

Abstract
Liver regeneration after partial hepatectomy is a clinically important process that is impaired by adaptation to chronic alcohol intake. We focused on the initial time points following partial hepatectomy (PHx) to analyze genome-wide binding activity of NF-κB, a key immediate early regulator. We investigated the effect of chronic alcohol intake on immediate early NF-κB genome-wide localization, in the adapted state as well as in response to partial hepatectomy, using chromatin immunoprecipitation followed by promoter microarray analysis. We found many ethanol-specific NF-κB binding target promoters in the ethanol-adapted state, corresponding to regulation of biosynthetic processes, oxidation-reduction and apoptosis. Partial hepatectomy induced a diet-independent shift in NF-κB binding loci relative to the transcription start sites. We employed a novel pattern count analysis to exhaustively enumerate and compare the number of promoters corresponding to the temporal binding patterns in ethanol and pair-fed control groups. The highest pattern count corresponded to promoters with NF-κB binding exclusively in the ethanol group at 1h post PHx. This set was associated with regulation of cell death, response to oxidative stress, histone modification, mitochondrial function, and metabolic processes. Integration with the global gene expression profiles to identify putative transcriptional consequences of NF-κB binding patterns revealed that several of ethanol-specific 1h binding targets showed ethanol-specific differential expression through 6h post PHx. Motif analysis yielded co-incident binding loci for STAT3, AP-1, CREB, C/EBP-β, PPAR-γ and C/EBP-α, likely participating in co-regulatory modules with NF-κB in shaping the immediate early response to PHx. We conclude that adaptation to chronic ethanol intake disrupts the NF-κB promoter binding landscape with consequences for the immediate early gene regulatory response to the acute challenge of PHx.
Introduction
The liver tissue has a unique ability to regenerate in response to injury. Liver regeneration involves a sequence of complex biochemical and physiological activities leading to cell proliferation, mass recovery and tissue structure reconstruction. Partial hepatectomy (PHx) is a widely used model to study initiation and progression of liver regeneration1–4. After PHx, various pathways are activated to maintain the liver functions. In the priming phase, hepatocytes exit the quiescent G0 phase and enter the pre-replicative G1 phase. Cell proliferation occurs in both parenchymal and non-parenchymal cells though over different time phases1,5,6. The hepatocyte replication preceding non-parenchymal replication phase is followed by the termination phase by which liver recovers most of its original mass. Among the initial responses post acute liver damage is the release of various inflammatory factors and cytokines leading to the expression of growth factors and hormonal modulators7. Earlier studies showed an increase in transcriptional activity of NF-κB p65 during the immediate early phase of the regenerative process3,8–15, leading to the activation of several genes associated with immune response, inflammation, adhesion, proliferation, oxidative stress and liver homeostasis3,12. Failure of NF-κB activation can result in decreased hepatocyte proliferation13,16,17 and impaired regeneration in the liver18–20.
Various external agents can cause hepatotoxicity leading to impaired regeneration. Chronic ethanol intake is known to activate multiple stress response pathways of the liver causing dysregulation of liver repair mechanisms by disrupting metabolic pathways21–26. This may have clinical significance relevant to alcoholic liver disease, one of the leading causes of mortality around the world9,12. Delayed cellular replication leading to inhibition of regeneration was observed in partially hepatectomized livers after ethanol adaptation with almost complete suppression of hepatocyte proliferation27,28. The molecular regulatory networks underlying this phenomenon remain poorly understood; however, it has been suggested that part of the ethanol-induced maladaptation suppressing regeneration is mediated through changes to NF-κB regulation of target genes29. Chronic ethanol exposure is associated with a sustained increase in NF-κB activity leading to the activation of pro-inflammatory genes8,9,12,30. It has been shown that ethanol mediated activation of NF-κB in primary hepatocytes can contribute to the accelerated progression of the liver diseases31. However, the specific role of NF-κB in the ethanol-adapted liver continues to be a subject of investigation.
Several small-scale studies and large-scale time-series expression studies have been carried out to investigate the dynamic changes of multiple factors during liver regeneration32–36. In an earlier study, we found an increase in NF-κB activation at 1 h post PHx, followed by a decrease to near baseline levels at 4 h post PHx and further increase at 6 h post PHx, suggesting a dynamic role of NF-κB during the early phase of regeneration34. ChIP-chip/seq techniques have been widely utilized to find binding sites of various transcription factors in response to perturbations37,38. These techniques have been informative in analyzing the genome-wide localization profiles of NF-κB to gain insight into the functional role of NF-κB in humans and rats39–46. We analyzed changes in genome-wide NF-κB binding activity during the immediate early phase of rat liver regeneration. Our results revealed a dynamic switch of NF-κB binding across the genome with putative consequences for hepatocyte entry into cell cycle39. It has been unclear whether adaptation to ethanol interferes with such a dynamic switch as a putative mechanism underlying ethanol-mediated dysregulation of the response to PHx. Our analysis of cell type-specific localization of NF-κB revealed an upward shift in distribution of NF-κB in hepatocytes47. However, studies thus far have not addressed the combinatorial effects of chronic ethanol intake diet and acute perturbation of PHx on genome-wide NF-κB binding patterns.
We addressed this issue by exploring the genome-wide NF-κB bound loci in whole liver tissue, using ChIP-chip technique. We analyzed the effect of chronic ethanol intake alone on the genome-wide NF-κB binding profile the ethanol-adapted state. We then employed a novel pattern analysis to evaluate global alterations in NF-κB p65 promoter binding as a result of the combined effect of chronic ethanol intake followed by 2/3rd PHx. This approach allowed us to account for the presence of multiple factors driving common and altered NF-κB binding responses in a time and diet dependent manner. In contrast to conventional approaches, our method provides the capability of analyzing the data in a systematic manner, by identifying the dominant patterns and masking them to uncover more subtle patterns. We integrated the ChIP-chip results with a time series gene expression data set to identify the NF-κB promoter binding targets that showed differential gene expression changes at the baseline-adapted condition as well as after PHx. We identified a set of co-incident motifs of NF-κB binding that were specific to the ethanol and pair-fed control groups. Our novel analysis technique followed by computational analysis to identify statistically significant loci, coincident motifs, and integration with gene regulation, enabled us to examine the extent to which the immediate early activation of NF-κB manifested as dynamic changes in the binding activity at the target promoters in the liver as a result of the combined perturbations of ethanol and PHx.
Experimental and analytical methods
Animals and diet
Adult Sprague-Dawley rats were held in a climate controlled, 12-hr day/night cycle in accordance with accepted animal handling practices. Animals were fed using the Lieber-DeCarli pair-feeding model48 in which rats were fed a nutritionally adequate liquid diet containing 36% of total calories derived from ethanol for 5 weeks (Ethanol group), with the pair-fed calorie-matched littermate controls receiving liquid diets in which ethanol calories were replaced by maltose dextran (Carbohydrate group). Rats (275-350 g) were anesthetized and subjected to 2/3rd PHx by surgical removal of left lateral and median lobes (LLM) as previously described6. The remnant liver was allowed to regenerate and the liver samples were harvested at 1 and 6h post PHx (Fig. S1, ESI†). The excised liver samples at t=0 served as within-animal controls. Collected liver samples were freeze-clamped in liquid nitrogen-cooled aluminium clamps for preparation of tissue lysates. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Thomas Jefferson University.
Chromatin Immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) assays were performed using liver tissue to map in-vivo distribution of NF-κB DNA interactions as per standard protocol using a Magna ChIP G kit (Fig. S2, ESI†). We used Rabbit polyclonal NF-κB p65 antibody (Abcam Inc., Cat#ab7970) and a negative control IgG antibody (Santa Cruz Biotechnology, Cat #sc-2026). Approximately 50 μg of liver tissue was fixed for 10 minutes with 1% formaldehyde to cross-link DNA and chromatin binding proteins for co-immunoprecipitation. The cross-linked chromatin was neutralized with glycine to quench the unreacted formaldehyde. Chromatin was sheared via sonication to generate fragments of 200-1000 bp size. The fragment size range was confirmed by a 1% agarose gel electrophoresis. The immune selection was performed using ChIP grade NF-κB antibody in combination with Protein G conjugated solid support matrix magnetic beads. This NF-κB antibody has been validated extensively in previous studies to detect NF-κB proteins in Western blots and immunohistochemistry assays49–51.
ChIP-chip using Roche Nimblegen Promoter Array
Purified ChIP DNA was amplified with GenomePlex Complete Whole Genome Amplification (WGA) kit from Sigma that generated nearly 500-fold amplification of genomic DNA using OmniPlex Library molecules flanked by universal priming sites. The promoter array analysis was performed with whole genome amplified ChIP DNA samples using the Roche Nimblegen Promoter array platform (Rat ChIP-chip 3x720K RefSeq Promoter Arrays with 3 identical arrays per glass slide with 72,000 probes per array) to cover the whole genome (Roche NimbleGen, Inc., 504 South Rosa Road, Madison, WI). Experimental and total DNA samples were labeled using 9-mer primers that have Cy3 and Cy5 dyes attached and Klenow added. The labelled experimental IP and total DNAs were co-hybridized to the array for 16 - 20 hours at 420C, washed, and scanned (Agilent scanner, Agilent Technologies) following manufacturer instructions. Array images were used for data extraction as pair files; genomic feature format files were then produced for visualization of scaled log2-ratio data. The intensity ratio of immunopreciptated to total DNA (not taken through immunoprecipitation steps) was plotted versus genomic position to identify regions where increased signal (i.e. DNA fragment enrichment) was observed relative to the control sample. Peak files identifying statistically significant binding/modification sites were generated from the scaled log2-ratio data, and peaks were mapped to the transcription start site of each gene. Roche NimbleGen's proprietary, light-mediated synthesis process produced high-density microarrays of long oligonucleotide probes (50-75mer). These long oligo arrays, when used in combination with high-stringency hybridization protocols, produced results of high sensitivity and specificity. In addition, because Roche NimbleGen performs ChIP-chip experiments in a two-color protocol, where control and test samples were co-hybridized to the same array, inter-array variation was eliminated. As a result, NimbleGen ChIP-chip service readily detects enrichment as low as two-fold of the target-binding site in a ChIP sample.
Genome-wide mapping and peak detection: Identification of binding sites
Using NimbleScan software, peak data files were generated from the scaled log2 ratio data. The peaks that correspond to the binding targets were detected if 4 or more probes shows signal above the cut-off value (percentage of the hypothetical maximum) between 15%-90% using 500bp-sliding window. Each peak was assigned a false discovery rate (FDR), which was estimated based on the probability of false positives determined by randomizing the ratio data. An initial FDR cut-off of <= 0.2 was used to identify the actual binding regimes (“peaks”) corresponding to the binding sites. To further reduce false positives, a more stringent false discovery rate (FDR) cut-off of 0.15 was used (Fig. S2, ESI†). Additionally, to be identified as bound, NF-κB binding had to be observed in 3 out of 4 biological replicates (peaks belonging to 60% of biological replicates) at a particular time point. Applying these filtering schemes, we identified 54127 NF-κB bound regions from all replicates in both dietary groups over the time course, resulting in 10083 high confidence peaks.
