

Abstract
Causes of the excess incidence rates of chronic kidney disease in the African American population have long been under study. Recently, polymorphisms in the nonmuscle myosin heavy chain 9 gene (MYH9) have been associated with nondiabetic kidney diseases in African- and European-derived populations. Risk variants in MYH9 contribute to approximately 70% of nondiabetic forms of ESRD in African Americans and 40 to 45% of all ESRD in this ethnic group, with lesser effects in European Americans. It is clear that MYH9 polymorphisms have a significant impact on the incidence rates of kidney disease in African Americans. This article describes the current spectrum of biopsy-proven MYH9-associated kidney diseases, along with potential effects of MYH9 on ethnic differences in clinical outcome. MYH9 risk variants exhibit the most impressive association with any common complex kidney disease yet identified.
The search for genes underlying susceptibility to nondiabetic, potentially hypertension-associated forms of ESRD arose in part from the controversy over whether mild to moderate essential hypertension commonly initiated chronic kidney disease (CKD) (1,2). The epidemiology of ESRD in the US Renal Data System database may not be fully accurate (3). Hypertension-associated ESRD reportedly accounts for >34% of incident African American dialysis cases and nearly 25% of European American cases (4). African Americans clearly develop nondiabetic forms of ESRD more often than do European Americans (4); however, relatively few patients with mild to moderate essential hypertension ultimately develop CKD (5,6), and the disorder labeled hypertensive ESRD aggregates in select African American families. These factors suggested an inherited basis for hypertensive nephrosclerosis (HN).
Complicating the issue of whether high BP initiates nephropathy, it was reported that physician bias led to a diagnosis of HN far more often in African Americans (7), individuals with a diagnosis of HN were typically seen by nephrologists late in their course when it was difficult to identify initiators of kidney disease (8), and renal biopsy studies in African American patients did not support that the vascular change “arteriolar nephrosclerosis” correlated with systemic BP (9,10). An important clue that the disease process labeled HN differs between European Americans and African Americans arose from kidney biopsy studies. Those studies revealed different renal lesions in African American and European Americans who were given the same clinical diagnosis “HN” (11). African American patients typically had focal global glomerulosclerosis (FGGS) with marked interstitial fibrosis, whereas European American patients demonstrated intimal medial thickening of small intrarenal arterioles with resultant reduced renal perfusion and ischemic glomerular collapse. European American patients with HN often demonstrate stabilization of kidney function and albuminuria with successful treatment of high BP, hyperlipidemia, and smoking cessation (12–15). Treatment of these cardiovascular risk factors is expected to improve renal microvascular disease, the process seen in European Americans with HN. In contrast, the primary site of renal injury in African American patients labeled with HN does not seem to be the microvasculature. The African American Study of Kidney Disease and Hypertension (AASK) demonstrated that strict BP control, including the use of angiotensin-converting enzyme inhibitors, failed to halt progression of the kidney disease that had been attributed to hypertension (16).
These phenomena, coupled with familial clustering of HN in African Americans, led to a search for nondiabetic nephropathy susceptibility genes. Other potential explanations for the higher frequency of severe kidney disease in African Americans include lower socioeconomic status, lack of access to adequate health care, and more severe hypertension (17). The recently detected major contribution of risk variants in the nonmuscle myosin heavy chain 9 gene (MYH9) to ESRD susceptibility in African Americans clearly demonstrates that inherited factors make major contributions to the aforementioned ethnic disparities (18 –20).
MYH9 and Susceptibility to CKD
Familial aggregation of the common forms of CKD, including diabetic nephropathy and purported HN, had been recognized for 20 years (21–27). Thirty to 40% of individuals with type 1 and type 2 diabetes are at risk for the development of nephropathy. As commonly seen in HN, diabetic nephropathy clusters in select families. This observation shifted our understanding of CKD risk factors. Instead of the prevailing concept that patients with the most severely elevated BP and blood sugar levels were more likely to develop nephropathy, it become apparent that CKD susceptibility from systemic disorders could have an inherited basis.
Familial aggregation of ESRD is strongest in the African American population and has been observed with additional forms of CKD, including FSGS, HIV-associated nephropathy (HIVAN), and systemic lupus erythematosus. (28,29) The observations that familial clustering was independent of socioeconomic factors (28,30) and that multiple causes of CKD often clustered in African American families suggested that an overarching “renal failure susceptibility gene” was present (31).
