria Box hits with a Pb-Luc IC50 of less than 10 μM are shown with their respective IC50 values. Hits were also selected to demonstrate a HepG2 IC50 and a Luc IC50 of more than 10 μM. A line at 1 μM highlights that almost half of the exoerythrocytic-stage active hits display nanomolar potency. Error bars represent the 95% confidence interval.
Screening the Broad Diversity-Oriented Synthesis Library
To assess performance in an unbiased screening library, two sets of diversity-oriented synthesis (DOS)-derived compounds were screened (outlined in Figure S3). These diverse compounds combine the stereochemical and skeletal complexity of the entire ensemble of natural products and the efficiency of high-throughput synthesis.22,23 This is an attractive validation library because multiple stereoisomers of each structural type are included, which permits a unique type of structure–activity relationship measurements. The first set included 9886 compounds selected to represent the structural diversity of the scaffolds from the Broad Institute’s 100 000 DOS compound library. This “informer set” was tested as a naïve library with respect to activity against Plasmodium. The second set comprised 89 compounds previously shown to be active in a P. falciparum erythrocytic-stage assay (IC50 < 2 μM, Nobutaka Kato et al., unpublished results). Compounds that inhibited the luciferase signal >75% were scored as hits. From the informer set (hit rate = 0.6%), 60 hits and 4 inconclusive (only active in one replicate) were identified, and 12 hits were identified from the erythrocytic-stage active set (hit rate = 13.4%). All available compounds (58) plus 25 additional weak actives (60–74% inhibition) from the informer set were tested in dose response in the primary exoerythrocytic-stage assay and also in a SYBR Green erythrocytic-stage assay.24 All of the compounds from the erythrocytic-stage active set were retested at a dose with IC50 < 5 μM, and 10 of these compounds had an IC50 < 1 μM. In all, 72% (60 compounds) of the naïve informer set hits retested with IC50 < 5 μM. A third of these had an IC50 < 1 μM (see additional file 2 for full screening results). Finally, stereoisomers for selected hits were also examined in dose response. These data (Figure 4a) showed stereoselective inhibition; for example, only the S,S,S stereoisomer, BRD0326, is active (IC50 = 0.152 μM) in a set of eight stereoisomers tested.
Figure 4.

DOS compounds exhibit stereoselective inhibition of Pb-Luc exoerythrocytic-stage parasite growth. (a) Representative compounds and activity profiles with activity against P. berghei in HepG2 cells. Stereocenters (Cn) are listed below the corresponding chemical structure. Pb-Luc exoerythrocytic-stage activity was measured for each of the eight possible stereoisomers (SRR, SSR, SRS, SSS, RRR, RSR, RRS, and RSS) of each compound. Three of the eight possible stereoisomers of BRD9781 have exoerythrocytic-stage activity, two with potent activity (SRR and SSS, IC50 < 0.1 μM) and another with moderate activity (RSS, IC50 < 1 μM). One stereoisomer of BRD0326 (SSS) is active (IC50 < 1 μM). Two stereoisomers of BRD47390 have significant exoerythrocytic-stage activity (SSR, IC50 < 0.1 μM; SRS, IC50 < 0.1 μM). (b) Compounds were tested in dose in the P. berghei/HepG2 assay, a Dd2 erythrocytic-stage assay, and in a mammalian cell cytotoxicity assay. Compounds from three scaffold libraries are shown. Compounds were tested twice in the exoerythrocytic-stage assay; values from the second assay are shown in parentheses.
MMV Malaria Box and Broad Library Exoerythrocytic-Stage Active Chemical Clustering Identifies Active Scaffolds and Important Targets
To cross-validate these data, we first identified scaffolds that were enriched for compounds with exoerythrocytic-stage activity (relative to erythrocytic stage or no antimalarial activity) using compound clustering. Given the small size of our initial compound set, we combined the MMV Malaria Box compounds with a GNF library consisting of 4422 compounds that were previously screened for exoerythrocytic-stage activity in P. berghei.14 Merging the libraries allowed us to determine if there was any overlap in scaffold hits between the two compound sets. The 4822 compounds were clustered using a hierarchical clustering method based on substructure similarity. To define scaffold groups, we separated clusters based on a minimum Tanimoto coefficient requirement of 0.65, resulting in 2335 total clusters that ranged in size from 1 to 45 compounds. DOS library compounds were also included, but given that the library was designed to eliminate structural redundancy, it was expected that DOS compounds would not contribute significantly to any scaffold clusters.
We identified 15 scaffold series that showed specific enrichment in exoerythrocytic-stage activity with rates higher than expected by chance (p < 0.001) (Figure 5). The scaffold clustering showed multiple scaffold families already known to be active. One is the diaminotriazine scaffold (1228) similar to the diaminopyrimidine found in antifolate drugs such as pyrimethamine (probability of enrichment by chance = 4.29 × 10–7). Another is a tetracyclic benzothiazepine scaffold, cluster 1096, that has shown to target the Q0 site of cytochrome bc1.25 Several enriched quinolone compounds (GNF-Pf-2549, GNF-Pf-4577, GNF-Pf-5037; cluster 342) were similar to ELQ300, a possible Q1 site cytochrome bc1 inhibitor26 with known causal prophylactic activity in mouse models of malaria.27 In addition, cluster 1096 may contain inhibitors of the electron transport chain (DHOD or cytochrome bc1) as do the three 4-quinolinol scaffolds (clusters 1122, 1613, and 2061). The compounds that were the precursors of the imidazolopiperazine compound in clinical trials, KAF156,28 were also found in a cluster of three compounds (GNF-Pf-5069, GNF-Pf-5179, GNF-Pf-5466; cluster 849). These compounds work by an unknown mechanism of action, but resistance is conferred by mutations in the P. falciparum cyclic amine resistance locus (Pfcarl).14 Additionally, compounds in cluster 2045 have some structural similarity to P. falciparum histone methyltransferase inhibitors.29
Figure 5.

Exoerythrocytic-stage active MMV compounds display unique chemical scaffold clustering. Compounds of the GNF and MMV Malaria Box were clustered by their substructure similarity, binning sets based on main common substructure (Tanimoto average compound similarity ≥0.85). Out of 2335 cluster sets, 15 were significantly enriched for exoerythrocytic-stage active compounds (depicted above). GNF Malaria Box compounds are shown as circle nodes, and MMV Malaria Box compounds as square nodes. Active compounds are indicated in red and blue for MMV and GNF compounds, respectively. Dark red/blue signifies IC50 < 1 μM, while light red/blue signifies IC50 < 10 μM. Inactive compounds are shown in black, and base scaffolds are shown in gray. It is important to note that of the 16 MMV compounds represented in the scaffold clustering, 13 of them were structurally identical to compounds also screened by the Novartis library using high-content imaging (TC = 1).