Annotation
The peaks were annotated with candidate target genes with the assumption that the distance between the center of a binding peak and the transcription start site (TSS) of the gene was shorter than the threshold cut-off. We defined these “gene regions” as spanning from ∼ 5kb (4280 bp) upstream of the transcription initiation site to ∼1.0kb (1070 bp) downstream of the end of transcription. The peak files were annotated with Ensembl version 5.0 (Rnor_5.0) transcript genes using a 5000 base pair cut-off distance from the TSS using the Chip Peak Anno Bioconductor package in R52,53 and the characteristic genomic features were retrieved using COMPLETEMotifs54.
Digitized dynamic patterns representing the comparative binding evolution
To study the alterations in NF-κB binding as a result of adaptation to the ethanol diet, we created combinations of digitized dynamic patterns (COMPACT) between control and ethanol binding data. Each row and column of the COMPACT matrix consisted of the number of genes corresponding to a dynamic pattern. This matrix provided a gene-centric view of the relative presence of the targets in the dietary groups. These digitized patterns consisted of gene lists reflecting the comparative dynamics of the control and ethanol data over the time course. This enabled us to study the responses in an ordered manner and helped narrow down the most relevant gene sets corresponding to the major and minor response patterns in each dietary group at particular time points. The layered approach also aided in systematically exploring some of the subtle, but relevant combinatorial patterns.
We used a circular representation generated by CIRCOS v0.6755 to visualize the above dynamic correlation patterns. This representation helped us visualize the patterns by comparing similar responses in both dietary groups drawn symmetrically on both sides of the circle. The patterns were arranged in a circular diagram in such a way as to separate the baseline binding from post PHx binding and were represented by differently colored ribbons, with their width proportional to the number of genes. Thus, each row and column of the COMPACT matrix was represented as segments, and the size of the ribbon connecting them encoded the shared number of genes. Relative row, column and overall total of each segment were shown in outer circular patches.
Integration with expression data
Gene expression was determined by using Affymetrix GeneChip Rat Gene 1.0 ST Arrays (Affymetrix, Santa Clara, CA) at the Thomas Jefferson Nucleic Acid Core Facility. Briefly, 10 μg of each of the total RNA samples for the 6-h time point as well as their biological control LLM samples, for both ethanol and carbohydrate diets, as described above, were further purified with the Qiagen RNeasy Mini kit (Qiagen, Germantown, MD) followed by ethanol precipitation. RNA samples were analyzed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) to ensure quality prior to labelling and hybridization. Data were RMA normalized with the Affymetrix Expression Console (Affymetrix). Genes were considered expressed above background if they had a log2 normalized signal of 5 and above in at least one sample. MIAME (Minimum Information About a Microarray Experiment)-compliant microarray data were deposited in the GEO database, no. GSE3378556. Functionally significant targets of NF-κB were found by integrating the binding data for each of the patterns with the time series microarray gene expression data obtained from the liver samples (12 replicates per dietary group for 1h and 6hr post PHx, average fold change >=1.5, q value<=0.2) at 1 and 6h post PHx.
Functional association using GO Pathway Analysis
We identified the statistically significant pathways associated with the binding and expression patterns using DAVID v6.7 (Database for Annotation, Visualization and Integrated Discovery), a software package for biological pathways and functional annotations57. DAVID provides a comprehensive set of functional annotation tools to identify biological functions associated with long gene lists using gene ontology (GO) terms and other annotation sources. A clustering p-value cut-off of 0.05 was used to filter the pathway list to obtain the highly enriched functions.
Motif discovery
To identify the binding sites enriched by NF-κB, we used multiple de novo motif discovery programs. We used DME (Discriminating Motif Enumerator), a de novo motif discovery program based on an enumerative algorithm that identifies optimal motifs from a discrete space of matrices with a specific lower bound on information content. DME is suited to analyze very large data sets58. String lengths ranging from 9 to 14 nucleotides were used to scan for motifs. We used PAINT v4.159, which interfaces the TRANSFAC Pro® transcription factor binding site database and the associated MATCH® pattern matching tool60, to scan the promoter peak sequences and to find matches for profiles of transcription factors and retrieve potential TREs. We computed a sum of both error rates to find cut-offs that gave an optimal number of false positives and false negatives. We further used a clustering program STAMP to scan the discovered de novo motifs with the known TF binding database, which returned high scoring motifs and visualized the motifs as logos61. We predicted regulatory modules based on the co-occurring binding sites within the peak regions of NF-κB binding loci.
Results
Chronic ethanol intake alters the NF-κB genome-wide binding profile leading to several ethanol-specific targets
ChIP-chip NF-κB binding data obtained from liver samples (3-4 biological replicates per dietary group – Carbohydrate control diet and Ethanol diet) were quality filtered and aligned to the rat genome and statistically significant peak regions were identified. As a first step, we explored the binding changes and functional differences associated with the chronic ethanol intake alone. Our analysis revealed that introduction of ethanol diet did not result in a significant increase in the number of NF-κB binding loci (Fig. 1A). To elucidate the diet-specific changes, we separated NF-κB bound peaks in the adapted state into Novel (binding only in the Ethanol group), Common (binding in both dietary groups) and Missing (binding only in the Carbohydrate group) groups. Analyzing the diet-specific changes revealed that the ethanol adaptation caused a shift in NF-κB binding to a novel set of targets (Fig. 1A).
Fig. 1. Analysis of the NF-κB binding profile in the ethanol adapted state.

(A) Comparison of number of NF-κB binding targets between ethanol and control groups. (B) Statistically overrepresented functional annotation associated with the diet-specific and common target promoters. (C) The distribution of NF-κB binding sites relative to nearest transcription start site. Statistically overrepresented functional annotations associated with the peaks of the distribution in the ethanol (red) and control (green) groups are indicated. (D) Comparison of peak scores corresponding to the common NF-κB binding targets shows a relatively stronger binding in the ethanol group. (E) Differentially expressed genes in the ethanol adapted state, categorized according to the NF-κB binding profile.
We wondered if the Novel binding targets in the ethanol-adapted state were associated with specific cellular functions. For this, we analyzed the genes corresponding to the NF-κB target promoters for statistically over-represented biological functions within the three groups of Novel, Common and Missing loci (Fig. 1C) using DAVID v6.7 (Database for Annotation, Visualization and Integrated Discovery) with a p-value cut-off of 0.05. The gene set corresponding to the ethanol-specific Novel loci included pro-inflammatory cytokine receptor (Ifngr1), Tgf-β receptor (Tgfβ-r3) and Histone deacetylase 1 (Hdac1). Hdac1 interacts with p65 directly to serve as a repressor by de-acetylating p65, thus reducing the trans activating potential of p6562,63. The statistically over-represented biological functions modulated by NF-κB binding in the adapted state included G protein coupled receptor protein signalling pathways (195 genes), cell surface receptor linked signal induction (238 genes), detection of chemical stimulus (142 genes) and cell-cell signalling (43 genes), in addition to immune response, cell death, cell adhesion and regulation of protein kinase activity (Fig. 1C). Our results were consistent with previous studies that reported a NF-κB mediated inflammatory gene activation in ethanol-adapted liver8,9.
By contrast, the Common and Missing groups did not contain any binding loci corresponding to the genes in the above pathways pointing to the potential ethanol specific activation of NF-κB mediated pathways. Loci showing similar NF-κB binding activity between ethanol and control groups (Common group) were enriched for genes participating in mitochondrial functions, cellular response to stress, regulation of phosphate metabolic process, regulation of catabolic process and apoptosis. Even within the Common binding group of genes, the NF-κB binding activity shifted towards higher levels in the ethanol-adapted group compared to the pair-fed controls (Fig. 1D). We interpret this result as an ethanol-induced increase in the number of cells with significant NF-κB binding at these loci. The Missing group of genes, i.e., loci with ethanol-induced attenuation of NF-κB binding activity, was significantly over-represented for cellular macromolecular catabolic process, cellular response to stress, negative regulation of apoptosis and ion binding.
We analyzed whether NF-κB activity was preferentially localized relative to the transcription start sites (TSS). The distribution of putative NF-κB binding regions with respect to TSS of target genes showed that the peaks were enriched within 1000bp upstream/downstream of TSS (Fig. 1B). The distribution did not indicate a preferential bias for the upstream versus downstream binding of NF-κB (Fig. 1B). TSS binding studies on eukaryotic promoters have shown that focused promoters have well positioned nucleosomes compared to broad promoters that had fuzzy nucleosome and weak signals64. Another study on human cell lines that used a transient transfection approach to detect mutant transcriptional activity, showed that functional TF binding sites (FDR <0.025) tended to be closer to TSS (∼50bp) than those with unknown functions65. In our study, we analyzed the genes associated with NF-κB binding within a 200 bp window around the peaks of this distribution. Our results indicate that proximal promoter binding of NF-κB corresponds to functionally distinct classes of targets depending on whether NF-κB binding occurs upstream or downstream of TSS. The genes with downstream binding in the ethanol-adapted group participated in a broader category of processes such as detection of stimulus, cell projection and stress response genes, while those with upstream binding were associated with cell-cell signalling, receptor metabolic process, cell death, cell differentiation and regulation of kinase activity (Fig. 1B).
We investigated whether the observed differences in NF-κB binding were associated with differential gene expression in the ethanol-adapted state. The gene expression data was obtained from the Gene Expression Omnibus resource (GEO accession: GSE33785). A total of 40 genes with novel ethanol-only NF-κB binding showed differential expression in the ethanol group compared to that of the carbohydrate controls. Inspection of the top ranked genes obtained by integrating with the expression data revealed that the NF-κB bound genes with increased expression are associated with biological processes such as phosphorylation (Dapk1, Pdk4), regulation of protein kinase activity (Efna1, Sphk2, Trib3), oxidation reduction (Aldh1a1, Aldh1a7, Cyb5r3), and cellular membrane localized components (Cd14, Abhd1, Avpr1a, P2ry2, Slc2ba2). Some of these NF-κB targets have been discussed in the context of liver injury. For example, Trib3 activation is known to block the Akt pathway thereby inhibiting insulin responses in liver66. SphK2-/- mice rapidly developed fatty livers on a high fat diet67. Cd14 deficient mice showed reduced ethanol induced injury68. NF-κB bound genes whose expression was down regulated in the ethanol-adapted state were associated with regulation of apoptosis, negative regulation of biosynthetic process and ion binding (Bcl6, Aph1b, Eif2ak3, Herpud1, Zbtb16, Il6a).
We examined the expression changes of the NF-κB binding target genes common to the ethanol and control groups (Fig. 1E). A higher resolution version of Fig. 1E is shown in Fig. S3, ESI†. The genes Arnt1, Cabc1, Igfbp1, Ppm1k and Rnf125 showed increased expression with chronic ethanol intake whereas Erlin1, Ppplr3c, Ste2, Tob1 (Transducer Of Erbb2, anti-proliferative factors) and Xdhhc23 showed down regulation. We identified a set of genes with strongest binding and expression signals based on a more stringent threshold for the binding peak score (>0.8), FDR (< 0.015) and expression fold change (>=1.5) (Table S1, ESI†). These included genes with novel ethanol-only response (Cabc2, Igfbp1, Rnf124, PPp1r3c, Arnt1, Ptp4a1 and Tob1) and likely contribute to increased protein transport and translocation. Our result is consistent with the previous finding by other investigators that the acute-phase response caused by a hepatocyte-specific mutation of NF-κB p65 shows alteration of protein synthesis, transport and localization69.