Results of linkage analyses and candidate gene association studies in nondiabetic ESRD have been reported in African Americans; however, it was not until the application of admixture mapping or mapping by admixture linkage disequilibrium that MYH9 was identified. Mapping by admixture linkage disequilibrium is a useful method to detect gene variants that contribute to diseases in admixed populations in which the ancestral populations have markedly different disease frequencies (32). African Americans are known to have a fourfold increase in risk for all cause ESRD, relative to European Americans (4). Diseases such as HIVAN occur up to 70 times more often in the African American population. Assuming that risk variants in kidney failure genes were derived from Africa, it was possible to compare the frequencies of approximately 1500 ancestry-informative markers spaced evenly across the genome in African Americans with nondiabetic renal diseases (FSGS and HIVAN) with those in African Americans with normal kidney function (18,19). Admixture mapping analyses demonstrated a 10% excess frequency of African ancestry on chromosome 22 with a major peak overlying the MYH9 gene. Follow-up genotyping identified a four–single-nucleotide polymorphism (SNP) haplotype (G/C/C/T; rs4821480, rs2032487, rs4821481, and rs3752462) in MYH9 that was strongly associated with FSGS, HIVAN, and the kidney disease historically attributed to HN in African Americans (Figure 1) (18). The risk haplotype that was referred to in the article by Kopp et al. (18) as E-1 (for Extended-1) has a frequency of approximately 60% in African Americans and 4% in European Americans with odds ratios (ORs) for association with FSGS of 5.0 and 7.7, respectively. These effect sizes are extremely large for common, complex human diseases.
Figure 1.

MYH9 structure and location of the E-1 haplotype SNPs. The first 40 kb of MYH9, with exons in boxes and the promoter regions in gray, is shown. The location of the four SNPs that make up the E-1 haplotype are indicated. Gene structure was developed using Haploview (52) with infotrack downloaded from http://www.hapmap.org.
Subsequent analyses demonstrated that the population-attributable risk from possessing MYH9 risk alleles approximates 70% in nondiabetic ESRD in African Americans; this effect was computed using the E-1 haplotype as the surrogate risk variant (95% confidence interval 55 to 81%) (19). Stated another way, replacement of MYH9 risk variants in the African American population with protective or neutral variants (as are often seen in the European American population) would reduce the rates of nondiabetic ESRD by a staggering 70%. Fine mapping studies are under way in an attempt to identify causative MYH9 SNPs. Additional studies will then be necessary to understand the population risk incurred by these MYH9 polymorphisms. It is also clear that additional factors beyond genotype must be important in the initiation of the MYH9-associated spectrum of kidney disease (20).
It is generally accepted that common complex diseases are not the result of mutation in a single gene but rather the cumulative effect of multiple genetic and environmental factors. For example, although nearly 40 type 2 diabetes susceptibility genes have been identified, in aggregate, they contribute to <10% of the total risk for developing diabetes (33). The strongest and most replicated diabetes genes, such as TCF7L2, contribute approximately a 40% increase in risk for each risk variant that is inherited (34). In contrast, MYH9 contributes a far stronger effect on the risk for kidney disease in African American and European-derived populations.
MYH9 encodes the nonmuscle myosin heavy chain 9, which, with other subunits, forms myosin II (35). Myosin II is a motor protein that binds actin and is involved in cellular motility. MYH9 is expressed in the podocyte, as well as in mesangial cells and arteriolar and peritubular capillaries (36). We postulate that a mutation in the MYH9 gene product could produce podocyte injury with FSGS, although other mechanisms, including interactions between glomerular cells and abnormal platelets, remain possible and warrant further study. MYH9 mutations had been implicated in a number of autosomal dominant syndromes: May-Hegglin, Sebastian, Fechtner, and Epstein (37,38). These syndromes are platelet disorders that are characterized by thrombocytopenia and leukocyte inclusions, with Fechtner and Epstein syndromes resulting in nephritis to varying degrees. The mutations identified in these cases are nonsense or missense mutations that result in significant structural mutations in MYH9 and platelet abnormalities. Despite the structural mutations, few of these patients develop renal failure. It is unclear why structural defects would not necessarily result in renal failure in the autosomal dominant syndromes, whereas the variants that are associated with ESRD in African Americans are intronic and confer significant risk. Functional studies on these variants will be an important future step in understanding the role of MYH9 in ESRD.
“Second Hits” in MYH9-Associated Nephropathy
Initial reports revealed that approximately 35% of the general African American and 1% of the European American population are homozygous for the MYH9 E-1 risk haplotype (18 –20); however, approximately 4% of African Americans who are homozygous for the MYH9 E-1 risk haplotype will develop idiopathic FSGS, whereas 20% of HIV-infected E-1 homozygous African Americans develops HIVAN (Jeffrey Kopp, personal communication, November 2009). Because African Americans with two MYH9 risk variants do not uniformly develop nephropathy, co-factors for initiation of renal disease must exist (Figure 2). It is clear that viral infections can contribute to risk, on the basis of the fivefold excess frequency of nephropathy in HIV-infected MYH9 risk homozygotes. Nonetheless, four of five HIV-infected African Americans who are homozygous for MYH9 risk variants do not develop nephropathy. This led us to hypothesize that “second hits,” or additional factors, are required to initiate kidney disease.