Although most of the over-represented scaffolds have been investigated as starting points of antimalarial drug discovery in recent years, there were also novel notable singleton molecules whose hepatic stage activity had not been previously described. For example, MMV666693 (the most potent compound from the exoerythrocytic-stage screen of the MMV Malaria Box) also strongly inhibits erythrocytic-stage P. falciparum with IC50 values reported below 100 nM (Table S1 and additional file 1). This compound was previously identified as an allosteric inhibitor of P. falciparum kinesin-5, a microtubule cross-linking enzyme required for cell division.4 It is therefore interesting to speculate that targeting this enzyme may be an efficient means to inhibit parasite replication across multiple developmental stages.
Compounds with Exclusive Activity against Exoerythrocytic Stages
Compounds that are active only against exoerythrocytic stages, but not erythrocytic stages, may represent new opportunities for development of drugs for which resistance acquisition may be less of a problem. While 42 of the 63 exoerythrocytic-stage DOS compounds were active in both the exoerythrocytic- and erythrocytic-stage assays, others could be starting points for such drugs. Several of the compounds that were identified in the DOS library unbiased screen are highlighted in Figure 4a. In particular, cyanoazetidine and a bicyclic azetidine series are shown. Although BRD7539 is active in both the exoerythrocytic- and asexual erythrocytic-stage assays, BRD9781 (its stereoisomer) is only active in the exoerythrocytic-stage assay. Although BRD7539 targets the Plasmodium DHODH enzyme,30 based on its profile across life cycle stages, we suspect that BRD9781 has a different target. These results suggest the important role of stereochemistry in inhibiting different biological targets. BRD47390 appears to result in specific activity in the exoerythrocytic-stage assay, whereas the DOS phenylalanine–tRNA ligase inhibitor is active against blood and exoerythrocytic stages. BRD0326, which has a stereochemistry identical to that of BRD7539 but different functionality at the azetidine nitrogen, also has no erythrocytic-stage activity and may have yet another target. Further testing in a DHODH assay31 confirmed that BRD0326 and BRD9781 are inactive (data not shown). BRD0326, BRD9781, and BRD47390 were all retested at dose along with all of their stereoisomers. With the exception of BRD7539, which targets DHODH, these compounds displayed stereoselective activity (Figure 4b). Overall, these data show that up to a third of the hits in a screen of an unbiased library might have exclusive exoerythrocytic activity, targeting either unique EEF parasite targets or the host factors needed to support parasite replication.
Cross-Validation Using Phenotypic Assays To Assess Time of Action during Exoerythrocytic-Stage Development
We sought to further confirm exoerythrocytic-stage activity for the three most potent MMV Malaria Box hits by an orthogonal and complementary assay. This assay utilizes a previously described flow-cytometry-based method32,33 to measure four specific metrics during exoerythrocytic-stage development: (1) sporozoite traversal, (2) sporozoite invasion, (3) EEF frequency, and (4) EEF development. The assay uses P. berghei expressing GFP (Pb-GFP) infection of Huh7.5.1 cells (Figure 6), a related exoerythrocytic cell line that can also be used to study EEF development (Figure S4), and measurements are taken at 2 and 48 h postinfection.
Figure 6.

Validation of exoerythrocytic-stage activity using an established flow-cytometry-based assay. (a) Flow cytometry plots measuring traversal and invasion (at 2 h postinfection) and EEF frequency and development (at 48 h postinfection) of exoerythrocytic-stage malaria parasites in Huh7.5.1 cells as previously described for three of the most potent compounds. Cytochalasin D was used as a positive control for traversal and invasion; KDU691 was used as a positive control for EEF frequency, and atovaquone was used as a positive control for EEF frequency and development. While traversal was measured by the percentage of rhodamine–dextran single-positive cells, invasion was measured by the percentage of Pb-GFP single-positive cells at 2 h postinfection. At 48 h postinfection, EEF frequency was measured by the percentage of Pb-GFP-positive cells, and EEF development was measured by the relative mean fluorescence intensity (MFI). Representative flow cytometry plots are shown. Atovaquone was tested at 1 μM (due to slight cytotoxicity at 10 μM in Huh7.5.1 cells, Figure S4), and cytochalasin D and KDU691 were tested at 10 μM. (b) Mean and SEM are shown graphically from the traversal/invasion, EEF frequency, and EEF size control experiments shown in panel (a) (cytochalasin D, atovaquone, and KDU691, respectively). Values are normalized to the DMSO control. (c) Exoerythrocytic-stage traversal, invasion, EEF frequency, and EEF development are shown for MMV666693, MMV007160, and MMV665916. Mean and SEM from three replicate experiments are shown. MMV66693 was tested at 1 μM (due to slight cytotoxicity at 10 μM in Huh7.5.1 cells, Figure S4), and MMV007160 and MMV665916 were tested at 10 μM.
At 2 h postinfection, the number of hepatocytes that have been traversed and invaded is measured. Sporozoite traversal is inferred based on the observation that traversal temporarily ruptures the plasma membrane of hepatocytes, allowing high molecular weight rhodamine–dextran to stain cells that have been traversed but not invaded.34 Sporozoite invasion is measured by the percentage of cells expressing GFP and not rhodamine–dextran, as the parasite enters the cells via a moving tight junction, excluding rhodamine–dextran.34 Double-positive cells, expressing both GFP and rhodamine–dextran, likely represent a population of nonproductively infected cells or cells in the process of being traversed by parasite and are therefore not used in the measurements of traversal and invasion.33 Cytochalasin D, a potent inhibitor of actin polymerization, is used as a positive control for traversal and invasion inhibition at 2 h postinfection as it has been previously demonstrated to reduce sporozoite motility (Figure 6a).33
At 48 h postinfection, the frequency and development (size) of exoerythrocytic-stage EEFs can be measured. The frequency of EEFs is determined by the percentage of cells expressing GFP at 48 h postinfection. KDU691, which inhibits Plasmodium phosphatidylinositol 4-kinase (PI(4)K), an enzyme that phosphorylates its phosphoinositide substrate to regulate intracellular signaling and trafficking,35 was used as a positive control because it was shown to significantly decrease the number of exoerythrocytic-stage EEFs (Figure 6a). At the same time, the assay measures EEF development by reporting GFP mean fluorescence intensity), an indicator of EEF size. Here, atovaquone serves as a positive control (Figure 6a).
As predicted, all compounds led to changes in cell populations that were detectable by flow cytometry (Figure 6b). At 48 h postinfection, all of the compounds tested led to a significant decrease in the EEF size, but not frequency, similar to that of atovaquone (Figure 6b). In addition, MMV666693 also appears to have a slight effect on sporozoite traversal of hepatocytes at 2 h postinfection (Figure 6b). Somewhat unexpectedly, sporozoite invasion at 2 h postinfection was not affected by MMV666693 but was even slightly increased. Unlike KDU691, none of these compounds affected EEF frequency. While further investigation is needed to better understand the specific mode of action, these results suggest that these compounds are acting primarily during EEF development. An important caveat of the flow-cytometry-based assay is that there may be prolonged fluorescence well after parasites have lost viability.36
To provide further validation of EEF development inhibition, MMV666693, MMV007160, and MMV665916 were characterized in an exoerythrocytic-stage time of action assay and compared to compounds with known activity in hepatic stages. This included atovaquone, which targets mitochondrial cytochrome bc1 complex and therefore inhibits the parasite’s electron transport chain during all developmental stages5 (Figure 7b). Pyrimethamine inhibits dihydrofolate reductase and thus the synthesis of purines and pyrimidines required for DNA synthesis.37 We also included several compounds in development such as DDD107498, which inhibits Plasmodium translation elongation factor 2 (eEF2),38 as well as KDU691.35 A third compound, GNF179, whose target has not been elucidated but which has potent causal prophylactic activity in mice and resistance mediated by mutations in the aforementioned Pfcarl gene, was also included.14 Compounds were added in 12-point serial dilutions (10 μM to 56 pM) or washed out to be present during specific time frames throughout exoerythrocytic-stage development. Their activity was measured using a modified 384-well version of our high-throughput luciferase assay, and bioluminescence was recorded at 48 h postinfection.