Key genes that showed upregulation in the Missing group were Ces6, Cyp51, Sqle (involved in cellular membrane related functions), Gsta2, Gsta3 (xenobiotic metabolic process), JunD, Fzd4, Prkcdbp, Smp2a and Yc2 (Fig. 1E). We found promoters of core circadian clock proteins (Per2, Per3, Dbp) to be targets of NF-κB only in the carbohydrate control group. These genes were upregulated in the control group relative to the chronic ethanol-adapted state. Previous studies on mouse embryo fibroblasts revealed that NF-κB (RelB subunit) regulates circadian transcription by directly interacting with core clock factors Bmal1 and Clock that bind to promoters of clock controlled genes such as Dbp, Per1 and Cry70. Dbp, Per1 and Per2 were altered in livers from mice fed an ethanol-containing diet71.
Partial hepatectomy induced significant dynamic changes in the NF-κB promoter binding landscape
We examined the peak distribution of NF-κB post PHx, with respect to TSS (Fig. 2). Previous genome wide bindings studies reported shifts in promoter binding with respect to TSS. A shift in binding was reported for EKLF factor during erythrocyte differentiation72 and for genome-wide NF-κB/RelA binding for human pulmonary epithelial cells indicating a slightly higher peak enrichment for downstream binding of the TSS of target genes46. To investigate the change in peak distribution of the same genes over the time course, we analyzed the binding loci distribution of NF-κB targets that are persistently expressed at baseline and at 1h post PHx. The genes specifically targeted in the carbohydrate control diet (Missing in ethanol) showed significant asymmetry relative to TSS in their dynamic NF-κB binding. The peak location distribution showed a shift towards downstream binding by 1h post PHx (Fig. 2A). This was less evident in the Common group where we observed NF-κB binding peaks both upstream and downstream of TSS. However, the Common group also showed higher enrichment upstream of TSS at baseline and downstream at 1h post PHx (Fig. 2B). The ethanol diet-specific Novel NF-κB binding group showed almost symmetric upstream and downstream binding with respect to TSS at baseline as well as after PHx. (Fig. 2C).
Fig. 2. NF-κB binding peak distribution relative to TSS for genes with binding activity at baseline and at 1h post PHx.

(A) Ethanol-specific targets, i.e., Novel group, showed similar NF-κB binding distribution upstream and downstream of TSS. (B) Common targets in the carbohydrate and ethanol groups followed the same trend as the Missing set. (C) Carbohydrate-specific targets, i.e., Missing in the ethanol group, showed increased likelihood of NF-κB binding upstream to TSS in the adapted state, but shifted towards downstream of TSS at 1h post PHx.
We observed a surge in NF-κB bound regions at 1h post PHx compared to the baseline state followed by a decline in binding by 6h post PHx, with fewer binding loci in the ethanol group (Fig. S4, ESI†). We observed that at 1h post PHx, ethanol specific targets (3076) were much higher in number compared to the carbohydrate specific targets (1164) (Fig. S4A, ESI†). However, at 6h post PHx, this trend was reversed with the carbohydrate group containing a higher number of genes (1571), compared to the ethanol group (689) (Fig. S4, ESI†). Comparison between the time points (1h versus 6h post PHx) revealed that both dietary groups had higher number of genes with NF-κB binding at 1h compared to 6h post PHx (carbohydrate: 3176 versus 2003; ethanol: 5369 versus 1411) (Fig. S4B, ESI†). The ethanol group showed persistent 1h and 6h NF-κB binding at a smaller fraction of the loci compared to control (570 versus 860; Fig. S4B, ESI†).
Comparative pattern counting analysis revealed key dynamic patterns of NF-κB binding post PHx
We developed a novel unbiased approach, termed Comparative Pattern Counts (COMPACT) approach, to exhaustively evaluate the comparisons between the dietary groups across various time points. In this approach, we evaluated the differential binding at each time point relative to the appropriate, likely time point-specific, control conditions. The time series binding data was averaged across replicates within each sample group, and discretized into a binary value indicating presence or absence of binding based on the statistical significance. Within each set, the discretized time series binding data was collated for each gene into a pattern vector. The number of genes corresponding to each pattern within the Ethanol and Carbohydrate groups yields a univariate distribution for that dietary group. The intersection between the Ethanol versus Carbohydrate pattern count distributions exhaustively considers all possible comparative patterns, yielding a COMPACT matrix. This approach helps to exhaustively evaluate the genome wide binding effects of a comparative pair (ethanol versus carbohydrate) at multiple levels of factors (e.g.: time points). Genes showing a common response are represented by the diagonal of this matrix, while the off-diagonal elements correspond to altered binding response. Such an organization naturally divides the gene sets into novel, common and altered binding patterns between the dietary groups.
At each time point, for each dietary group, we discretized the NF-κB binding activity into a ‘1′ or a ‘0′ at each locus based on the statistical significance of binding. This yielded a total of 8 potential temporal binding patterns within each dietary group (2 discrete binding levels at each of the three time points = 2*2*2 = 8 patterns). We categorized the promoters as corresponding to these patterns and evaluated the corresponding genes for statistically significant functional associations (Fig. S5, ESI†). In the carbohydrate group, more loci (645 + 1605 + 2710 = 4960) showed binding post PHx, compared to baseline binding (1269 + 457 + 398 + 215 = 2339). Similarly, in the ethanol group, more loci showed binding post PHx (309 + 1145 + 4230 = 5684) than at the baseline (773 + 1139 + 266 + 261 = 2439). Our analysis also revealed that transient NF-κB binding at 1h was the dominant pattern in both the ethanol and control groups. However, the pattern-wise comparison between the dietary groups revealed that a significant difference in the number of promoter targets occurred in the transient 1h-binding pattern (ethanol: 4230 versus control: 2710), as well as the baseline and early 1h binding (ethanol: 1139 versus control: 457). However, the early and persistent group showed reduced number of target promoters in the ethanol group compared to the carbohydrate group (ethanol: 309 versus control: 645). This lower number of binding loci in the ethanol group was also evident at 6h post PHx (ethanol: 1145 versus control: 1605). These results suggest that chronic ethanol intake induced an overall higher activity of NF-κB binding at 1h but not by 6h post PHx (Fig. S5, ESI†). Such a dramatic ethanol-dependent alteration in the dynamic NF-κB binding post PHx indicates that NF-κB may regulate novel functions following the combined stress of ethanol adaptation and PHx.
We used the COMPACT approach to understand the detailed differences in the dynamic NF-κB binding patterns between ethanol and control groups. We exhaustively evaluated all the possible combinations of binding patterns between control and ethanol groups and categorized the promoters accordingly. We represented the number of promoters corresponding to each of the 64 combinational patterns (8 patterns X 2 dietary groups) (COMPACT) matrix (Fig. 3A). The element at the ith row and jth column of the 8x8 matrix contains the number of genes that show ith-binding pattern in Carbohydrate and jth binding pattern in Ethanol. We computed the COMPACT matrices for various FDR threshold values and found that the relative rank of the dominant and subtle patterns were consistent across different FDR threshold values (Fig. S2, ESI†). We chose the results corresponding to FDR < 0.15 in subsequent analysis. This data-driven unbiased approach enabled us to uncover dominant as well as subtle differences in NF-κB binding between ethanol and control groups.
Fig. 3. Comparison of dynamic NF-κB binding patterns between the ethanol and carbohydrate control groups.

(A) COMPACT matrix showing the number of NF-κB binding targets for each combination of dynamic binding patterns between the ethanol and carbohydrate groups. The element at the ith row and jth column of the COMPACT matrix contains the number of genes that show ith NF-κB binding pattern in carbohydrate group and jth NF-κB binding pattern in ethanol group. The diagonal of the matrix represents those genes showing a common NF-κB binding response and the off-diagonal elements represent the genes showing an altered temporal NF-κB binding response between the dietary groups. The matrix was partitioned such that the patterns representing “no NF-κB binding” and “baseline NF-κB binding” separate the NF-κB binding in response to PHx. (B) A chord diagram visualization of the COMPACT matrix in A. The length of the arcs is proportional to the number of genes showing the corresponding NF-κB binding pattern. The width of the ribbons connecting the arcs corresponds to the comparative pattern count, i.e., the number of genes showing the corresponding NF-κB binding patterns in the two dietary groups. The arcs are arranged such that the “no NF-κB binding” pattern separates the patterns with and without NF-κB binding at baseline (lower and upper segements, respectively). (C-F) Select patterns are highlighted in the chord diagram to illustrate the visual analysis approach – NF-κB binding occurring only at 6h post PHx (C), only 1hr post PHx (D), at baseline (E), and at baseline and 1hr post PHx (F).
We visualized the COMPACT matrix in such a way that the null pattern representing “No binding” broadly separates the baseline binding from absence of baseline binding (Fig. 3A). Partitioning the matrix based on “no binding” and baseline-binding patterns provides a systematic hierarchical view of the patterns. The matrix revealed a significant number of genes in the quadrant representing absence of baseline binding. All the elements except one of this quadrant were distributed with binding patterns containing more than 40 genes. The significantly higher number of genes participating in binding post PHx with no baseline binding (6227 genes out of 10083 total) showed that irrespective of the diet, PHx turned on the binding of a new set of NF-κB target genes. It was interesting to note that majority of them (sum of off-diagonal elements was 4112 genes) showed ethanol-altered response. The largest contributions of all the patterns correspond to no binding (2784 in Carbohydrate and 2000 in Ethanol) and transient binding (2710 in Carbohydrate and 4230 in Ethanol). The COMPACT matrix also revealed significant contributions from only baseline binding group (1269 in Carbohydrate Vs. 733 in Ethanol).
We found that the most dominant comparative pattern contained 1720 promoters showing transient 1h binding in ethanol group and no binding in the control. The next largest comparative pattern contained 1478 promoters showing similarly transient 1h binding between ethanol and control groups. We wanted to highlight different aspects of these results, particularly in interpreting the number of promoters in each of comparative patterns in terms of their corresponding proportions of targets in ethanol and control groups. For this, we visualized the COMPACT matrix using a circular chord diagram that has been typically employed to compare genome sequence synteny (Fig. 3B). In this visual scheme, each arc represents a temporal binding pattern, with the size of the arc corresponding to the number of promoters. The arcs are connected by chords corresponding to the comparative patterns (elements of the COMPACT matrix), with the width of the chords based on the corresponding number of promoters. Such a visual representation readily demonstrated that within each of the temporal binding patterns, NF-κB binding responses were characterized by various combinatorial patterns, such as no binding, baseline, 1hr transient, baseline and early, baseline and late, early and persistent and late binding between the dietary groups. However, the majority of the response was characterized by Novel to ethanol group, Common to both dietary groups, and Missing in the ethanol group. For instance, highlighting the arc corresponding to the 1h transient binding pattern with 4230 promoters in ethanol showed that a majority of this pattern was spanned by the chords connected to no binding in control (Novel: 1720 promoters) and transient 1h binding (Common: 1478 promoters) in the control (Fig. 3C). A similar trend was observed when highlighting the arc corresponding to 1h transient binding in the control group. The largest set corresponded to transient 1h binding in ethanol and control groups, followed by a set connected to no binding in the ethanol group (Missing: 754 promoters) (Fig. 3C). We observed a similar underlying organization based on the Novel-Common-Missing scheme, and also uncovered ethanol-induced temporal shifts for the other binding activity patterns (Fig. 3D-F). An analysis of these subgroups is presented in the following section.