Figure 2.
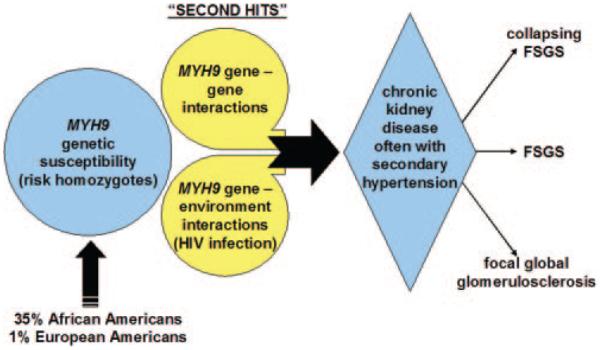
Proposed pathogenesis of MYH9-associated nephropathy.
On the basis of the role of HIV infection in MYH9-associated HIVAN (39), we postulate that gene– environment interactions could produce renal disease in MYH9 risk homozygotes. Several non-HIV viral infections are associated with CKD, including BK virus and Parvovirus B19 (40,41). We postulate that these or other viruses that can infect renal cells or lymphocytes contribute to development of MYH9-associated nephropathy in the manner of HIV1 viral infection.
In addition, it remains possible that gene– gene interactions may be factors. Polymorphisms in podocin, ex actinin-4, and TRPC6 have been shown to contribute to glomerulosclerosis (42– 44). It is conceivable that heterozygous mutations in these or other genes coupled with MYH9 risk homozygosity may ultimately produce susceptibility to FSGS. For example, disruption of MYH9 alone may not be sufficient to result in significant podocyte structural malfunction; however, mutation of other cytoskeletal components, such as ex actinin-4, in addition to MYH9 may substantially damage the structure of the podocyte and impair its function as a filtration barrier. Studies to investigate these possibilities are actively under way.
Essential Hypertension and Initiation of MYH9-Associated Nephropathy
As discussed in the beginning of this article, although hypertension is often listed as a cause of ESRD in African Americans, familial clustering of other forms of CKD, including diabetic nephropathy, chronic glomerulonephritis, lupus nephritis, and HIVAN in families with index cases who have “hypertensive ESRD,” suggested the presence of an overarching kidney failure susceptibility gene (1,24). An association analysis of 696 African Americans who reportedly had hypertensive ESRD revealed the important contribution made by MYH9 polymorphisms, with SNP association OR as high as 3.4 (20). In addition, multiple regions in MYH9 seem to produce independent susceptibility, including the major E-1 haplotype (OR 2.4), the recently identified L-1 haplotype (A/G/C/T; rs7078, rs12107, rs735853, and rs5756129; OR 1.9), and one additional SNP, rs5756152 (OR 2.3). This suggests that the disease labeled HN resides in the FSGS family (2).
An association analysis in the NHLBI-Hypertension Genetics Study (HyperGEN) demonstrated weak association between albuminuria and MYH9 polymorphisms (45). Of greater importance, the frequency of the E-1 risk haplotype was not different in the HyperGEN African American population with hypertension when contrasted to the control populations in previous reports (18 –20). This suggests that MYH9 does not play a major role in susceptibility to essential hypertension.
We recently reported that putative HN in AASK participants was strongly MYH9 associated (46). MYH9 E-1 haplotype SNP rs4821481 had an OR of 1.99 (P = 7 × 10−6, recessive) in those with serum creatinine concentrations ≥2 mg/dl, increasing to OR 2.7 with serum creatinine concentrations >3 mg/dl. Thus, FGGS in AASK falls within the spectrum of MYH9-associated disorders. Patients with FGGS in AASK were similar to those with “hypertensive ESRD” in the Wake Forest cohort (20), considering the risk imparted by MYH9.