Figure 7.

Exoerythrocytic-stage active compounds display unique potencies during exoerythrocytic-stage EEF development. (a) Diagram illustrating the major stages of malaria parasite exoerythrocytic-stage development, including invasion, parasitophorous vacuole membrane (PVM) remodeling, trophozoite development, and EEF schizont development. During the first 4 h after sporozoite invasion, the parasite dramatically remodels its parasitophorous vacuole membrane by degrading host-cell-derived proteins and at the same time inserting its own parasite-derived proteins.6 During the next 18 h, the sporozoites transform from their elongated motile form to round, nonmotile, and metabolically active trophozoites. The trophozoites undergo impressive nuclear replication starting at around 24 h postinfection, displaying one of the fastest replication rates known to eukaryotic organisms to develop into mature EEFs.6 Drug treatments 1–6, corresponding to compound incubation during the exoerythrocytic developmental stages indicated, are shown. (b) Pb-Luc IC50 data for established antimalarial compounds (atovaquone and pyrimethamine), antimalarials in development (GNF179, KDU691, DDD107498), and the three MMV Malaria Box compounds (MMV666693, MMV007160, and MMV665916) added during Pb-Luc exoerythrocytic-stage development in a modified 384-well luciferase-based assay (discussed in Materials and Methods) are shown. Likewise, the Pb-Luc IC50 fold changes normalized to the 2–50 h drug-treated controls are shown and colored based on the indicated heat map.
The assay supported the flow cytometry data and showed that MMV666693 and MMV007160 inhibit parasite replication during all time points during exoerythrocytic-stage EEF development and seem to be most potent when added during trophozoite development 6–24 h postinfection (Figure 7b). Unlike the other compounds tested, MMV665916 may need longer incubation in vitro in order to achieve optimal activity against exoerythrocytic-stage parasites, as all of the developmental time points tested resulted in a more than 10-fold IC50 change compared to the 2–50 h postinfection control (Figure 7b). These data highlight how exoerythrocytic-active compounds may be further classified and how this assay may reveal information about a compound’s mechanism of action.
Conclusions
The assays described here provide a high-throughput approach to identify scaffolds or scaffold families that will have causal prophylactic activity. Although our assay depends on rodent malaria parasites, which are not infectious to humans, they have an advantage over human parasites because mosquitoes infected with P. berghei or P. yoelii can be handled and shipped more easily. In addition, the number of sporozoites per mosquito is high (∼20 000), enabling higher-throughput methods and evaluation of more starting points. This reduces cost (20 cents per well) as mosquito production and mosquito dissection are very labor-intensive. One concern is that activity tests using rodent malaria parasites might not translate into activity against human parasites. In cases where a compound is also active against P. falciparum erythrocytic stages, this is less likely to be a concern. In the small number of cases where compounds are not active in erythrocytic stages, additional testing using exoerythrocytic stages of P. falciparum or P. vivax may be warranted. Compounds of this class could be particularly interesting as leads for drugs that could be used in malaria elimination and eradication campaigns because they could be developed into drugs that could provide long-acting protection and which would not have the same resistance-development liabilities as compounds that act against the billions of erythrocytic-stage parasites that teem in an infected human. There is also the possibility that P. falciparum- or P. vivax-specific hits may be lost; however, this is likely a small number of compounds given the screening throughput capacity of the assay.
An unanswered question is whether leads identified with this assay will have radical cure activity. Primaquine and tafenoquine are the only two compounds that can provide radical cures, and both behave poorly in cellular assays such as those described here because they depend on host organismal metabolism. These compounds are not even particularly active in assays that involve primary hepatocytes.39 Likewise, we have recently tested a number of MMV Malaria Box screening hits in an ex vivo P. cynomolgi model of hypnozoite development;40 however, only one compound, MMV007224, had moderate activity (at 10 μM) against small or large forms in the assay (Table S2). Interestingly, this was also the only compound of the set tested with any pharmacokinetic exposure in vivo.41 This highlights the utility of using hepatoma cells rather than primary hepatocytes for screening purposes, as compounds with less than favorable pharmacokinetic properties can be evaluated for activity. As exoerythrocytic-stage active compounds identified in this report will also likely include inhibitors of Plasmodium exoerythrocytic-stage hypnozoites, it will be important to address compound metabolic stability in vitro and bioavailability in vivo during the development of novel hypnozoite models. This class of compounds will represent an important starting point for the development of novel treatments capable of providing a malaria radical cure.
Materials and Methods
Compound Libraries
MMV Validation Set of Antimalarials
This collection of 50 known antimalarial powders were obtained from the MMV and are all commercially available and active primarily against erythrocytic-stage malaria parasites.
MMV Malaria Box
This open source compound library comprises 400 diverse open source compounds with proven antimalarial activity. Two hundred of these compounds are described by MMV as “drug-like” and 200 as “probe-like” compounds.18 They have been distilled down from ∼20 000 hits generated from a screening campaign of 4 million compounds from the libraries of St. Jude’s Children’s Hospital, Novartis, and GSK. All compounds are commercially available, and the library is also available for free from MMV as long as the resulting data are published and placed in the public domain.
Broad Diversity-Oriented Synthesis Library
Two sets of compounds were tested from the DOS library of 100 000 compounds. The first set included 9886 compounds selected to represent the structural diversity of all of the scaffolds. This “informer” set was used as a naïve library to screen for compounds with unknown activity against Plasmodium. The second set comprised 89 compounds previously shown to be active in a P. falciparum erythrocytic-stage assay (IC50 < 2 μM, Nobutaka Kato, unpublished results).
Parasites
P. berghei-ANKA-GFP-Luc-SMCON (Pb-Luc)42 and P. berghei-GFP (Pb-GFP)35 sporozoites were obtained by dissection of infected Anopheles stephensi mosquito salivary glands. Dissected salivary glands were homogenized in a glass tissue grinder and filtered twice through nylon cell strainers (20 μm pore size, Millipore SCNY00020) and counted using a Neubauer hemocytometer. The sporozoites were kept on ice until needed. Both Pb-Luc- and Pb-GFP-infected A. stephensi mosquitoes were obtained from the Insectary Core Facility at New York University.