NF-κB transient binding showed higher sensitivity to combined stimuli of ethanol and PHx
The major dynamic effects of ethanol intake followed by PHx on NF-κB during the priming phase happened at 1hr post PHx as indicated by largest number of promoters, (1720 targets) in the Novel (ethanol specific) group (Fig. 4A). Within the early transient binding profiles, promoters with Common (1478) and Missing (754) binding in ethanol group were the next two predominant fractions of the overall total number of binding targets. We were interested in identifying the extent to which the alteration in transient binding translates to potential regulatory mechanisms associated with a new cellular state. We used Gene Ontology based Pathway Enrichment Analysis to identify the statistically over-represented biological functions and annotations associated with the target genes in the Novel, Missing and Common groups (Fig. 4B). Most of the genes associated with apoptosis did not show NF-κB binding in the adapted state, but showed a transient NF-κB binding response in the ethanol group after PHx. However, the similar number of positive (42) and negative (41) regulators of cell death indicated the presence of active compensatory mechanisms regulating apoptosis, post PHx.
Fig. 4. Analysis of transient NF-κB binding dynamics at 1h post PHx.

(A) COMPACT matrix highlighting the elements corresponding to transient binding in either dietary group. (B) Statistically overrepresented Gene Ontology terms for the ethanol-specific (Novel), common, and carbohydrate-specific (Missing in ethanol) 1h transient NF-κB binding groups. (C) Gene Set Enrichment Analysis comparing Novel (red) against Missing (green), illustrating distinct pathways and biological processes between these subgroups (p <= 0.05, q-value <= 0.05, overlap coefficient >= 0.3). Visual representation was generated using the Cytoscape software for network analysis. In this visual, the enriched functional annotations are represented as nodes. The size of the nodes is proportional to the relative number of genes associated with the corresponding annotation in the Novel or Missing groups. Edge thickness is proportional to the fraction of genes that overlap between two connected nodes.
A large set of genes (754) did not show NF-κB binding in ethanol group (Missing), but showed transient NF-κB binding in the control group. Associated over-represented pathways included homeostatic process, calcium signalling pathways, ion transport and regulation of cell death. As in the case of Novel group, the Missing set also comprised of the same number (∼18) of pro-apoptotic (e.g., Cdk4, Dedd, Cidec) and anti-apoptotic (E.g.: Thy1, Cx3cr1, Vegfa) targets. Key genes belonging to this category included Nod2 (an intracellular pattern recognition receptor that can induce NF-κB activation), Ccl11 (a cytokine involved in the response to wounding), Klf4, Tnf and Nr4a1 (intracellular receptors expressed in macrophages) and Crem, Csad, Gata1, and Smad7 (other regulators of signalling and transcription) (Fig. 4A).
We performed gene set enrichment analysis using the Enrichment Map tool73 to characterize and visualize pathways statistically over-represented in the gene sets, identified from the COMPACT analysis. The gene set enrichment map separated Novel and Missing groups into minimally overlapping sets of functions (Fig. 4C). Cell cycle (67 genes), apoptosis (50), regulation of cell death (78), RNA metabolic process (129) and response to oxidative stress (32) were enriched in the Novel group, whereas the immune response (27), homeostatic process (44) and circadian regulation (5) were enriched in the Missing group, revealing that distinct pathways may be targeted by NF-κB in the ethanol versus control groups in response to PHx.
The large set of NF-κB target genes (1478) with transient binding activity at 1h post PHx in both control and ethanol groups (Common group) were associated with several biological functions and processes including cell proliferation, positive regulation (43) and negative regulation (31), mitochondrial function, protein complex assembly, MAPK signalling pathway, response to nutrient, protein biosynthesis, RNA processing, lipid catabolic process, and glucose metabolic process. This set included a few key liver-associated genes such as Cebpb, Hes1, Egr1, Smad3, Foxm1, Ccrn4l, Npas2, Zpf36, Jun, Junb, Btg2, Pim3, Gata4 and Per1 (Fig. 4A), most of which are involved in regulation of transcription and biosynthetic processes.
Our COMPACT analysis highlighted other early transient patterns in the ethanol group including those that showed later binding at 6h post PHx in control group (163 genes). This set consisted of 68 cell cycle genes and 16 homeostatic process related genes, indicating that NF-κB may be targeting these genes in the ethanol group during the earlier phase of the response to PHx than in the control group. The set of genes (348) that were delayed by ethanol intake were associated with ion binding (63), cell cycle, apoptosis (24), negative regulation of cell death (12), cellular response to stress (15) and regulation of transcription (42).
Chronic ethanol intake caused a decline in late 6h binding compared to 1hr post PHx. At 6h post PHx, pair-fed carbohydrate samples showed higher NF-κB promoter binding (1605 genes), compared to ethanol (1145 genes) out of which 567 genes were shared between the dietary groups (Fig. 3A). The “Novel” late binding group consisted of 330 genes. Ethanol intake resulted in larger number of NF-κB targets showing late and persistent binding post PHx. The Novel patterns with a large number of genes (49 genes) did not show any significant functional associations in the pathway enrichment analysis. A persistent binding over time was observed for similar number of genes (261 versus 215). The “Missing” sets identified in the COMPACT analysis represent the promoters targeted only in the control group after PHx (Fig. 3A). Of these genes, a significant subset showed late binding (541) or persistent binding (115). However, these genes were associated with pathways/functions such as detection of chemical stimulus, olfactory signals, sensory perception and GPCR signaling, making it difficult to interpret these findings, as nearly all of these genes were not expressed in the liver to a significant degree (Fig. S6, ESI†). Our previous studies on genome-wide NF-κB binding in rat liver showed that NF-κB binds to many genes within the functional category sensory perception of smell, even prior to hepatectomy39. We speculated that these signalling pathways, might act as chemical sensors to modify liver behavior. This may also serve as a sink for NF-κB binding to prevent spurious hepatocyte priming but are possibly made unavailable for NF-κB binding following a sustained challenge such as partial hepatectomy. Recent studies in mice showed olfactory receptors in kidneys may act as chemical sensors that can modulate glomerular filtration rate74. It is not yet clear whether these receptors play a functional role during liver regeneration.
Fig. 5 shows a comprehensive summary of the pathway analysis of the dynamic patterns with the distribution of functional associations of the targets across dietary groups and time points after PHx. One caveat, shared by nearly all of the focused as well as genome-wide binding activity studies, is the difficulty in interpreting the functional consequences of the observed binding activity differences. In the next section, we tried to relate the COMPACT-highlighted gene sets to the differential gene expression patterns.
Fig. 5.

Number of genes associated with the statistically overrepresented functional annotations in various COMPACT highlighted NF-κB binding target gene groups.
Integration of NF-κB binding with expression data reveals strong correlation between aberrant transient binding and differential expression at 6h post PHx
In order to gain better functional understanding of the observed changes in NF-κB promoter binding, we investigated the expression changes in the COMPACT-highlighted NF-κB binding targets. Functionally significant targets of NF-κB were found by integrating the binding profile with the microarray gene expression data obtained from the liver samples (GEO accession GSE33785)56. We discretized the differential gene expression data at each of the 1h and 6h time points as 1 or 0, based on statistically significant differential regulation (q-value<=0.2, average fold change <= 1.5) or no change. We organized the genes based on a set of integrated patterns that combined the binding activity patterns at 1 and 6h together with the differential expression patterns at these time points, yielding a total of 16 integrated patterns for each dietary group (Fig. 6A). We utilized the COMPACT analysis to compare the integrated binding and expression profiles between ethanol and control groups (Fig. 6A). We also employed a circular matrix visualization of the COMPACT matrix for a visual approach that highlights relative sizes of gene sets more readily (Fig. 6B). We organized the integrated binding and expression patterns into three groups: Group (a) showed correlation leading to similar temporal patterns between binding and expression at 1h and 6h post PHx. Group (b) has overlapping temporary profiles between binding and expression, and Group (c) corresponded to genes showing expression changes without NF-κB binding or showing NF-κB binding without any differential gene expression.
Fig. 6. Comparative pattern count analysis of the integrated NF-κB binding and gene expression data.
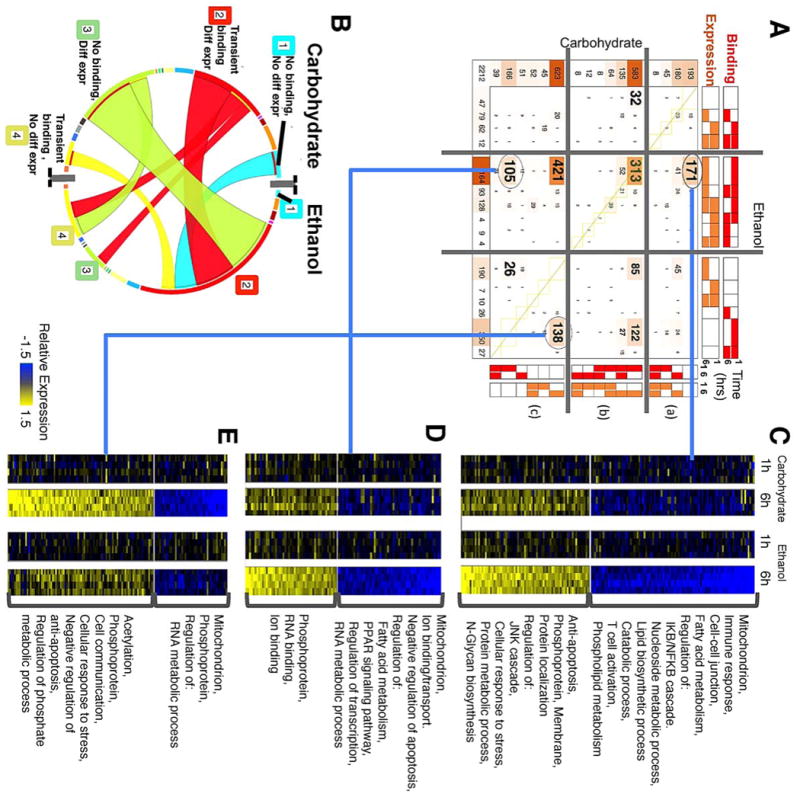
(A) COMPACT matrix for combined patterns of NF-κB promoter binding and differential gene expression. The matrix is separated into segments based on whether NF-κB binding and differential expression follow the same temporal pattern, occur within the 6h time frame post PHx, or are mutually exclusive, within each dietary group. In the resultant 9 segments, a few dominant patterns with relatively high number of genes are highlighted. (B) A chord diagram visualization of the patterns highlighted in A. (C-E) Differential gene expression profiles corresponding to select comparative patterns in A. (C) novel ethanol-specific binding at 1h and novel ethanol-specific differential expression, (D) common diet-independent binding at 1h and novel ethanol-specific differential expression, and (E) novel ethanol-specific binding at 1h and missing (i.e., carbohydrate-specific) differential expression, at 6h post PHx.