Collapsing FSGS: The Most Aggressive Lesion in the Spectrum
FSGS is a disorder of podocyte depletion (47). In contrast, HIVAN is manifested by abnormal extensive proliferation of podocytes, cells that are normally terminally differentiated. Recent results showed that HIV can directly infect the podocyte, and a series of interrelated pathways leads to loss of the differentiated phenotype with extensive proliferation and resultant glomerular collapse (39). HIV1-specific gene products may be involved in this process and interact with MYH9. It also remains possible that other environmental and genetic co-factors are second hits necessary for the development of podocyte proliferation. It recently became clear that mesangial immune complex deposition can also trigger the collapsing variant of MYH9 nephropathy. HIV-negative African Americans with mesangial C1q-containing immune complexes have developed collapsing glomerulopathy in the presence of MYH9 risk variants (48). This broadens the spectrum of collapsing FSGS to include immune complex–mediated diseases and effects of viral infection. The effects of MYH9 risk variants on development of the idiopathic variant of collapsing FSGS, the most common cause, is unknown; however, there is no reason to suspect that MYH9 does not underlie this form of collapsing FSGS. This is an important renal lesion that warrants additional analysis.
The Evolving Spectrum of MYH9-Associated Nephropathy
The association of risk variants in MYH9 with nondiabetic forms of nephropathy in African Americans and European-derived populations demonstrates among the strongest association with any common complex human kidney disease. In addition, although the OR for MYH9 association with type 2 diabetic nephropathy in African Americans is 1.4 (far lower than for FSGS, HIVAN, and FGGS), approximately 16% of African Americans who receive a clinical diagnosis of type 2 diabetes–associated ESRD have disorders in the MYH9 spectrum (49). We believe that it is most likely that these patients actually have FSGS with coincident diabetes, although kidney biopsy studies remain to be performed to demonstrate whether the classic histologic changes that are seen in diabetic nephropathy are present. Nonetheless, MYH9 contributes to 40 to 45% of all ESRD in the African American population, as well as FSGS and CKD in Europeans and European Americans.
The emerging spectrum of MYH9 nephropathy clearly links FGGS (the disease historically attributed to hypertension), idiopathic FSGS, and collapsing variants of FSGS secondary to HIV infection and C1q immune complex deposition (Figure 3). This disease spectrum explains the presence of multiple causes of CKD in single African American families, a phenomenon rarely observed in non–African American families. This finding has important implications, including an inherited mechanism of disease in the African American population, although the contribution of environmental risk factors to initiation of kidney scarring should not be downplayed.
Figure 3.
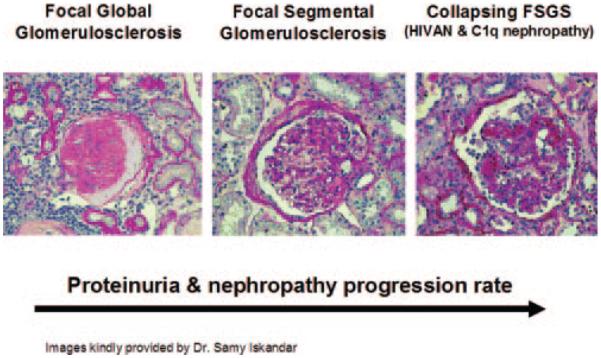
The spectrum of MYH9-associated nephropathy.
The implications and novel questions raised by this observation extend beyond the epidemiology of CKD (Table 1). The unexplained poorer allograft survival of kidneys that are donated by African Americans may be attributable, in part, to MYH9 mutations. Deceased-donor kidneys from African Americans survive less well than kidneys that are donated by European Americans, whether transplanted into African American or European American recipients (50,51). It is possible that the combined effects of cold ischemia time, nephrotoxic calcineurin inhibitors, and donor–recipient genotype interactions may interact with MYH9 to explain this phenomenon.
Table 1.
Issues under investigation in MYH9-associated nephropathy
| Clinical |
| value of genetic screening to identify individuals who are at risk for future nephropathy |
| influence of donor genotypes on outcomes after kidney transplantation |
| potential influence on ethnic differences in outcomes after dialysis initiation |
| assessment of renal histology: variants in FSGS on the basis of genotype lesions in African Americans with clinically diagnosed diabetic nephropathy |
| detection of environmental co-factors that may interact to initiate renal disease |
| determine natural history of disease and effect on rates of nephropathy progression |
| determine response to conventional and novel therapies for FSGS and related disorders |
| Basic |
| identify the disease-causing polymorphism(s) |
| determine mechanisms of podocyte injury, and develop preventive therapies |
| detect additional genes that interact to initiate renal disease |
| identify the factor(s) on the African continent that selected for nephropathy risk variants |
The rapidly expanding spectrum of MYH9-associated kidney diseases has been a major breakthrough in our understanding of the epidemiology of ESRD, particularly in the African American population. The effects of various second hits may dictate the type of MYH9-associated renal lesion. For example, HIV infection most commonly yields collapsing glomerulopathy, whereas other co-factors might produce FGGS or classic FSGS. The role of these co-factors and resultant renal histologic patterns remain important to determine. It is abundantly clear that mild to moderate forms of essential hypertension are insufficient to initiate kidney disease among MYH9 risk homozygotes. The majority of African Americans who have low-level proteinuria and are labeled as having HN and hypertension-associated ESRD have an incorrect diagnosis. We remain optimistic that identification and treatment of environmental second hits, as well as drug therapies directed at maintaining the normal podocyte cytoskeletal architecture, offer hope for the prevention of CKD in the African American population. MYH9 encodes a motor protein involved in the movement of actin filaments in the podocyte and is also expressed in other renal cells. We believe that alterations in podocyte architecture lead to the spectrum of MYH9-associated nephropathies, because FSGS is commonly associated with other gene polymorphisms that interrupt the podocyte cytoskeleton and glomerular filtration barrier.