Cell Lines
HepG2-A16-CD81EGFP20 cells stably transformed to express a GFP-CD81 fusion protein were cultured at 37 °C and 5% CO2 in DMEM (Invitrogen, Carlsbad, USA) supplemented with 10% FCS, 0.29 mg/mL glutamine, 100 units of penicillin, and 100 μg/mL streptomycin. Huh7.5.1 cells were cultured at 37 °C and 5% CO2 in DMEM (Invitrogen, Carlsbad, USA) supplemented with 10% FCS (Corning cat# 35-011-CV), 200 U/mL penicillin, 200 μg/mL streptomycin (Invitrogen cat# 15140-122), 10 mM Hepes (Invitrogen cat# 15630-080), 1× Glutamax (Invitrogen cat# 35050-061), and 1× nonessential amino acids (Invitrogen). During infection, cell medium was supplemented with 50 μg/mL gentamycin and 50 μg/mL neomycin. After infection, the antimycotic 5-fluorocytosine at a final concentration of 50 μg/mL was added to the media.
High-Content Imaging
The high-content imaging experiments were performed as previously described.14 Briefly, we used the HepG2-A16-CD81EGFP host cells and either the P. yoelii or Pb-Luc rodent malaria parasites. We seeded the cells in 384-well plates and infected them with a ratio of 2:1 (cells/sporozoites). After the parasites were stained with a polyclonal mouse anti-PyHSP70 antibody, these data were acquired (image analysis) with the PerkinElmer Opera. Average parasite size per well served as the primary readout for compound effectiveness.
Luciferase-Based High-Throughput Screening
Sporozoite Infection
For the Pb-Luc high-throughput screen, we utilized P. berghei because its higher infection rates of immortal human hepatocyte cell lines are more conducive to high-throughput screening than the infection rates of human malaria parasites. P. berghei is able to infect human hepatocarcinoma HepG2 cells expressing the tetraspanin CD81 receptor.20 HepG2-A16-CD81EGFP cells (3 × 103) in 5 μL of medium (2 × 105 cells/mL, 5% FBS, 5× Pen/Strep/Glu) were seeded into 1536-well, white, solid-bottom plates (ref# 789173-F, Greiner Bio-One) 20–26 h prior to the actual infection. Eighteen hours prior to infection, 50 nL of compound in DMSO (0.5% final DMSO concentration per well) was transferred with a PinTool (GNF Systems) into the assay plates (10 μM final concentration). Atovaquone (12-point serial dilution starting at 10 μM) and 0.5% DMSO were used as positive and negative controls, respectively. Pb-Luc sporozoites were freshly dissected, and their concentration was adjusted to 200 sporozoites/μL. Penicillin and streptomycin were added at 5× concentration for a final 5× concentration in the well. The increased antibiotic concentration did not interfere with parasite or HepG2-A16-CD81EGFP growth. The HepG2-A16-CD81EGFP cells were then infected with 1 × 103 sporozoites per well (5 μL) using a single tip bottle valve liquid handler (GNF), and the plates were centrifuged for 3 min at room temperature and at 330g (Eppendorf 5810 R centrifuge) on lowest acceleration and brake setting. The plates were then incubated at 37 °C for 48 h in 5% CO2 with high humidity to minimize media evaporation and edge effect.
Bioluminescence Quantification of Exoerythrocytic Forms
After incubation, the parasite EEF growth was quantified by bioluminescence measurement. Medium was removed by spinning the inverted plates at 150g for 30 s. Two microliters of BrightGlo reagent (Promega) was dispensed with the MicroFlo liquid handler (BioTek). Immediately after addition of the luminescence reagent, the plates were vortexed at median intensity setting for 10 s and read by an EnVision Multilabel plate reader (PerkinElmer). IC50 values were obtained using measured bioluminescence intensity and a nonlinear variable slope four-parameter regression curve fitting model in Prism 6 (GraphPad Software Inc.).
Bioluminescence Quantification of HepG2 Cytotoxicity
After incubation, the HepG2 cytotoxicity was assessed by removing the media through an inverted spin of the plates at 150g for 30 s and addition of 2 μL of CellTiterGlo reagent (Promega diluted 1:2 with deionized water) per well using the MicroFlo liquid handler (BioTek). Immediately after addition of the luminescence reagent, the plates were vortexed for 10 s and read with an EnVision Multilabel reader (PerkinElmer).
Bioluminescence Quantification of Compound Luciferase Inhibition
After a 3 h incubation period, 2 μL of BrightGlo (Promega) was added to the wells with the MicroFlo liquid handler (BioTek). Immediately after addition, the plates were read by an EnVision Multilabel reader (PerkinElmer).
Bioinformatic Analysis of EEF Inhibition, HepG2 Cytotoxicity, and Luciferase Inhibition
For the first screening round, the luminescence reads from each 1536-well plate were analyzed separately. Briefly, a lack of inhibition was defined as the average DMSO readings (64 wells) minus the baseline inhibition readings. For both the first and second round of screening, the baseline for the EEF inhibition was defined as the average of the five highest atovaquone concentrations (10 wells); the baseline for the HepG2 cytotoxicity was defined as the average of the highest puromycin concentrations (8 wells), and the baseline of the luciferase inhibition assay was defined as the average of 48 wells with 500 μM resveratrol. For the first round of screening, we determined the inhibition percentage relative to the normalized well concentrations for each compound. This analysis was repeated for every 1536-well plate. For the second reconfirmation round of screening, IC50 values were obtained using the average normalized bioluminescence intensity of 4 wells per concentration and plate (96 wells in total for each compound) and a nonlinear variable slope four-parameter regression curve fitting model in Prism 6 (GraphPad Software Inc.).
Culturing Asexual Erythrocytic-Stage Parasites
P. falciparum parasites were cultured in complete medium containing 5% hematocrit in a low-oxygen atmosphere composed of 1% oxygen, 3% carbon dioxide, and 96% nitrogen at 37 °C. Complete medium consists of RPMI medium 1640 (with l-glutamine, without phenol red, Thermo Fisher Scientific) supplemented with 4.3% heat-inactivated O+ human serum, 0.2% AlbuMAX II lipid-rich BSA, 0.014 mg/mL hypoxanthine, 3.4 mM NaOH, 38.4 mM Hepes, 0.2% glucose, 0.2% sodium bicarbonate, and 0.05 mg/mL gentamicin.