COMPACT analysis highlighted a small set of integrated patterns containing a large number of genes (Fig. 6A). Most of the Novel 1h transient NF-κB targets in the ethanol group showed differential gene expression at 6h post PHx (Fig. 6A, B). The largest patterns in each dietary group corresponded to “Transient binding at 1h along with differential expression at 6h”, “Transient binding at 1h with no differential expression”, and “Differential expression at 6h with no binding activity” (longest arcs in Fig. 6B). Within the COMPACT matrix, the dominant sets corresponded to seven of the nine comparative patterns formed by intersecting the above three largest patterns from each dietary group. We interpret these temporally shifted correlations as indicating that NF-κB binding at 1h likely leads to differential expression of the target genes by 6h post PHx. A set of 421 genes showed differential expression at 6h in ethanol and control groups, but with NF-κB binding occurring only in the ethanol group at 1h. Another set of 313 genes showed similar 1h NF-κB binding and 6h differential expression between ethanol and control groups (Mitochondrion, acetylation, lipid biosynthetic process, cholesterol biosynthesis, oxyreductase, methyltransferase). Also of interest were patterns with ethanol-specific NF-κB binding and differential expression (171 genes, Fig. 6C), ethanol-specific NF-κB binding without differential expression and no NF-κB binding with differential expression in the control (138 genes, Fig. 6E), similar NF-κB binding between ethanol and control groups but differential expression only in ethanol group (105 genes, Fig. 6D). These sets correspond to cases where NF-κB binding likely has dietary group-specific effects on differential expression of the binding targets.
(I) Correlation of differential gene expression with transient novel binding revealed opposing regulation of apoptotic and anti-apoptotic signals
Integrating the NF-κB binding with gene expression data revealed that 171 genes showed novel differential expression in the ethanol group at 6h post PHx (96 down regulated genes, and 75 upregulated genes) (Fig. 6C). As opposed to the ethanol adapted state, the transient state turns off relatively few pathways and turns on multiple pathways. The upregulated genes were associated with anti-apoptosis, regulation of JNK cascade, apoptosis, cellular response to stress and the down regulated genes were associated with mitochondrial functions, immune response, fatty acid metabolism and cell-cell junction. GO pathway analysis suggests the upregulated expression of a small set of anti-apoptotic genes possibly as a result of stress response induced due to the chronic ethanol diet indicating a compensatory mechanism mediated by NF-κB activation. DAVID pathway analysis of the novel set of genes emphasized the response triggered by NF-κB binding targeting members of key signalling pathways such as oxidative stress response, xenobiotic pathway, p53/PTEN signalling, immune response, GPCR signalling etc.
NF-κB appears to switch its role from a pro-apoptotic to an anti-apoptotic function. Such a shift was supported by the Gene Set Enrichment Analysis visualizing the over-represented pathways shared between the differentially bound NF-κB target genes (Fig. S7, ESI†). The overlapping pathway annotation network enabled us to distinguish the distinct pathways associated with upregulated and down regulated genes. Some of the down regulated genes were previously established NF-κB targets such as Blc1075, Keap176, Ndflp177 and Map4k3. The down regulated genes were enriched for fatty acid metabolism, oxidation, and mitochondrion network and may point to mediation of oxidative stress in the hepatocytes, driven by ethanol, post PHx. Genes with upregulated expression were found to be involved in protein transport, RNA processing and protein kinase cascade. These were relatively broader responses indicating the higher metabolic and immune response driven by activation of NF-κB. These ethanol specific genes appeared to balance the increase in oxidative stress contributing to the metabolic load with activation of genes that maintain tissue functions during regeneration.
(II) A set of 105 genes (of which 49 were upregulated and 56 were down regulated), that showed novel differential expression in the ethanol group, were found to have commonly NF-κB binding in both dietary groups at 1h (Fig. 6D). These genes were bound by NF-κB at 1h and remained bound irrespective of the dietary change indicating they are essential for maintaining tissue function after acute damage. Genes whose expression was down regulated were associated with fatty acid metabolism, PPAR signalling, regulation of transcription and RNA metabolic process while the upregulated genes are known to modulate phosphoprotein and RNA binding pathways.
(III) Among the novel bound genes, a group of 138 genes showed anti-correlated behavior between binding and expression (Fig. 6E). It is possible that NF-κB binding leads to suppression of differential regulation of these target genes. Within this set, 92 genes showed upregulation post PHx in the control, and were associated with acetylation, cellular response to stress, apoptosis, cell communication, etc. The down regulated genes (46 genes) were associated with mitochondrion related functions such as oxidation-reduction, oxidative phosphorylation or ATP binding and RNA metabolic processes. If these changes reflect a reduction in ATP and GSH levels, they can drive signalling pathways that mediate anti-proliferative effects leading to a delay in regeneration.
Integration with expression data revealed that, while there were significant changes in NF-κB binding activity due to ethanol, the functional consequences on differential gene expression might also be dietary group specific, shaping the system response to PHx. Several genes associated with apoptosis showed baseline NF-κB binding in control, but showed a post PHx NF-κB binding response in the ethanol group. The majority of these were transiently bound at 1h, while some showed persistent binding after PHx. A relatively high number of anti-apoptotic genes showed binding at 1h post PHx and upregulated expression at 6h in the ethanol group. This suggests that in the ethanol-induced liver, by the end of the priming phase, NF-κB begins to drive increased production of metabolic and proliferative genes required for hepatocyte replication. These results indicated an ethanol-altered stress response dynamics, potentially mediated through changes in promoter localization of NF-κB. Our results suggest an intriguing idea that part of ethanol-altered NF-κB response acts as a compensatory process triggered after PHx.
Identification of potential co-regulatory modules containing NF-κB post PHx
Specificity of transcriptional regulation is determined by the organization of regulatory elements distributed along the genome and their interaction with NF-κB. Rel/NF-κB binds to 9-10 base pair DNA sites as dimers (with the general consensus sequence: GGGRNNYYCC)78. We performed an exhaustive search for enriched de novo motifs on the genomic locations corresponding to the dynamic patterns of genome-wide NF-κB promoter binding in control and ethanol data, using motif discovery and analysis tools MEME79 and DME58. We also employed PAINT 4.159, as a convenient interface to the TRANSFAC Pro transcription factor binding site database and the associated MATCH pattern matching tool60 to scan the promoter peak sequences and to find matches for profiles of transcription factors and retrieve potential TREs. We computed a sum of both error rates to find cut-offs that gave an optimal number of false positives and false negatives. To quantify the enrichment, total coverage was calculated for uniquely enriched sequences from both DME and PAINT for each of the dynamic patterns independently. Most of the dynamic patterns had more than, or equal to 75% of their target loci containing canonical NF-κB binding sites (Fig. 7A). Overall, we found more NF-κB enriched sequences in the ethanol group than in the control. Interestingly, sequences corresponding to the major comparative groups Novel, Common and Missing at 1h post PHx did not show very high NF-κB occupancy.
Fig. 7. Bioinformatics analysis of transcription factor binding sites within the peak NF-κB binding loci.

(A) Coverage of NF-κB binding across the comparative patterns in Fig. 3A, showing mostly high fraction of genes with TRANSFAC-predicted NF-κB binding sites, except in the case of baseline and 1h transient binding patterns. (B) Network containing transcription factors corresponding to top ranked motifs predicted using DME, and their targets in Novel, Common and Missing patterns corresponding to the 1h transient NF-κB binding groups in Fig. 4B. (C) The percentage of loci with the indicated TF binding site modules calculated after an exhaustive search for transcription factor binding motifs using DME and PAINT.
NF-κB is known to synergistically interact with other factors to co-ordinately regulate transcription. Sets of such factors that co-occur in the top ranked sequences likely contribute to transcriptional regulatory modules. NF-κB p65 interaction with adjacent cis-regulatory modules partially contributes to its stimulus-specific binding. We used PAINT software to analyze the promoter sequences of various gene sets from our COMPACT analysis and performed an unbiased analysis of binding sites for other transcription factors that may be enriched nearby the NF-κB binding peaks (Table S2, ESI†). We carried out an exhaustive analysis using hierarchical clustering to identify the candidate cofactors for each of the COMPACT-highlighted patterns, using DME and PAINT. The dynamic patterns were significantly enriched for binding sites of factors STAT3, AP-1, HNF4, GATA, C/EBPβ, CREB and EGR-1. We retrieved the putatively co-regulated genes with promoters containing STAT, AP-1, C/EBP and CREB binding sites, and calculated the corresponding statistical enrichments in the Novel, Common and Missing 1h transient NF-κB binding patterns (Fig. 7B). The fraction of promoters with these modules did not significantly vary between the Novel, Common and Missing comparative patterns. Our results revealed that a large majority (∼97%) of the NF-κB targets are likely under combinatorial regulation by three or more transcription factors considered here. Coordinated regulation of NF-κB with other transcription factors has been previously investigated in the context of liver regeneration. For example, the interaction between STAT3 and NF-κB pathways in the liver inflammatory response has been described80. In our study, STAT3 binding sites occurred in nearly all of the NF-κB target loci that showed a transient increase in NF-κB binding at 1h post PHx in either dietary group (Common). NF-κB activity at some target genes was limited by direct interaction of p65 with the CREB81. In contrast to the STAT motifs, CREB binding sites were only present in 19-23% of the NF-κB target loci at 1h. AP-1 family transcription factors play an important role along with NF-κB in regulating the priming for liver regeneration. Coordinated regulation of NF-κB with C/EBP has been predicted during the priming phase of liver regeneration82. Our results predicted that nearly half the target loci containing AP-1 and C/EBP binding sites were within the peak regions of NF-κB binding.
We conducted a search using DME to retrieve the top ranked motifs in Novel, Common and Missing 1h transient NF-κB binding patterns (Fig. 7C). All three of the dynamic patterns were enriched for motifs such as HSF, LF-A1, SMAD4 and NRF-2. HSF is known to prevent the inhibition of NF-κB binding post acute stress83. Our results indicated the existence of a positive feedback loop between NF-κB and HSF. Crosstalk between NF-κB and NRF-2 that suppressed NF-κB activation had been observed in early stages of cancer84. We found some motifs (TGIF, HNF-4, RAP-1 and SREBP-1) to be enriched in the Novel as well as the Missing groups. One mechanism by which ethanol may disrupt the balance of lipid metabolism is through increased activity of the transcription factor SREBP-185. A few motifs (SMAD-3, AP-2, TFII-1 and LXR) showed overlap between Common and Missing groups. A different set of motifs (RBP-Jκ, p53, EGR-3 and ISGF-3) was enriched for Novel and Common groups. C/EBP-α, IRF-α and PPAR-γ were enriched only in the promoters with Novel NF-κB binding in the ethanol group, whereas SRF and LBP-1 were found to be enriched only in the promoters with NF-κB binding uniquely occurring in the carbohydrate group (Fig. S8, ESI†).
Discussion
By performing a time course study using ChIP-chip experiments, we analyzed the in-vivo NF-κB p65-DNA interactions in the whole liver tissue to identify the potential targets of NF-κB across dietary and time conditions. Our primary goal was to investigate how the perturbations due to ethanol intake affected NF-κB binding in the liver post PHx. Chronic ethanol diet shifted the global binding profile of NF-κB, but did not have any effect on the total number of the targets compared to carbohydrate diet. However, the combined effect of ethanol diet followed by PHx significantly altered the NF-κB binding landscape. We found an overall shift towards downstream in the NF-κB peak distribution with respect to TSS, post PHx. This novel finding suggests that the perturbation due to PHx can potentially induce global level alterations in genome-wide binding. COMPACT analysis showed an increase in immediate early NF-κB binding at 1h post PHx followed by a decrease at 6h post PHx. Gene expression integration revealed that a majority of the 1h binding targets were differentially expressed at 6h post PHx. Pathway analysis associated these genes with key biological functions such as stress response, apoptosis, cell proliferation, cell cycle and metabolic processes. Motif analysis identified potential co-incident transcription factor binding motifs such as those corresponding to STAT-3, CREB, AP-1 and C/EBP.