It is our opinion that screening for MYH9 risk variants may eventually be useful in high-risk populations such as African Americans with close relatives who have ESRD or in those who wish to donate a kidney; however, the high frequency of MYH9 risk variants in African-derived individuals could render this a less useful screening test. It is important that the additional triggers, or “second hits,” required for initiation of renal disease be identified: Either gene– gene or gene– environment interactions. MYH9 genotypes may be more useful to clinicians for screening purposes and ultimately for selecting therapies, when second hits are identified. For example, if viral infections or other environmental exposures interact with MYH9 to produce nephropathy, then immunization against infection or avoidance of exposures may be protective from kidney disease even in those who harbor risk variants. At present, novel therapies for slowing or preventing MYH9-associated nephropathy and use of MYH9 genotypes for screening purposes require further study.
Conclusions
Genetic methods have contributed to the detection of a newly recognized spectrum of related kidney diseases. MYH9-associated nephropathy is clearly the most common form of CKD in African Americans.
Acknowledgments
This work was supported in part by National Institutes of Health grants RO1 DK 070941, RO1 DK 071891, and RO1 DK084149 (B.I.F.).
Footnotes
Disclosures
None.
References
- 1.Freedman BI, Iskandar SS, Appel RG. The link between hypertension and nephrosclerosis. Am J Kidney Dis. 1995;25:207–221. doi: 10.1016/0272-6386(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 2.Freedman BI, Sedor JR. Hypertension-associated kidney disease: Perhaps no more. J Am Soc Nephrol. 2008;19:2047–2051. doi: 10.1681/ASN.2008060621. [DOI] [PubMed] [Google Scholar]
- 3.Zarif L, Covic A, Iyengar S, Sehgal AR, Sedor JR, Schelling JR. Inaccuracy of clinical phenotyping parameters for hypertensive nephrosclerosis. Nephrol Dial Transplant. 2000;15:1801–1807. doi: 10.1093/ndt/15.11.1801. [DOI] [PubMed] [Google Scholar]
- 4.US Renal Data System . USRDS 2008 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda: 2008. [Google Scholar]
- 5.Shulman NB, Ford CE, Hall WD, Blaufox MD, Simon D, Langford HG, Schneider KA. Prognostic value of serum creatinine and effect of treatment of hypertension on renal function: Results from the hypertension detection and follow-up program. The Hypertension Detection and Follow-up Program Cooperative Group. Hypertension. 1989;13:I80–I93. doi: 10.1161/01.hyp.13.5_suppl.i80. [DOI] [PubMed] [Google Scholar]
- 6.Walker WG, Neaton JD, Cutler JA, Neuwirth R, Cohen JD. Renal function change in hypertensive members of the Multiple Risk Factor Intervention Trial: Racial and treatment effects. The MRFIT Research Group. JAMA. 1992;268:3085–3091. [PubMed] [Google Scholar]
- 7.Perneger TV, Whelton PK, Klag MJ, Rossiter KA. Diagnosis of hypertensive end-stage renal disease: Effect of patient’s race. Am J Epidemiol. 1995;141:10–15. doi: 10.1093/oxfordjournals.aje.a117338. [DOI] [PubMed] [Google Scholar]
- 8.Schlessinger SD, Tankersley MR, Curtis JJ. Clinical documentation of end-stage renal disease due to hypertension. Am J Kidney Dis. 1994;23:655–660. doi: 10.1016/s0272-6386(12)70275-5. [DOI] [PubMed] [Google Scholar]
- 9.Freedman BI, Iskander SS, Buckalew VM, Jr, Burkart JM, Appel RG. Renal biopsy findings in presumed hypertensive nephrosclerosis. Am J Nephrol. 1994;14:90–94. doi: 10.1159/000168695. [DOI] [PubMed] [Google Scholar]
- 10.Fogo A, Breyer JA, Smith MC, Cleveland WH, Agodoa L, Kirk KA, Glassock R. Accuracy of the diagnosis of hypertensive nephrosclerosis in African Americans: A report from the African American Study of Kidney Disease (AASK) Trial. AASK Pilot Study Investigators. Kidney Int. 1997;51:244–252. doi: 10.1038/ki.1997.29. [DOI] [PubMed] [Google Scholar]