Asexual Erythrocytic-Stage Screening in a 1536-Well Plate format
Pathogenic asexual erythrocytic-stage parasites were screened using a modified fluorescence-based proliferation assay described previously.10 Briefly, P. falciparum 3D7 parasites were cultured until a parasitemia of 3–6% was reached. The level of erythrocytic-stage parasitemia was determined by microscopic inspection of Giemsa-stained blood smears for the presence of parasites. A parasite suspension with 0.3% parasitemia and 4% hematocrit was prepared in screening medium consisting of RPMI medium 1640 (with l-glutamine, without phenol red) supplemented with 0.4% AlbuMAX II lipid-rich BSA, 0.014 mg/mL hypoxanthine, 3.4 mM NaOH, 38.4 mM Hepes, 0.2% glucose, 0.2% sodium bicarbonate, and 0.05 mg/mL gentamicin. The parasite culture was gassed with 1% oxygen, 3% carbon dioxide, and 96% nitrogen and stored at 37 °C until used. Next, 3 μL of screening medium was dispensed into 1536-well, black, clear-bottom plates (ref# 789092-F, Greiner Bio-One, Kremsmünster, Austria) using the MultiFlo microplate dispenser (BioTek Instruments, VT, USA). For determining the IC50 values, 50 nL of compounds dissolved in DMSO (12-point serial dilutions (1:3) starting at 10 mM) was transferred into the assay plates (62.5 μM final drug concentration, 0.625% final DMSO concentration) using the Biomek FXP Laboratory Automation Workstation (Beckman Coulter, CA, USA) with a PinTool (V&P Scientific, CA, USA). Artemisinin and DMSO were included as background and baseline controls, respectively. Next, 5 μL of prepared parasite suspension was dispensed into the 1536-well plates, resulting in a final parasitemia of 0.3% and a final hematocrit concentration of 2.5% (MultiFlo microplate dispenser). The assay plates were transferred into Ziploc bags and gassed with a gas mixture of 1% oxygen, 3% carbon dioxide, and 96% nitrogen. After a 72 h incubation at 37 °C, 2 μL of detection solution consisting of 10× SYBR Green I (Thermo Fisher Scientific) in lysis buffer (20 mMTris/HCl, 5 mM EDTA, 0.16% saponin, 1.6% Triton X-100) was added to the plates (MultiFlo microplate dispenser) and incubated for 24 h at room temperature in the dark. After 24 h, fluorescence signals were measured at 530 nm with a 485 nm excitation from the bottom using the 2104 EnVision Multilabel reader (PerkinElmer, MA, USA). After the signal of the highest concentration of artemisinin (background) was subtracted from all output values and the values to the average DMSO signal normalized, the IC50 values were calculated by using a nonlinear variable slope regression curve-fitting model in GraphPad Prism (GraphPad Software Inc.).
Computational Compound Clustering
To evaluate clustering of exoerythrocytic-stage hits and enrichment of compound groups, the 400 compound MMV set was coclustered with a set of >4000 compounds that had previously been evaluated in a P. yoelii high-content imaging assay.14 Briefly, SMILESL (simplified molecular-input line-entry system) strings were loaded into R, and the maximum common substructure Tanimoto coefficient (MCS-TC) was calculated using the fmcsR package.43 The compounds were then subsequently hierarchically clustered with the hclust package, using the ward.D2 agglomeration method pair clusters. To create compound bins, the tree branches were separated at a maximum pairwise distance of 0.4. Hypergeometric mean statistical tests were applied to each compound bin to identify sets where exoerythrocytic-stage activity was enriched.
Flow Cytometry Assay
Exoerythrocytic-stage Pb-GFP traversal, invasion, and schizont development were measured using a previously established flow-cytometry-based method.33 Briefly, 24 h prior to infection, 1.75 × 105 Huh7.5.1 cells were seeded in 24-well plates in 1 mL of DMEM hepatocyte culture medium for the traversal and invasion assay, as well as for the quantitation of EEF size and frequency. Pb-GFP sporozoites were freshly isolated from infected Anopheles stephensi mosquitoes as described above, and 3.5 × 104 or 7.0 × 104 sporozoites were added to the cells for the traversal/invasion assay or EEF quantitation assay, respectively, and incubated for 2 h. Rhodamine–dextran was added to the wells at a final concentration of 1 mg/mL for the traversal and invasion assay. The cells were washed after the 2 h infection and assayed using flow cytometry for rhodamine–dextran and GFP signal (traversal and invasion, respectively) or incubated for 48 h and assayed by flow cytometry for GFP frequency and MFI (EEF frequency and size, respectively). Data were analyzed using the FlowJo software.
Pb-Luc Time of Action Assay
For the Pb-Luc time-course assay, we seeded 1 × 104 Huh7.5.1 cells in 30 μL of hepatocyte culture media per well in a 384-well plate (Greiner Bio) 24 h before infection. Pb-Luc sporozoites were freshly dissected from infected Anopheles stephensi mosquitoes, and 5 × 103 sporozoites in 30 μL were added to each well. The plates were centrifuged for 5 min at 330g and incubated for 2 h at 37 °C and 5% CO2. After incubation, medium was removed and 50 μL of fresh culture media was added. The 12-point serial dilutions of compound in DMSO were added and removed from the plates at the indicated time points postinfection. At 48 h postinfection, medium was removed from the plates and 20 μL of BrightGlo reagent (Promega) was added to each well. Luciferase light units were measured by bioluminescence using an EnVision Multilabel plate reader (PerkinElmer).
P. cynomolgi Liver Assay
The P. cynomolgi assay was performed as previously reported by Zeeman et al.40 All Rhesus macaques (Macaca mulatta) used in this study were bred in captivity for research purposes and were housed at the Biomedical Primate Research Centre (BPRC; AAALAC-certified institute) facilities under compliance with the Dutch law on animal experiments, European directive 2010/63/EU, and with the “Standard for Humane Care and Use of Laboratory Animals by Foreign institutions” identification number A5539-01, provided by the Department of Health and Human Services of the U.S. National Institutes of Health. The local independent ethical committee first approved all protocols.
Acknowledgments
We thank our colleagues from The Genomics Institute of the Novartis Research Foundation, in particular, Richard Glynne, Purvi Sanghvi, Annie Mak, and Jason Matzen. They provided insight, expertise, and assistance that greatly assisted the research. The mosquitoes were supplied by the Insectary Core Facility at New York University School of Medicine (http://microbiology-parasitology.med.nyu.edu/research/parasitology/insectary-core-facility-and-parasite-culture). Infection of mosquitoes was performed by bite of mice infected with the same parasite in blood stage. This procedure was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of New York University School of Medicine (Protocol Number 150816), which is fully accredited by the Association For Assessment and Accreditation Of Laboratory Animal Care International (AAALAC). The authors acknowledge the Medicines for Malaria Venture (MMV) for access to the MMV Malaria Box. S.M., J.S., C.R., and Y.A. were supported by MMV. V.C. was supported by the Bill and Melinda Gates Foundation (OPP104040). E.A.W. was funded by the Bill and Melinda Gates Foundation (OPP1054480), MMV, and NIH (R01AI090141 and R01AI103058). M.M. was supported by the National Science Foundation (NSF) GRPF (DGE1144152), and N.K., E.C., C.S., and S.L.S. were supported by the Bill and Melinda Gates Foundation (OPP1032518). S.L.S. is an investigator at the Howard Hughes Medical Institute. C.M., K.G., and M.I. were funded by the Welcome Trust (WT078285). A.M.Z. and C.H.M.K. were funded by the Wellcome Trust (WT078285) and received additional funding from MMV. The authors would like to acknowledge Omar Vandal for assistance with grant management, and Paula Maguina for administrative assistance.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.5b00143.
Author Present Address
¶ California Institute for Biomedical Research (Calibr), San Diego, CA 92037, USA.