The shift in NF-κB binding relative to TSS points to the potential role of distinct regulatory elements depending on the genomic locus and the experimental condition. Conventionally, functional transcription factor binding sites for cis-regulation have been identified within the proximal upstream promoters as well as within the first exon and intron downstream to TSS. However, it is not straightforward to differentiate the functional effects solely based on the upstream versus downstream binding activity. Consistent with this expectation, in our analysis, we did not find up- and down-regulation of gene expression to preferentially correlate with upstream versus downstream binding. Our results revealed that even though the overall number of binding targets and their distribution with respect to TSS are similar between ethanol-adapted and control groups, their functional roles are significantly different. Chronic ethanol intake was found to activate some of the initial damage response pathways mediated by NF-κB. However, the results indicate that the 5-week Lieber-DeCarli ethanol feeding model may not lead to significant inflammatory responses in the liver driven by NF-κB induced pathways in the absence of other pro-inflammatory stimuli, in agreement with the extensive body of literature on this chronic ethanol feeding model.
In order to analyze the time series binding data, we developed a novel pattern analysis approach, termed COMPACT analysis, where we examined the combinations of NF-κB binding targets across the dietary and time conditions. In addition to identifying the most significant dynamic patterns, this approach helped us unravel subtle, yet functionally significant groups of genes. COMPACT analysis enabled us to derive better clues to the underlying regulatory changes mediated by chronic ethanol intake by partitioning the ethanol specific effects that act along particular pathways. We interpret our results as likely evidence for similarity of regulatory processes that drive similar extent of the binding response leading to similar regulation of cellular functions.
Previous studies showed activation of NF-κB signalling with ethanol intake8,86,87. We wondered whether the absence of a significant alteration in the binding after long-term adaptation to the ethanol diet was due to the activation of damage controlling compensatory mechanisms making the system less sensitive to perturbations. To investigate the differential gene expression associated with alterations in NF-κB binding, we combined ChIP-chip data with microarray gene expression data. Functional analysis of NF-κB bound and differentially expressed genes indicated that our results are consistent with the observed role of NF-κB as a stress signal response factor. A comparative analysis of the dietary groups suggested that NF-κB could play a compensatory functional role mediating the initial damage response pathways in the ethanol-adapted state compared to that of the controls.
Primary analysis of NF-κB bound regions and the novel repertoire of target genes revealed that the combined effects of chronic ethanol intake and PHx alter the binding dynamics of NF-κB significantly. The significant increase in transient NF-κB occupancy post PHx in the ethanol-adapted state suggested the possibility of a change in role of this early transcription factor was a result of acute perturbation. The alteration in NF-κB binding could be correlated with aberrant gene regulatory response in liver post PHx. This can result from the intrinsic cytoprotective response to prevent the progression of irreversible tissue damage or to help cellular proliferation after PHx. Such preconditioning of liver against subsequent ischemia-reperfusion (IR) injury is also known to increase NF-κB activity in mice88. The drastic reduction in NF-κB binding at 6h post PHx suggests that the increase in binding at 1h post PHx could be a transient compensatory behavior as an immediate early response to PHx. Several studies investigated the importance of the rapid induction of NF-κB after PHx 20,89,90. There is a robust induction of NF-κB within the first hour post PHx, playing a distinct role in different liver cell types. One of the early studies on NF-κB activation post PHx in female Fischer rat reported rapid activation followed by a decline of NF-κB p65 activity by 1h post PHx12. In the male Sprague-Dawley rat, the rise of NF-κB activity at 1h post PHx and then a fall to near baseline levels at 4h post PHx, followed by an increased activity at 6h post PHx indicated the dynamic role of NF-κB during the initial phase of regeneration34. DNA binding activity of NF-κB was shown to follow a similar dynamic profile after CCl4 exposure11. However these studies did not directly test the functional role of NF-κB induction in the liver regeneration process. Hepatocyte-specific deletion of inhibitor-of-kappaB-kinase-2 (IKK2) was shown to promote proliferation in the regenerating liver16. Other hepatocyte-specific NF-κB inhibition or knock out studies showed that liver mass recovery a day after partial hepatectomy was unaffected, potentially due to activation of compensatory pathways such as JNK and STAT391,92. In mice, Kupffer cell depletion led to diminished induction of NF-κB activity at 3 days post-PHx, leading to impaired regeneration93. However, none of these studies tested acute inhibition of NF-κB activity during the immediate early priming phase post-PHx. It is known that chronic ethanol adaption inhibits the acute induction of NF-κB post-PHx29, possibly in a cell type-specific manner with hepatocytes contributing to overall inhibition47. Our results on the ethanol-induced shift in the NF-κB genome-wide binding landscape during the immediate early phase post-PHx suggest a system-wide rewiring of NF-κB transcriptional regulatory network and are consistent with the expected effects of attenuated NF-κB induction on liver regeneration. It remains to be tested if other transcriptional regulators also show such an ethanol-induced shift in the genome-wide landscape during the immediate early phase response.
Our comparative pattern analysis revealed the novel ethanol group at 1h post PHx as the most occupied pattern indicating that it was the combined perturbation that caused the drastic increase in NF-κB binding. This unique approach helped us to unravel some of the other important sub groups of NF-κB binding patterns such as 6h binding only in carbohydrate diet, baseline binding only in ethanol diet, baseline and 1h binding only in ethanol diet. Integration with expression data revealed that most of the NF-κB target genes that showed transient binding at 1hr post PHx were differentially expressed at 6hr post PHx. Transcription factor binding at promoters can occur much earlier than changes observed in target gene expression. The 6h differential expression of the ethanol-specific NF-κB binding target genes suggests a delayed/persistent transcriptional regulation of these targets bound at 1h. Another possibility for the reduction in binding at 6h was the existence of a feedback mechanism triggered around 6h post PHx. An extensive time series analysis is required to characterize the precise correlation between binding and gene expression during the immediate early priming phase of liver regeneration.
We used GO pathway analysis to find the overrepresented biological processes and molecular functions for the differentially bound and expressed genes. Our aim was to make predictions of pathways potentially activated as a result of the dynamic adaptation of the liver due to changes in the environment, such as the log-term adaptation to ethanol. Functional analysis of transient NF-κB binding indicated that chronic ethanol consumption may be affecting the regenerative stimulus, leading to modulation of hepatocyte priming by regulating cell cycle entry and hence proliferative responses. In the normal liver, the vast majority of hepatocytes are arrested in the G0 phase of the cell cycle. Hepatocytes require stimulation by hormones and growth factors to traverse G1 and begin cellular replication. A previous study using isolated rat hepatocytes showed that inclusion of ethanol to the growth medium caused inhibition of hormone-stimulated DNA synthesis to reduce the proliferative response94. These results indicated that the signals required to induce completion of G1 were also affected by ethanol. Our results showed that most of the cell proliferation genes were associated with transient upregulation in both control and ethanol indicating that this pathway was not significantly affected by ethanol exposure. However, we found a number of targets associated with cellular replication as well as cell cycle related genes in chronic ethanol-fed rat liver samples compared to carbohydrate pair-fed samples. NF-κB appeared to switch its role from pro-apoptotic (transient) to anti-apoptotic (delayed). Additionally, we found NF-κB targets to be the participants in processes like histone modification, homeostasis and biosynthetic and metabolic processes post PHx. We understand that our binding data alone is insufficient to accurately predict the downstream apoptotic behavior of the regenerating liver. However, these pathways, particularly those governed by the immediate and transient binding give some clues as to how chronic adaptation to stresses, such as chronic alcohol consumption, may lead to disrupted liver regeneration. Chronic alcohol consumption impairs liver regeneration by suppressing the number of hepatocytes that enter the cell cycle following PHx27,28. Our results suggested a possibility that one of the combinatorial mechanisms through which chronic alcohol consumption impairs liver regeneration, was by reducing the factors available for hepatocyte entry into the cell cycle. This may be followed by activation of other non-parenchymal cells leading to increased extracellular matrix deposition causing increased growth factor sequestration and further limit the number of hepatocytes entering the cell cycle.
We found that irrespective of the dietary and time conditions; NF-κB occupied a large number of binding sites in the promoter region of the rat liver genome. This putative genome-wide targeting remained even after stringent statistical thresholds to minimize the expected false positive rate. However, such an extensive occupancy, while detected at statistically significant levels, may not necessarily translate into genome-wide functional activity in regulating the target gene expression. Moreover, binding data is insufficient to differentiate between activation and inhibition of the targets. We considered this issue by filtering the p65 target sets based on differential gene regulation data. The expression based filtering reduced the number of targets considerably in the adapted state as well as post PHx and identified NF-κB binding targets that showed increase or decrease in expression. An intriguing interpretation of the observed extensive p65 binding was that NF-κB could not only act as an activator/repressor regulating transcription, but could possibly also serve as a ‘pioneer’ factor binding at a number of genomic loci or potentially interact with other known pioneer factors in liver such as FOXA1 or GATA. This can in turn contribute to nuclear re-organization and modify acute responses in the chronic adapted state. A similar scenario was discussed in an earlier work by Yang et al. where NF-κB induced gene expression by promoter specific activation of histone-modifying co-activators, leading to chromatin remodelling46. We carried out extensive motif analysis and found that a majority (∼97%) of the NF-κB targets were likely under a combinatorial regulation by transcription factors like STAT-3, CREB, AP-1 and C/EBP, which are associated with inflammatory reactions following the acute phase response in liver. Coordinated transcriptional regulation of NF-κB with these factors has been independently reported80–82. Research conducted on other organ systems also showed coordinated behavior of these TFs in disease progression. Cell culture studies on Hodgkin's lymphoma (HL) identified binding motifs for the TFs AP-1, NF-κB, and STAT in HL-specific accessible chromatin demonstrating their role in HL biology95. Our study, for the first time reports the co-occurrence of these transcription factors with NF-κB in liver suggesting their modular organization with NF-κB during liver regeneration.
Our findings demonstrated that the genome-wide localization of NF-κB was altered as a response to acute perturbations. We interpret the altered distribution in ethanol diet as contributing to the defective early gene regulatory network response, thereby influencing the acute response of tissue injury and interfering with regeneration outcome. Based on these findings, we propose that the baseline increase in NF-κB activity due to chronic ethanol intake limits further changes in promoter binding at key genes, and this may underlie the adverse affects of chronic ethanol treatment on the liver regeneration response. For example, Stat3 is a known downstream target of NF-κB during normal liver regeneration96. Our results indicated that NF-κB binding at the Stat3 promoter was at higher levels in alcoholic livers, but the baseline target gene expression was similar across diets. This adaptive state may arise because of additional regulators negatively affecting Stat3 expression in alcoholic livers. An increase in Stat3 expression post PHx coupled with a lack of a concomitant increase in NF-κB binding activity at the Stat3 promoter indicated that Stat3 may not be under the control of NF-κB in the alcoholic liver. It will be interesting to study if and how the increased baseline NF-κB activity leads to loss of its control over key target genes in response to perturbations after chronic ethanol intake. Our results also point to the complexity of the response and diversity in which the consequences of increasing the NF-κB activity varied across target genes, and were dependent on the perturbation context with baseline changes acting distinctly from that in response to PHx.