- 11.Marcantoni C, Ma LJ, Federspiel C, Fogo AB. Hypertensive nephrosclerosis in African Americans versus Caucasians. Kidney Int. 2002;62:172–180. doi: 10.1046/j.1523-1755.2002.00420.x. [DOI] [PubMed] [Google Scholar]
- 12.Bleyer AJ, Chen R, D’Agostino RB, Jr, Appel RG. Clinical correlates of hypertensive end-stage renal disease. Am J Kidney Dis. 1998;31:28–34. doi: 10.1053/ajkd.1998.v31.pm9428448. [DOI] [PubMed] [Google Scholar]
- 13.Bianchi S, Bigazzi R, Caiazza A, Campese VM. A controlled, prospective study of the effects of atorvastatin on proteinuria and progression of kidney disease. Am J Kidney Dis. 2003;41:565–570. doi: 10.1053/ajkd.2003.50140. [DOI] [PubMed] [Google Scholar]
- 14.Phisitkul K, Hegazy K, Chuahirun T, Hudson C, Simoni J, Rajab H, Wesson DE. Continued smoking exacerbates but cessation ameliorates progression of early type 2 diabetic nephropathy. Am J Med Sci. 2008;335:284–291. doi: 10.1097/MAJ.0b013e318156b799. [DOI] [PubMed] [Google Scholar]
- 15.Shepherd J, Kastelein JJ, Bittner V, Deedwania P, Breazna A, Dobson S, Wilson DJ, Zuckerman A, Wenger NK. Treating to New Targets Investigators: Effect of intensive lipid lowering with atorvastatin on renal function in patients with coronary heart disease: The Treating to New Targets (TNT) Study. Clinical J Am Soc Nephrol. 2007;2:1131–1139. doi: 10.2215/CJN.04371206. [DOI] [PubMed] [Google Scholar]
- 16.Appel LJ, Wright JT, Jr, Greene T, Kusek JW, Lewis JB, Wang X, Lipkowitz MS, Norris KC, Bakris GL, Rahman M, Contreras G, Rostand SG, Kopple JD, Gabbai FB, Schulman GI, Gassman JJ, Charleston J, Agodoa LY. African American Study of Kidney Disease and Hypertension Collaborative Research Group: Long-term effects of renin-angiotensin system-blocking therapy and a low blood pressure goal on progression of hypertensive chronic kidney disease in African Americans. Arch Intern Med. 2008;168:832–839. doi: 10.1001/archinte.168.8.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McClellan W, Tuttle E, Issa A. Racial differences in the incidence of hypertensive end-stage renal disease (ESRD) are not entirely explained by differences in the prevalence of hypertension. Am J Kidney Dis. 1988;12:285–290. doi: 10.1016/s0272-6386(88)80221-x. [DOI] [PubMed] [Google Scholar]
- 18.Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI, Bowden DW, Oleksyk T, McKenzie LM, Kajiyama H, Ahuja TS, Berns JS, Briggs W, Cho ME, Dart RA, Kimmel PL, Korbet SM, Michel DM, Mokrzycki MH, Schelling JR, Simon E, Trachtman H, Vlahov D, Winkler CA. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008;40:1175–1184. doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kao WH, Klag MJ, Meoni LA, Reich D, Berthier-Schaad Y, Li M, Coresh J, Patterson N, Tandon A, Powe NR, Fink NE, Sadler JH, Weir MR, Abboud HE, Adler SG, Divers J, Iyengar SK, Freedman BI, Kimmel PL, Knowler WC, Kohn OF, Kramp K, Leehey DJ, Nicholas SB, Pahl MV, Schelling JR, Sedor JR, Thornley-Brown D, Winkler CA, Smith MW, Parekh RS. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet. 2008;40:1185–1192. doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freedman BI, Hicks PJ, Bostrom MA, Cunningham ME, Liu Y, Divers J, Kopp JB, Winkler CA, Nelson GW, Lange-feld CD, Bowden DW. Polymorphisms in the non-muscle myosin heavy chain 9 gene (MYH9) are strongly associated with end-stage renal disease historically attributed to hypertension in African Americans. Kidney Int. 2009;75:736–745. doi: 10.1038/ki.2008.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freedman BI, Tuttle AB, Spray BJ. Familial predisposition to nephropathy in African-Americans with non-insulin-dependent diabetes mellitus. Am J Kidney Dis. 1995;25:710–713. doi: 10.1016/0272-6386(95)90546-4. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson R, Grim CE, Opgenorth TJ. A familial risk of chronic renal failure among blacks on dialysis? J Clin Epidemiol. 1988;41:1189–1196. doi: 10.1016/0895-4356(88)90023-6. [DOI] [PubMed] [Google Scholar]