Author Present Address
# Samumed, San Diego, CA 92121, USA.
Author Contributions
E.A.W. wrote the manuscript and analyzed data. S.M. performed assays, analyzed data, and wrote the manuscript. J.S. wrote the manuscript, analyzed data, performed flow cytometry and time of action assays. V.C. analyzed data and created figures. C.A.S. wrote the manuscript, created figures, and provided compounds. N.K. performed erythrocytic-stage screens. E.C. and M.M. analyzed data. C.W.M. provided advice. D.P. performed assays and optimization. C.R. performed assays and wrote the manuscript. Y.A.K., S.K., K.H., and M.I. performed assays. S.L.S. wrote the manuscript. A.M.Z. and C.H.M.K. performed P. cynomolgi assays. B.C. provided advice and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Carter R.; Mendis K. N. (2002) Evolutionary and historical aspects of the burden of malaria. Clin. Microbiol. Rev. 15, 564–594. 10.1128/CMR.15.4.564-594.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow R. W.; Guerra C. A.; Noor A. M.; Myint H. Y.; Hay S. I. (2005) The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 434, 214–217. 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs J.; Malaney P. (2002) The economic and social burden of malaria. Nature 415, 680–685. 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- Sinka M. E.; Bangs M. J.; Manguin S.; Rubio-Palis Y.; Chareonviriyaphap T.; Coetzee M.; Mbogo C. M.; Hemingway J.; Patil A. P.; Temperley W. H.; Gething P. W.; Kabaria C. W.; Burkot T. R.; Harbach R. E.; Hay S. I. (2012) A global map of dominant malaria vectors. Parasites Vectors 5, 69. 10.1186/1756-3305-5-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery E. L.; Chatterjee A. K.; Winzeler E. A. (2013) Antimalarial drug discovery - approaches and progress towards new medicines. Nat. Rev. Microbiol. 11, 849–862. 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M.; Rodriguez A.; Mota M. M. (2006) The silent path to thousands of merozoites: the Plasmodium liver stage. Nat. Rev. Microbiol. 4, 849–856. 10.1038/nrmicro1529. [DOI] [PubMed] [Google Scholar]
- Wells T. N.; Burrows J. N.; Baird J. K. (2010) Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol. 26, 145–151. 10.1016/j.pt.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Clark I. A.; al Yaman F. M.; Jacobson L. S. (1997) Int. J. Parasitol. 27, 1237–1249. 10.1016/S0020-7519(97)00121-5. [DOI] [PubMed] [Google Scholar]
- Baker D. A. (2010) Malaria gametocytogenesis. Mol. Biochem. Parasitol. 172, 57–65. 10.1016/j.molbiopara.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delves M.; Plouffe D.; Scheurer C.; Meister S.; Wittlin S.; Winzeler E. A.; Sinden R. E.; Leroy D. (2012) The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS medicine 9, e1001169. 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan K. R.; Magill A. J.; Parise M. E.; Arguin P. M. (2011) Doxycycline for malaria chemoprophylaxis and treatment: report from the CDC expert meeting on malaria chemoprophylaxis. Am. J. Trop. Med. Hyg. 84, 517–531. 10.4269/ajtmh.2011.10-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde J. E. (2002) Mechanisms of resistance of Plasmodium falciparum to antimalarial drugs. Microbes Infect. 4, 165–174. 10.1016/S1286-4579(01)01524-6. [DOI] [PubMed] [Google Scholar]
- Ganesan S.; Chaurasiya N. D.; Sahu R.; Walker L. A.; Tekwani B. L. (2012) Understanding the mechanisms for metabolism-linked hemolytic toxicity of primaquine against glucose 6-phosphate dehydrogenase deficient human erythrocytes: evaluation of eryptotic pathway. Toxicology 294, 54–60. 10.1016/j.tox.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Meister S.; Plouffe D. M.; Kuhen K. L.; Bonamy G. M.; Wu T.; Barnes S. W.; Bopp S. E.; Borboa R.; Bright A. T.; Che J.; Cohen S.; Dharia N. V.; Gagaring K.; Gettayacamin M.; Gordon P.; Groessl T.; Kato N.; Lee M. C.; McNamara C. W.; Fidock D. A.; Nagle A.; Nam T. G.; Richmond W.; Roland J.; Rottmann M.; Zhou B.; Froissard P.; Glynne R. J.; Mazier D.; Sattabongkot J.; Schultz P. G.; Tuntland T.; Walker J. R.; Zhou Y.; Chatterjee A.; Diagana T. T.; Winzeler E. A. (2011) Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 334, 1372–1377. 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire E. R.; Mota M. M.; Clardy J. (2011) The next opportunity in anti-malaria drug discovery: the liver stage. PLoS Pathog. 7, e1002178. 10.1371/journal.ppat.1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reader J.; Botha M.; Theron A.; Lauterbach S. B.; Rossouw C.; Engelbrecht D.; Wepener M.; Smit A.; Leroy D.; Mancama D.; Coetzer T. L.; Birkholtz L. M. (2015) Nowhere to hide: interrogating different metabolic parameters of Plasmodium falciparum gametocytes in a transmission blocking drug discovery pipeline towards malaria elimination. Malar. J. 14, 213. 10.1186/s12936-015-0718-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire E. R.; Prudencio M.; Mota M. M.; Clardy J. (2012) Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc. Natl. Acad. Sci. U. S. A. 109, 8511–8516. 10.1073/pnas.1118370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg T.; Burrows J. N.; Kowalczyk P.; McDonald S.; Wells T. N.; Willis P. (2013) The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS One 8, e62906. 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse C. J.; Franke-Fayard B.; Mair G. R.; Ramesar J.; Thiel C.; Engelmann S.; Matuschewski K.; van Gemert G. J.; Sauerwein R. W.; Waters A. P. (2006) High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol. Biochem. Parasitol. 145, 60–70. 10.1016/j.molbiopara.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Silvie O.; Greco C.; Franetich J. F.; Dubart-Kupperschmitt A.; Hannoun L.; van Gemert G. J.; Sauerwein R. W.; Levy S.; Boucheix C.; Rubinstein E.; Mazier D. (2006) Expression of human CD81 differently affects host cell susceptibility to malaria sporozoites depending on the Plasmodium species. Cell. Microbiol. 8, 1134–1146. 10.1111/j.1462-5822.2006.00697.x. [DOI] [PubMed] [Google Scholar]