Follow-up studies may focus on determining whether the NF-κB targets show cell type-specific responses. Our immuno-histo chemistry study revealed that NF-κB activity showed a variable response to PHx in a cell type specific manner47. Other studies have demonstrated that hepatocytes from ethanol-fed animals showed higher constitutive NF-κB DNA binding leading to the activation of NF-κB signalling pathways8,97–100. Ethanol intake elevates iron levels, priming Kupffer cells for NF-κB activation leading to TNFα production in rats treated with a high fat and alcohol diet for 9 weeks101,102. It has also been shown that the combination of chronic ethanol intake and unsaturated fats triggers NF-κB activation and oxidative stress in Kupffer cells8,99 leading the activation of various inflammatory factors. A recent study showed that chronic ethanol feeding in mice increased the responsiveness of macrophages by sensitization to LPS, resulting from ethanol-induced alterations in intracellular signalling through NF-κB87. They showed that additional stimulation by LPS activated NF-κB in Kupffer cells in ethanol-fed mice. These results suggest ethanol treatment induces characteristic NF-κB binding responses in different liver cell types. Our study largely focused on hepatocytes, since the signal from whole-liver tissue ChIP is likely predominantly contributed by hepatocytes in contrast to non-parenchymal cells. Emerging techniques such as micro-ChIP and single cell ChIP methods potentially enable study of NF-κB genome-wide binding responses in non-parenchymal cells103–106.
Conclusions
We developed a novel comparative analysis method to classify NF-κB genome-wide binding in liver in the ethanol-fed and carbohydrate-fed control dietary groups. Our COMPACT approach highlighted subtle aspects corresponding to binding activity and expression patterns involving only a small number of genes that may otherwise have been ignored in conventional analyses of genome-wide binding and gene expression data. Partial hepatectomy induced a shift in the preferential proximal promoter binding loci of NF-κB towards downstream of TSS in carbohydrate specific missing group. Additionally, we found that chronic ethanol intake shifted the binding landscape of NF-κB post PHx. NF-κB target genes at 1h post PHx, specific to the chronic ethanol group participated in a select set of pathways associated with the initiation of an adaptive regenerative response. Integration with gene expression data revealed that the immediate early transient binding of NF-κB was associated with differential expression at a later time point post PHx. These genes were associated with key biological functions such as stress response, apoptosis, cell proliferation, cell cycle and metabolic processes. The system-wide shift in immediate early NF-κB targeting and co-incident binding sites serves as a signature of ethanol-mediated disrupted organization of transcriptional regulatory modules, potentially underlying the deficient regenerative response. Our results raise additional questions as to the general applicability of this phenomenon in which compensatory mechanisms that lead to an adaptive state in alcoholic rats play a role in suppressing the sensitivity of the system to certain perturbations. This could affect the ability of the liver to respond to damage and initiate effective repair in the face of additional environmental challenges.
Supplementary Material
Acknowledgments
The authors thank Daniel Cook for his help while preparing the manuscript. Financial support for this study was provided by National Institutes of Health grants from National Institute on Alcohol Abuse and Alcoholism R21 AA016919, R01 AA018873, R21 AA022417, T32 AA007463.
List of abbreviations
- ChIP-chip
Chromatin immunoprecipitation on chip
- PHx
Partial hepatectomy
- TF
Transcription factor. NF-κB, Nuclear factor kappa B, FDR, False discovery rate
Footnotes
Electronic supplementary information (ESI) is available.
Authors' contributions: RV conceived of and supervised all aspects of the study; RV, BP and JH designed the experiments. BP performed the experiments. LK analyzed the data. LK, JH and RV interpreted the results. LK wrote the manuscript. RV and JH critically reviewed the manuscript. We confirm that all authors have approved the final version of the manuscript and that all persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Availability of supporting data/Additional files: The data sets supporting the results of this article are available in the Gene Expression Omnibus (GEO) repository (Accession number: GSE74273) and are publically available at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74273)
References
- 1.Taub R. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 2.Fausto N, Campbell JS, Riehle KJ. Hepatology. 2006;43:S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 3.Kurinna S, Barton MC. Int J Biochem Cell Biol. 2011;43:189–97. doi: 10.1016/j.biocel.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michalopoulos GK. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michalopoulos GK. Am J Pathol. 2010;176:2–13. doi: 10.2353/ajpath.2010.090675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crumm S, Cofan M, Juskeviciute E, Hoek JB. Hepatology. 2008;48:898–908. doi: 10.1002/hep.22421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurinna S, Barton MC. Prog Mol Biol Transl Sci. 2010;97:201–227. doi: 10.1016/B978-0-12-385233-5.00007-6. [DOI] [PubMed] [Google Scholar]
- 8.Nanji AA, Jokelainen K, Rahemtulla A, Miao L, Fogt F, Matsumoto H, Tahan SR, Su GL. Hepatology. 1999;30:934–943. doi: 10.1002/hep.510300402. [DOI] [PubMed] [Google Scholar]
- 9.Zeldin G, Yang SQ, Yin M, Lin HZ, Rai R, Diehl aM. Alcohol Clin Exp Res. 1996;20:1639–45. doi: 10.1111/j.1530-0277.1996.tb01710.x. [DOI] [PubMed] [Google Scholar]
- 10.May MJ, Ghosh S. Semin Cancer Biol. 1997;8:63–73. doi: 10.1006/scbi.1997.0057. [DOI] [PubMed] [Google Scholar]
- 11.Salazar-Montes A, Ruiz-Corro L, Sandoval-Rodriguez A, Lopez-Reyes A, Armendariz-Borunda J. World J Gastroenterol. 2006;12:5995–6001. doi: 10.3748/wjg.v12.i37.5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cressman DE, Greenbaum LE, Haber Ba, Taub R. J Biol Chem. 1994;269:30429–35. [PubMed] [Google Scholar]
- 13.Yang L, Magness ST, Bataller R, Rippe RA, Brenner DA. Am J Physiol Gastrointest Liver Physiol. 2005;289:G530–G538. doi: 10.1152/ajpgi.00526.2004. [DOI] [PubMed] [Google Scholar]
- 14.FitzGerald M, Webber E. Cell Growth Differ. 1995;6:417–27. [PubMed] [Google Scholar]
- 15.Sakuda S, Tamura S, Yamada A, Miyagawa J, Yamamoto K, Kiso S, Ito N, Higashiyama S, Taniguchi N, Kawata S, Matsuzawa Y. J Hepatol. 2002;36:527–533. doi: 10.1016/s0168-8278(01)00310-5. [DOI] [PubMed] [Google Scholar]
- 16.Malato Y, Sander LE, Liedtke C, Al-Masaoudi M, Tacke F, Trautwein C, Beraza N. Hepatology. 2008;47:2036–2050. doi: 10.1002/hep.22264. [DOI] [PubMed] [Google Scholar]
- 17.Malato Y, Ehedego H, Al-Masaoudi M, Cubero FJ, Bornemann J, Gassler N, Liedtke C, Beraza N, Trautwein C. Gastroenterology. 2012;143:1597–1608. doi: 10.1053/j.gastro.2012.08.030. [DOI] [PubMed] [Google Scholar]
- 18.Yamada Y, Kirillova I, Peschon JJ, Fausto N. Proc Natl Acad Sci U S A. 1997;94:1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatano E, Bennett BL, Manning AM, Qian T, Lemasters JJ, Brenner DA. Gastroenterology. 2001;120:1251–1262. doi: 10.1053/gast.2001.23239. [DOI] [PubMed] [Google Scholar]
- 20.Iimuro Y, Nishiura T, Hellerbrand C, Behrns KE, Schoonhoven R, Grisham JW, Brenner DA. J Clin Invest. 1998;101:802–811. doi: 10.1172/JCI483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang SQ, Lin HZ, Yin M, Albrecht JH, Diehl AM. Am J Physiol JID - 0370511. 1998;275:G696–G704. doi: 10.1152/ajpgi.1998.275.4.G696. [DOI] [PubMed] [Google Scholar]
- 22.Koteish A, Yang S, Lin H, Huang J, Diehl AM. Alcohol Clin Exp Res. 2002;26:1710–1718. doi: 10.1097/01.ALC.0000036923.77613.59. [DOI] [PubMed] [Google Scholar]
- 23.Orrego H, Crossley IR, Saldivia V, Medline A, Varghese G, Israel Y. J Lab Clin Med. 1981;97:221–230. [PubMed] [Google Scholar]
- 24.Wands JR, Carter EA, Bucher NL, Isselbacher KJ. Gastroenterology. 1979;77:528–531. [PubMed] [Google Scholar]
- 25.Zhang B, Horsfield BP, Farrell GC. J Clin Invest. 1996;98:1237–1244. doi: 10.1172/JCI118907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diehl aM, Chacon M, Wagner P. Hepatology. 1988;8:237–42. doi: 10.1002/hep.1840080208. [DOI] [PubMed] [Google Scholar]
- 27.Diehl AM, Wells M, Brown ND, Thorgeirsson SS, Steer CJ. J Clin Invest. 1990;85:385–390. doi: 10.1172/JCI114450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diehl a. Clin Liver Dis. 1998;2:723–738. doi: 10.1016/s1089-3261(05)70366-4. [DOI] [PubMed] [Google Scholar]
- 29.Yang SQ, Lin HZ, Yin M, Albrecht JH, Diehl AM. Am J Physiol. 1998;275:G696–G704. doi: 10.1152/ajpgi.1998.275.4.G696. [DOI] [PubMed] [Google Scholar]
- 30.Kim WH. FASEB J. 2001:2551–2553. doi: 10.1096/fj.01-0217. [DOI] [PubMed] [Google Scholar]
- 31.Gao B, Seki E, Brenner Da, Friedman S, Cohen JI, Nagy L, Szabo G, Zakhari S. Am J Physiol Gastrointest Liver Physiol. 2011;300:G516–25. doi: 10.1152/ajpgi.00537.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su AI, Guidotti LG, Pezacki JP, Chisari FV, Schultz PG. Proc Natl Acad Sci U S A. 2002;99:11181–11186. doi: 10.1073/pnas.122359899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fukuhara Y, Hirasawa A, Li XK, Kawasaki M, Fujino M, Funeshima N, Katsuma S, Shiojima S, Yamada M, Okuyama T, Suzuki S, Tsujimoto G. J Hepatol. 2003;38:784–792. doi: 10.1016/s0168-8278(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 34.Juskeviciute E, Vadigepalli R, Hoek JB. BMC Genomics. 2008;9:527–552. doi: 10.1186/1471-2164-9-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White P, Brestelli JE, Kaestner KH, Greenbaum LE. J Biol Chem. 2005;280:3715–22. doi: 10.1074/jbc.M410844200. [DOI] [PubMed] [Google Scholar]
- 36.Xu C, Chen X, Chang C, Wang G, Wang W, Zhang L, Zhu Q, Wang L, Zhang F. Dev Genes Evol. 2010;220:263–274. doi: 10.1007/s00427-010-0345-1. [DOI] [PubMed] [Google Scholar]