- 23.Seaquist ER, Goetz FC, Rich S, Barbosa J. Familial clustering of diabetic kidney disease: Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med. 1989;320:1161–1165. doi: 10.1056/NEJM198905043201801. [DOI] [PubMed] [Google Scholar]
- 24.Freedman BI, Spray BJ, Tuttle AB, Buckalew VM., Jr The familial risk of end-stage renal disease in African Americans. Am J Kidney Dis. 1993;21:387–393. doi: 10.1016/s0272-6386(12)80266-6. [DOI] [PubMed] [Google Scholar]
- 25.Freedman BI, Wilson CH, Spray BJ, Tuttle AB, Olorenshaw IM, Kammer GM. Familial clustering of end-stage renal disease in blacks with lupus nephritis. Am J Kidney Dis. 1997;29:729–732. doi: 10.1016/s0272-6386(97)90126-8. [DOI] [PubMed] [Google Scholar]
- 26.Freedman BI, Soucie JM, Stone SM, Pegram S. Familial clustering of end-stage renal disease in blacks with HIV-associated nephropathy. Am J Kidney Dis. 1999;34:254–258. doi: 10.1016/s0272-6386(99)70352-5. [DOI] [PubMed] [Google Scholar]
- 27.Bergman S, Key BO, Kirk KA, Warnock DG, Rostant SG. Kidney disease in the first-degree relatives of African-Americans with hypertensive end-stage renal disease. Am J Kidney Dis. 1996;27:341–346. doi: 10.1016/s0272-6386(96)90356-x. [DOI] [PubMed] [Google Scholar]
- 28.Lei HH, Perneger TV, Klag MJ, Whelton PK, Coresh J. Familial aggregation of renal disease in a population-based case-control study. J Am Soc Nephrol. 1998;9:1270–1276. doi: 10.1681/ASN.V971270. [DOI] [PubMed] [Google Scholar]
- 29.Freedman BI, Volkova NV, Satko SG, Krisher J, Jurkovitz C, Soucie JM, McClellan WM. Population-based screening for family history of end-stage renal disease among incident dialysis patients. Am J Nephrol. 2005;25:529–535. doi: 10.1159/000088491. [DOI] [PubMed] [Google Scholar]
- 30.Song EY, McClellan WM, McClellan A, Rajyalakshmi G, Hadley AC, Krisher J, Clay M, Freedman BI. Effect of community characteristics on familial clustering of end-stage renal disease. Am J Nephrol. 2009;30:499–504. doi: 10.1159/000243716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freedman BI. End-stage renal failure in African Americans: Insights in kidney disease susceptibility. Nephrol Dial Transplant. 2002;17:198–200. doi: 10.1093/ndt/17.2.198. [DOI] [PubMed] [Google Scholar]
- 32.Smith MW, Patterson N, Lautenberger JA, Truelove AL, McDonald GJ, Waliszewska A, Kessing BD, Malasky MJ, Scafe C, Le E, De Jager PL, Mignault AA, Yi Z, De The G, Essex M, Sankale JL, Moore JH, Poku K, Phair JP, Goedert JJ, Vlahov D, Williams SM, Tishkoff SA, Winkler CA, De La Vega FM, Woodage T, Sninsky JJ, Hafler DA, Altshuler D, Gilbert DA, O’Brien SJ, Reich D. A high-density admixture map for disease gene discovery in African Americans. Am J Hum Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCarthy MI. What will genome-wide association studies mean to the clinical endocrinologist? >J Clin Endocrinol Metab. 2009;94:2245–2246. doi: 10.1210/jc.2009-0403. [DOI] [PubMed] [Google Scholar]
- 34.Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 35.Marini M, Bruschi M, Pecci A, Romagnoli R, Musante L, Candiano G, Ghiggeri GM, Balduini C, Seri M, Ravazzolo R. Non-muscle myosin heavy chain IIA and IIB interact and co-localize in living cells: Relevance for MYH9-related disease. Int J Mol Med. 2006;17:729–736. [PubMed] [Google Scholar]
- 36.Arrondel C, Vodovar N, Knebelmann B, Grunfeld JP, Gubler MC, Antignac C, Heidet L. Expression of the non-muscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol. 2002;13:65–74. doi: 10.1681/ASN.V13165. [DOI] [PubMed] [Google Scholar]