- Ignowski J. M.; Schaffer D. V. (2004) Kinetic analysis and modeling of firefly luciferase as a quantitative reporter gene in live mammalian cells. Biotechnol. Bioeng. 86, 827–834. 10.1002/bit.20059. [DOI] [PubMed] [Google Scholar]
- Dandapani S.; Marcaurelle L. A. (2010) Grand challenge commentary: Accessing new chemical space for ’undruggable’ targets. Nat. Chem. Biol. 6, 861–863. 10.1038/nchembio.479. [DOI] [PubMed] [Google Scholar]
- Nielsen T. E.; Schreiber S. L. (2008) Towards the optimal screening collection: a synthesis strategy. Angew. Chem., Int. Ed. 47, 48–56. 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D.; Brinker A.; McNamara C.; Henson K.; Kato N.; Kuhen K.; Nagle A.; Adrian F.; Matzen J. T.; Anderson P.; Nam T. G.; Gray N. S.; Chatterjee A.; Janes J.; Yan S. F.; Trager R.; Caldwell J. S.; Schultz P. G.; Zhou Y.; Winzeler E. A. (2008) In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. U. S. A. 105, 9059–9064. 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. K.; Urgaonkar S.; Cortese J. F.; Gamo F. J.; Garcia-Bustos J. F.; Lafuente M. J.; Patel V.; Ross L.; Coleman B. I.; Derbyshire E. R.; Clish C. B.; Serrano A. E.; Cromwell M.; Barker R. H. Jr.; Dvorin J. D.; Duraisingh M. T.; Wirth D. F.; Clardy J.; Mazitschek R. (2011) Identification and validation of tetracyclic benzothiazepines as Plasmodium falciparum cytochrome bc1 inhibitors. Chem. Biol. 18, 1602–1610. 10.1016/j.chembiol.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capper M. J.; O’Neill P. M.; Fisher N.; Strange R. W.; Moss D.; Ward S. A.; Berry N. G.; Lawrenson A. S.; Hasnain S. S.; Biagini G. A.; Antonyuk S. V. (2015) Antimalarial 4(1H)-pyridones bind to the Qi site of cytochrome bc1. Proc. Natl. Acad. Sci. U. S. A. 112, 755–760. 10.1073/pnas.1416611112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen A.; LaCrue A. N.; White K. L.; Forquer I. P.; Cross R. M.; Marfurt J.; Mather M. W.; Delves M. J.; Shackleford D. M.; Saenz F. E.; Morrisey J. M.; Steuten J.; Mutka T.; Li Y.; Wirjanata G.; Ryan E.; Duffy S.; Kelly J. X.; Sebayang B. F.; Zeeman A. M.; Noviyanti R.; Sinden R. E.; Kocken C. H.; Price R. N.; Avery V. M.; Angulo-Barturen I.; Jimenez-Diaz M. B.; Ferrer S.; Herreros E.; Sanz L. M.; Gamo F. J.; Bathurst I.; Burrows J. N.; Siegl P.; Guy R. K.; Winter R. W.; Vaidya A. B.; Charman S. A.; Kyle D. E.; Manetsch R.; Riscoe M. K. (2013) Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl. Med. 5, 177ra37. 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhen K. L.; Chatterjee A. K.; Rottmann M.; Gagaring K.; Borboa R.; Buenviaje J.; Chen Z.; Francek C.; Wu T.; Nagle A.; Barnes S. W.; Plouffe D.; Lee M. C.; Fidock D. A.; Graumans W.; van de Vegte-Bolmer M.; van Gemert G. J.; Wirjanata G.; Sebayang B.; Marfurt J.; Russell B.; Suwanarusk R.; Price R. N.; Nosten F.; Tungtaeng A.; Gettayacamin M.; Sattabongkot J.; Taylor J.; Walker J. R.; Tully D.; Patra K. P.; Flannery E. L.; Vinetz J. M.; Renia L.; Sauerwein R. W.; Winzeler E. A.; Glynne R. J.; Diagana T. T. (2014) KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob. Agents Chemother. 58, 5060–5067. 10.1128/AAC.02727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmquist N. A.; Moss T. A.; Mecheri S.; Scherf A.; Fuchter M. J. (2012) Small-molecule histone methyltransferase inhibitors display rapid antimalarial activity against all blood stage forms in Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 109, 16708–16713. 10.1073/pnas.1205414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N.; Sakata-Kato T.; Maetani M.; Bastien J.; Corey V.; Derbyshire E. R.; Dornan G.; Duffy S.; Eckley S.; Karin M. J.; Koolen T. A. L.; Lukens A. K.; Lund E.; Riera S.; Bennett C.; Meier J. M.; Branko M.; Moss E. L.; Sayes M.; VanGessel Y.; Wawer M. J.; Yoshinaga T.; Zeeman A.-M.; Avery V. M.; Bhatia S. N.; Burke J. E.; Catteruccia F.; Clardy J. C.; Clemons P. A.; Dechering K. J.; Duvall J. R.; Foley M. A., Gusovsky F.; Kocken C. H. M.; Morningstar M. L.; Munoz B.; Neafsey D. E.; Winzeler E. A.; Wirth D. F.; Scherer C. A.; Schreiber S. L.. Diversity synthesis yields multistage antimalarial inhibitors including of a novel target that results in low single-dose cures in mice. Manuscript in review.
- Ross L. S.; Gamo F. J.; Lafuente-Monasterio M. J.; Singh O. M.; Rowland P.; Wiegand R. C.; Wirth D. F. (2014) In vitro resistance selections for Plasmodium falciparum dihydroorotate dehydrogenase inhibitors give mutants with multiple point mutations in the drug-binding site and altered growth. J. Biol. Chem. 289, 17980–17995. 10.1074/jbc.M114.558353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M.; Rodrigues C. D.; Ataide R.; Mota M. M. (2008) Dissecting in vitro host cell infection by Plasmodium sporozoites using flow cytometry. Cell. Microbiol. 10, 218–224. 10.1111/j.1462-5822.2007.01032.x. [DOI] [PubMed] [Google Scholar]
- Sinnis P.; De La Vega P.; Coppi A.; Krzych U.; Mota M. M. (2012) Quantification of sporozoite invasion, migration, and development by microscopy and flow cytometry. Methods Mol. Biol. 923, 385–400. 10.1007/978-1-62703-026-7_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota M. M.; Pradel G.; Vanderberg J. P.; Hafalla J. C.; Frevert U.; Nussenzweig R. S.; Nussenzweig V.; Rodriguez A. (2001) Migration of Plasmodium sporozoites through cells before infection. Science 291, 141–144. 10.1126/science.291.5501.141. [DOI] [PubMed] [Google Scholar]