- 37.Kim TH, Barrera LO, Zheng M, Qu C, Singer Ma, Richmond Ta, Wu Y, Green RD, Ren B. Nature. 2005;436:876–80. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 39.Cook DJ, Patra B, Kuttippurathu L, Hoek JB, Vadigepalli R. Front Physiol. 2015;6:1–14. doi: 10.3389/fphys.2015.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim CA, Yao F, Wong JJY, George J, Xu H, Chiu KP, Sung WK, Lipovich L, Vega VB, Chen J, Shahab A, Zhao XD, Hibberd M, Wei CL, Lim B, Ng HH, Ruan Y, Chin KC. Mol Cell. 2007;27:622–635. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 41.Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, Weissman S, Snyder M. Proc Natl Acad Sci U S A. 2003;100:12247–52. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rao NS, McCalman MT, Moulos P, Francoijs KJJ, Chatziioannou A, Kolisis FN, Alexis MN, Mitsiou DJ, Stunnenberg HG. Genome Res. 2011;21:1404–1416. doi: 10.1101/gr.118042.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xing Y, Yang Y, Zhou F, Wang J. Gene. 2013;526:142–149. doi: 10.1016/j.gene.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 44.Schreiber J, Jenner RG, Murray HL, Gerber GK, Gifford DK, Young RA. Proc Natl Acad Sci U S A. 2006;103:5899–5904. doi: 10.1073/pnas.0510996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, Hong MY, Karczewski KJ, Huber W, Weissman SM, Gerstein MB, Korbel JO, Snyder M. Science. 2010;328:232–235. doi: 10.1126/science.1183621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Mitra A, Dojer N, Fu S, Rowicka M, Brasier AR. Nucleic Acids Res. 2013;41:7240–59. doi: 10.1093/nar/gkt493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nilakantan H, Kuttippurathu L, Parrish A, Hoek JB, Vadigepalli R. PLoS One. 2015;10:e0140236. doi: 10.1371/journal.pone.0140236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lieber CS, Decarli LM. Methods Enzym. 1994;233:585–594. doi: 10.1016/s0076-6879(94)33061-1. [DOI] [PubMed] [Google Scholar]
- 49.Xing D, Gong K, Feng W, Nozell SE, Chen YF, Chatham JC, Oparil S. PLoS One. 2011;6:e24021. doi: 10.1371/journal.pone.0024021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burdelya LG, Brackett CM, Kojouharov B, Gitlin II, Leonova KI, Gleiberman AS, Aygun-Sunar S, Veith J, Johnson C, Haderski GJ, Stanhope-Baker P, Allamaneni S, Skitzki J, Zeng M, Martsen E, Medvedev A, Scheblyakov D, Artemicheva NM, Logunov DY, Gintsburg AL, Naroditsky BS, Makarov SS, Gudkov AV. Proc Natl Acad Sci U S A. 2013;110:E1857–1866. doi: 10.1073/pnas.1222805110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kasama Y, Mizukami T, Kusunoki H, Peveling-Oberhag J, Nishito Y, Ozawa M, Kohara M, Mizuochi T, Tsukiyama-Kohara K. PLoS One. 2014;9:e91373. doi: 10.1371/journal.pone.0091373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, Green MR. BMC Bioinformatics. 2010;11:237–247. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuttippurathu L, Hsing M, Liu Y, Schmidt B, Maskell DL, Lee K, He A, Pu WT, Kong SW. Bioinformatics. 2011;27:715–717. doi: 10.1093/bioinformatics/btq707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dippold RP, Vadigepalli R, Gonye GE, Hoek JB. Am J Physiol Gastrointest Liver Physiol. 2012;303:G733–G743. doi: 10.1152/ajpgi.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang DW, Sherman BT, Lempicki RA. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith AD, Sumazin P, Das D, Zhang MQ. Bioinformatics. 2005;21(Suppl 1):i403–12. doi: 10.1093/bioinformatics/bti1043. [DOI] [PubMed] [Google Scholar]
- 59.Vadigepalli R, Chakravarthula P, Zak DE, Schwaber JS, Gonye GE. OMICS. 2003;7:235–252. doi: 10.1089/153623103322452378. [DOI] [PubMed] [Google Scholar]
- 60.Matys V, Fricke E, Geffers R, Göβling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Münch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mahony S, Benos PV. Nucleic Acids Res. 2007;35:1–6. doi: 10.1093/nar/gkm272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashburner BP, Westerheide SD, Baldwin aS. Mol Cell Biol. 2001;21:7065–77. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhong H, May MJ, Jimi E, Ghosh S. Mol Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 64.Ambrosini G, Bucher P. Proc IWBBIO. 2014:695–706. [Google Scholar]
- 65.Whitfield TW, Wang J, Collins PJ, Partridge EC, Aldred SF, Trinklein ND, Myers RM, Weng Z. Genome Biol. 2012;13:R50. doi: 10.1186/gb-2012-13-9-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang YG, Shi M, Wang T, Shi T, Wei J, Wang N, Chen XM. World J Gastroenterol. 2009;15:2329–2335. doi: 10.3748/wjg.15.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M, Pandak WM, Dent P, Spiegel S, Shi R, Xu W, Liu X, Bohdan P, Zhang L, Zhou H, Hylemon PB. Hepatology. 2012;55:267–276. doi: 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yin M, Bradford BU, Wheeler MD, Uesugi T, Froh M, Goyert SM, Thurman RG. J Immunol. 2001;166:4737–4742. doi: 10.4049/jimmunol.166.7.4737. [DOI] [PubMed] [Google Scholar]
- 69.Quinton LJ, Blahna MT, Jones MR, Allen E, Ferrari JD, Hilliard KL, Zhang XL, Sabharwal V, Algul H, Akira S, Schmid RM, Pelton SI, Spira A, Mizgerd JP. J Clin Invest. 2012;122:1758–1763. doi: 10.1172/JCI59408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bellet MM, Zocchi L, Sassone-Corsi P. Cell Cycle. 2012;11:3304–3311. doi: 10.4161/cc.21669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Filiano AN, Millender-Swain T, Johnson R, Young ME, Gamble KL, Bailey SM. PLoS One. 2013;8:e71684. doi: 10.1371/journal.pone.0071684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pilon AM, Ajay SS, Kumar SA, Steiner La, Cherukuri PF, Wincovitch S, Anderson SM, Mullikin JC, Gallagher PG, Hardison RC, Margulies EH, Bodine DM. Blood. 2011;118:139–149. doi: 10.1182/blood-2011-05-355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pluznick JL, Zou DJ, Zhang X, Yan Q, Rodriguez-Gil DJ, Eisner C, Wells E, Greer CA, Wang T, Firestein S, Schnermann J, Caplan MJ. Proc Natl Acad Sci U S A. 2009;106:2059–2064. doi: 10.1073/pnas.0812859106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou H, Wertz I, O'Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. Nature. 2004;427:167–171. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]
- 76.Kim JE, You DJ, Lee C, Ahn C, Seong JY, Hwang JI. Cell Signal. 2010;22:1645–1654. doi: 10.1016/j.cellsig.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 77.Wang Y, Tong X, Ye X. J Immunol. 2012;189:5304–13. doi: 10.4049/jimmunol.1201445. [DOI] [PubMed] [Google Scholar]
- 78.Hayden MS, Ghosh S. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 79.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. Nucleic Acids Res. 2009:37. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.He G, Karin M. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parry GC, Mackman N. J Immunol. 1997;159:5450–5456. [PubMed] [Google Scholar]
- 82.Taub R. Int Hepatol Commun. 1995;3:S6. [Google Scholar]
- 83.Mandrekar P, Catalano D, Jeliazkova V, Kodys K. J Leukoc Biol. 2008;84:1335–1345. doi: 10.1189/jlb.0407256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bellezza I, Mierla AL, Minelli A. Cancers (Basel) 2010;2:483–497. doi: 10.3390/cancers2020483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.You M, Fischer M, Deeg MA, Crabb DW. J Biol Chem. 2002;277:29342–29347. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 86.Wheeler MD, Yamashina S, Froh M, Rusyn I, Thurman RG. J Leukoc Biol. 2001;69:622–630. [PubMed] [Google Scholar]
- 87.Maraslioglu M, Oppermann E, Blattner C, Weber R, Henrich D, Jobin C, Schleucher E, Marzi I, Lehnert M. Mediators Inflamm. 2014;2014 doi: 10.1155/2014/808695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Teoh N, Pena A, Farrell G. Hepatology. 2002;36:94–102. doi: 10.1053/jhep.2002.33134. [DOI] [PubMed] [Google Scholar]
- 89.Tewari M, Dobrzanski P, Mohn KL, Cressman DE, Hsu JC, Bravo R, Taub R. Mol Cell Biol. 1992;12:2898–2908. doi: 10.1128/mcb.12.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oakley F, Meso M, Iredale JP, Green K, Marek CJ, Zhou X, May MJ, Millward-Sadler H, Wright MC, Mann DA. Gastroenterology. 2005;128:108–120. doi: 10.1053/j.gastro.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 91.Chaisson ML, Brooling JT, Ladiges W, Tsai S, Fausto N. J Clin Invest. 2002;110:193–202. doi: 10.1172/JCI15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ringelhan M, Schmid RM, Geisler F. PLoS One. 2012;7:e46469. doi: 10.1371/journal.pone.0046469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Abshagen K, Eipel C, Kalff JC, Menger MD, Vollmar B. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1570–7. doi: 10.1152/ajpgi.00399.2006. [DOI] [PubMed] [Google Scholar]
- 94.Carter EA, Wands JR. Alcohol Clin Exp Res. 1988;12:555–562. doi: 10.1111/j.1530-0277.1988.tb00241.x. [DOI] [PubMed] [Google Scholar]
- 95.Küppers R. Nat Rev Cancer. 2009;9:15–27. doi: 10.1038/nrc2542. [DOI] [PubMed] [Google Scholar]
- 96.Cressman DE, Diamond RH, Taub R. Hepatology. 1995;21:1443–9. [PubMed] [Google Scholar]
- 97.Bowie A, O'Neill LA. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- 98.Jokelainen K, Thomas P, Lindros K, Nanji AA. Biochem Biophys Res Commun. 1998;253:834–836. doi: 10.1006/bbrc.1998.9863. [DOI] [PubMed] [Google Scholar]
- 99.Essani NA, McGuire GM, Manning AM, Jaeschke H. J Immunol. 1996;156:2956–2963. [PubMed] [Google Scholar]
- 100.Valles SL, Blanco AM, Azorin I, Guasch R, Pascual M, Gomez-Lechon MJ, Renau-Piqueras J, Guerri C. Alcohol Clin Exp Res. 2003;27:1979–1986. doi: 10.1097/01.ALC.0000099261.87880.21. [DOI] [PubMed] [Google Scholar]
- 101.Valerio LG, Parks T, Petersen DR. Alcohol Clin Exp Res. 1996;20:1352–61. doi: 10.1111/j.1530-0277.1996.tb01134.x. [DOI] [PubMed] [Google Scholar]
- 102.Tsukamoto H, Lin M, Ohata M, Giulivi C, French SW, Brittenham G. Am J Physiol. 1999;277:G1240–50. doi: 10.1152/ajpgi.1999.277.6.G1240. [DOI] [PubMed] [Google Scholar]
- 103.Inoue S, Kaji N, Kataoka M, Shinohara Y, Okamoto Y, Tokeshi M, Baba Y. Electrophoresis. 2011;32:3241–3247. doi: 10.1002/elps.201100261. [DOI] [PubMed] [Google Scholar]
- 104.Acevedo LG, Iniguez AL, Holster HL, Zhang X, Green R, Farnham PJ. Biotechniques. 2007;43:791–797. doi: 10.2144/000112625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Amatori S, Ballarini M, Faversani A, Belloni E, Fusar F, Bosari S, Pelicci PG, Minucci S, Fanelli M. Epigenetics Chromatin. 2014;7:18. doi: 10.1186/1756-8935-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Murphy SJ, Cheville JC, Zarei S, Johnson SH, Sikkink RA, Kosari F, Feldman AL, Eckloff BW, Karnes RJ, Vasmatzis G. DNA Res. 2012;19:395–406. doi: 10.1093/dnares/dss021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.