- 37.Heath KE, Campos-Barros A, Toren A, Rozenfeld-Granot G, Carlsson LE, Savige J, Denison JC, Gregory MC, White JG, Barker DF, Greinacher A, Epstein CJ, Glucksman MJ, Martignetti JA. Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am J Hum Genet. 2001;69:1033–1045. doi: 10.1086/324267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seri M, Cusano R, Gangarossa S, Caridi G, Bordo D, Lo NC, Ghiggeri GM, Ravazzolo R, Savino M, Del Vecchio M, d’Apolito M, Iolascon A, Zelante LL, Savoia A, Balduini CL, Noris P, Magrini U, Belletti S, Heath KE, Babcock M, Glucksman MJ, Aliprandis E, Bizzaro N, Desnick RJ, Martignetti JA. Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The May-Hegglin/Fechtner Syndrome Consortium. Nat Genet. 2000;26:103–105. doi: 10.1038/79063. [DOI] [PubMed] [Google Scholar]
- 39.Papeta N, Chan KT, Prakash S, Martino J, Kiryluk K, Ballard D, Bruggeman LA, Frankel R, Zheng Z, Klotman PE, Zhao H, D’Agati VD, Lifton RP, Gharavi AG. Susceptibility loci for murine HIV-associated nephropathy encode trans-regulators of podocyte gene expression. J Clin Invest. 2009;119:1178–1188. doi: 10.1172/JCI37131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanawattanacharoen S, Falk RJ, Jennette JC, Kopp JB. Parvovirus B19 DNA in kidney tissue of patients with focal segmental glomerulosclerosis. Am J Kidney Dis. 2000;35:1166–1174. doi: 10.1016/s0272-6386(00)70055-2. [DOI] [PubMed] [Google Scholar]
- 41.Celik B, Randhawa PS. Glomerular changes in BK virus nephropathy. Hum Pathol. 2004;35:367–370. doi: 10.1016/j.humpath.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 42.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 43.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodriguez-Perez JC, Allen PG, Beggs AH, Pollak MR. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 44.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 45.Freedman BI, Kopp JB, Winkler CA, Nelson GW, Rao DC, Eckfeldt JH, Leppert MF, Hicks PJ, Divers J, Langefeld CD, Hunt SC. Polymorphisms in the nonmuscle myosin heavy chain 9 gene (MYH9) are associated with albuminuria in hypertensive African Americans: The HyperGEN study. Am J Nephrol. 2009;29:626–632. doi: 10.1159/000194791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lipkowitz MS, Iyengar S, Molineros J, Langefeld CD, Comeau ME, Klotman PE, Bowden DW, Freedman RG, Khitrov G, Zhang W, Kao WH, Parekh RS, Choi M, Kopp JB, Winkler CA, Nelson G, Freedman BI, Bottinger EP, AASK Investigators Association analysis of the non-muscle myosin heavy chain 9 gene (MYH9) in hypertensive nephropathy: African American Study of Kidney Disease and Hypertension (AASK) [Abstract] J Am Soc Nephrol. 2009;20:56A. [Google Scholar]
- 47.Barisoni L, Kriz W, Mundel P, D’Agati V. The dysregulated podocyte phenotype: A novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 1999;10:51–61. doi: 10.1681/ASN.V10151. [DOI] [PubMed] [Google Scholar]
- 48.Reeves-Daniel AM, Iskandar SS, Bowden DW, Bostrom MA, Hicks PJ, Comeau ME, Langefeld CD, Freedman BI. Is collapsing C1q nephropathy another MYH9-associated kidney disease? A case report. Am J Kidney Dis. 2010 Jan 27; doi: 10.1053/j.ajkd.2009.10.060. [epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freedman BI, Hicks PJ, Bostrom MA, Comeau ME, Divers J, Bleyer AJ, Kopp JB, Winkler CA, Nelson GW, Langefeld CD, Bowden DW. Non-muscle myosin heavy chain 9 gene MYH9 associations in African Americans with clinically diagnosed type 2 diabetes mellitus-associated ESRD. Nephrol Dial Transplant. 2009;24:3366–3371. doi: 10.1093/ndt/gfp316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katznelson S, Gjertson DW, Cecka JM. The effect of race and ethnicity on kidney allograft outcome. Clin Transpl. 1995:379–394. [PubMed] [Google Scholar]
- 51.Callender CO, Cherikh WS, Miles PV, Hermesch A, Mad-dox G, Nash J, Hernandez A, Burston B. Blacks as donors for transplantation: Suboptimal outcomes overcome by transplantation into other minorities. Transplant Proc. 2008;40:995–1000. doi: 10.1016/j.transproceed.2008.03.063. [DOI] [PubMed] [Google Scholar]
- 52.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]