- McNamara C. W.; Lee M. C.; Lim C. S.; Lim S. H.; Roland J.; Nagle A.; Simon O.; Yeung B. K.; Chatterjee A. K.; McCormack S. L.; Manary M. J.; Zeeman A. M.; Dechering K. J.; Kumar T. R.; Henrich P. P.; Gagaring K.; Ibanez M.; Kato N.; Kuhen K. L.; Fischli C.; Rottmann M.; Plouffe D. M.; Bursulaya B.; Meister S.; Rameh L.; Trappe J.; Haasen D.; Timmerman M.; Sauerwein R. W.; Suwanarusk R.; Russell B.; Renia L.; Nosten F.; Tully D. C.; Kocken C. H.; Glynne R. J.; Bodenreider C.; Fidock D. A.; Diagana T. T.; Winzeler E. A. (2013) Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504, 248–253. 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corish P.; Tyler-Smith C. (1999) Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng., Des. Sel. 12, 1035–1040. 10.1093/protein/12.12.1035. [DOI] [PubMed] [Google Scholar]
- Peterson D. S.; Walliker D.; Wellems T. E. (1988) Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc. Natl. Acad. Sci. U. S. A. 85, 9114–9118. 10.1073/pnas.85.23.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baragana B.; Hallyburton I.; Lee M. C.; Norcross N. R.; Grimaldi R.; Otto T. D.; Proto W. R.; Blagborough A. M.; Meister S.; Wirjanata G.; Ruecker A.; Upton L. M.; Abraham T. S.; Almeida M. J.; Pradhan A.; Porzelle A.; Martinez M. S.; Bolscher J. M.; Woodland A.; Norval S.; Zuccotto F.; Thomas J.; Simeons F.; Stojanovski L.; Osuna-Cabello M.; Brock P. M.; Churcher T. S.; Sala K. A.; Zakutansky S. E.; Jimenez-Diaz M. B.; Sanz L. M.; Riley J.; Basak R.; Campbell M.; Avery V. M.; Sauerwein R. W.; Dechering K. J.; Noviyanti R.; Campo B.; Frearson J. A.; Angulo-Barturen I.; Ferrer-Bazaga S.; Gamo F. J.; Wyatt P. G.; Leroy D.; Siegl P.; Delves M. J.; Kyle D. E.; Wittlin S.; Marfurt J.; Price R. N.; Sinden R. E.; Winzeler E. A.; Charman S. A.; Bebrevska L.; Gray D. W.; Campbell S.; Fairlamb A. H.; Willis P. A.; Rayner J. C.; Fidock D. A.; Read K. D.; Gilbert I. H. (2015) A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 522, 315–320. 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembele L.; Gego A.; Zeeman A. M.; Franetich J. F.; Silvie O.; Rametti A.; Le Grand R.; Dereuddre-Bosquet N.; Sauerwein R.; van Gemert G. J.; Vaillant J. C.; Thomas A. W.; Snounou G.; Kocken C. H.; Mazier D. (2011) Towards an in vitro model of Plasmodium hypnozoites suitable for drug discovery. PLoS One 6, e18162. 10.1371/journal.pone.0018162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeman A. M.; van Amsterdam S. M.; McNamara C. W.; Voorberg-van der Wel A.; Klooster E. J.; van den Berg A.; Remarque E. J.; Plouffe D. M.; van Gemert G. J.; Luty A.; Sauerwein R.; Gagaring K.; Borboa R.; Chen Z.; Kuhen K.; Glynne R. J.; Chatterjee A. K.; Nagle A.; Roland J.; Winzeler E. A.; Leroy D.; Campo B.; Diagana T. T.; Yeung B. K.; Thomas A. W.; Kocken C. H. (2014) KAI407, a potent non-8-aminoquinoline compound that kills Plasmodium cynomolgi early dormant liver stage parasites in vitro. Antimicrob. Agents Chemother. 58, 1586–1595. 10.1128/AAC.01927-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Voorhis W. C.; Roberto Adelfio J. H. A.; Ahyong V.; Akabas M. H.; Alano P.; Alday A.; Resto Y. A.; Alsibaee A.; Alzualde A.; Andrews K. T.; Avery S. V.; Avery V. M.; Ayong L.; Baker M.; Baker S.; Mamoum C. B.; Bhatia S.; Bickle Q.; Bounaadja L.; Bowling T.; Bosch J.; Boucher L. E.; Boyom F. F.; Brea J.; Brennan M.; Burton A.; Caffrey C. R.; Camarda G.; Carrasquilla M.; Carter D.; Cassera M. B.; Cheng K. C.-C.; Chindaudomsate W.; Chubb A.; Colon B. L.; Colón-López D. C.; Corbett Y.; Crowther G. J.; Cowan N.; D’Alessandro S.; Dang N. L.; Delves M.; DeRisi J. L.; Du A. Y.; Duffy S.; Abd El-Salam El-Sayed S.; Ferdig M. T.; Fernández Robledo J. A.; Fidock D. A.; Florent I.; Patrick V. T.; Galstian A.; Javier Gamo F.; Gokool S.; Gold B.; Golub T.; Goldgof G. M.; Guha R.; Guiguemde W. A.; Gural N.; Kiplin G. R.; Hansen M.; Hanson K. K.; Hemphill A.; van Huijsduijnen R. H.; Horii T.; Horrocks P.; Hughes T. B.; Huston C.; Igarashi I.; Ingram-Sieber K.; Itoe M. A.; Jadhav A.; Jensen A. N.; Jensen L. T.; Jiang R. H. Y.; Kaiser A.; Keiser J.; Ketas T.; Kicka S.; Kim S.; Kirk K.; Kumar V.; Kyle D. E.; Lafuente M. J.; Landfear S.; Lee N.; Lee S.; Lehane A.; Li F.; Little D.; Liu L.; Llinás M.; Loza M. I.; Lubar A.; Lucantoni L.; Lucet I.; Maes L.; Mansour N. R.; March S.; McGowan S.; Vera I. M.; Meister S.; Mercer L.; Mestres J.; Mfopa A. N.; Misra R. N.; Moon A. B.; Moore J. P.; Müller J.; Muriana A.; Nakazawa S.; Hewitt M. S.; Nare B.; Nathan C.; Narraidoo N.; Nawaratna S.; Ojo K. K.; Ortiz D.; Panic G.; Papadatos G.; Parapini S.; Patra K.; Pham N.; Prats S.; Plouffe D. M.; Poulsen S.-A.; Pradhan A.; Quevedo C.; Quinn R. J.; Rice C. A.; Rizk M. A.; Ruecker A.; St.Onge R.; Samra J.; Natalie G.; Schlecht U.; Schmitt M.; Sinden R.; Silvestrini F.; Smith D. A.; Soldati T.; Spitzmüller A.; Stamm S. M.; Sullivan D. J.; Sullivan W.; Suresh S.; Suzuki B. M.; Suzuki Y.; Swamidass S. J.; Taramelli D.; Tchokouaha L. R. Y.; Thomas D.; Tonissen K. F.; Townson S.; Tripathi A. K.; Trofimov V.; Udenze K. O.; Ullah I.; Vallieres C.; Vigil E.; Vinetz J. M.; Vinh P. V.; Vu H.; Watanabe N.-a.; Weatherby K.; White P. M.; Winzeler E. A.; Wojcik E.; Wree M.; Wu W.; Yokoyama N.; Zollo P. H. A.; Abla N.; Blasco B.; Burrows J.; Laleu B.; Leroy D.; Spangenberg T.; Wells T.; Willis P.. Open-source drug discovery with the Malaria Box compound collection for neglected diseases and beyond. Manuscript in revew. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz F. P.; Martin C.; Buchholz K.; Lafuente-Monasterio M. J.; Rodrigues T.; Sonnichsen B.; Moreira R.; Gamo F. J.; Marti M.; Mota M. M.; Hannus M.; Prudencio M. (2012) Drug screen targeted at Plasmodium liver stages identifies a potent multistage antimalarial drug. J. Infect. Dis. 205, 1278–1286. 10.1093/infdis/jis184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava I. K.; Morrisey J. M.; Darrouzet E.; Daldal F.; Vaidya A. B. (1999) Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 33, 704–711. 10.1046/j.1365-2958.1999.01515.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.