

Abstract
Purpose of review
In chronic kidney disease (CKD), multiple factors contribute to the development of cardiac hypertrophy by directly targeting the heart or indirectly by inducing systemic changes such as hypertension, anemia and inflammation. Furthermore, disturbances in phosphate metabolism have been identified as non-classical risk factors for cardiovascular mortality in these patients. With declining kidney function, the physiologic regulators of phosphate homeostasis undergo changes in their activity as well as their circulating levels, thus potentially contributing to cardiac hypertrophy once they are out of balance. Recently, two of these phosphate regulators, fibroblast growth factor 23 (FGF23) and Klotho, have been shown to affect cardiac remodeling, thereby unveiling a novel pathomechanism of cardiac hypertrophy in CKD. Here we discuss the potential direct versus indirect effects of FGF23 and the soluble form of Klotho on the heart, and their crosstalk in the regulation of cardiac hypertrophy.
Recent findings
In models of CKD, FGF23 can directly target cardiac myocytes via FGF receptor 4 and induce cardiac hypertrophy in a blood pressure-independent manner. Soluble Klotho may directly target the heart via an unknown receptor thereby protecting the myocardium from pathologic stress stimuli that are associated with CKD, such as uremic toxins or FGF23.
Summary
Elevated serum levels of FGF23 and reduced serum levels of soluble Klotho contribute to uremic cardiomyopathy in a synergistic manner.
Keywords: Uremic Cardiomyopathy, Cardiac Hypertrophy, Fibroblast Growth Factor 23, Klotho
INTRODUCTION
The interrelation between reduced renal function and altered cardiac remodeling in patients with chronic kidney disease (CKD) is termed uremic cardiomyopathy. In CKD, cardiac hypertrophy is the most commonly diagnosed cardiovascular abnormality and the strongest independent predictor of cardiovascular mortality (1–5). The pathogenesis of uremic cardiomyopathy and underlying molecular pathways remain uncertain, and existing treatments improve outcomes only modestly (6–8). Although there is a high prevalence of conventional cardiovascular risk factors in the CKD population, such as hypertension, diabetes, obesity and lipid abnormalities, the relationship between these factors and outcome is less clear than in the general population (9, 10). Recent studies suggest that circulating molecules, including uremic toxins such as indoxyl sulfate, may directly target the myocardium and initiate hypertrophic growth of cardiac myocytes independently of mechanical stress (11, 12). Furthermore, disturbances of mineral metabolism, including hyperphosphatemia, secondary hyperparathyroidism, and vitamin D deficiency, have been identified as non-classical risk factors for cardiovascular mortality in patients with CKD (13–17). This review article focuses on circulating factors that are involved in the regulation of phosphate homeostasis and that are misregulated in CKD – namely Fibroblast Growth Factor 23 (FGF23) and Klotho - and their potential direct effects on the heart that might contribute to the development and progression of uremic cardiomyopathy (Figure 1).
Figure 1. Potential contributions of circulating phosphate, FGF23, soluble Klotho and vitamin D to uremic cardiomyopathy.
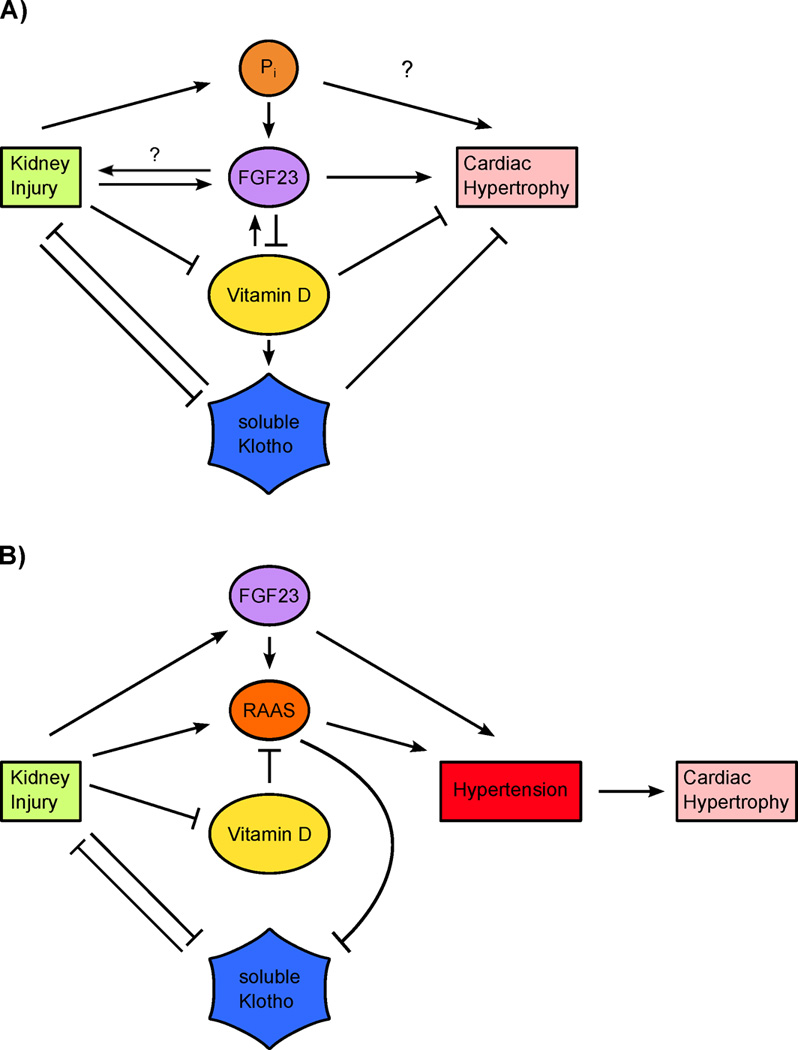
(A) Blood pressure-independent cardiac effects. Upon kidney injury, circulating levels of phosphate (Pi) and FGF23 are elevated whereas serum levels of vitamin D and soluble Klotho are reduced. By directly interfering with cardiac remodeling, all four events might independently contribute to cardiac hypertrophy. Direct effects of Pi on the myocardium are not well understood. By regulating each other’s activity and concentration, these four factors might also be able to amplify each other’s cardiac actions. This includes the increase of pro-hypertrophic FGF23 by Pi, the decrease of anti-hypertrophic vitamin D by FGF23, the increase of anti-hypertrophic soluble Klotho by vitamin D, and the decrease of anti-hypertrophic soluble Klotho by Pi (not depicted). (B) Blood pressure-dependent cardiac effects. Hypertension is a potent inducer of cardiac hypertrophy. FGF23, soluble Klotho and vitamin D might directly modulate blood pressure and thereby indirectly contribute to cardiac hypertrophy in the context of kidney injury. FGF23 may elevate blood pressure by stimulating the renin-angiotensin-aldosterone system (RAAS) as well as by renal sodium conservation. Reduced levels of anti-hypertrophic vitamin D cause an activation of RAAS, whereas RAAS reduces serum levels of anti-hypertrophic soluble Klotho.
TEXT OF REVIEW
Cardiac effects of FGF23
Produced by osteocytes, FGF23 is a hormone that regulates phosphate and vitamin D metabolism (18, 19). In the kidney and parathyroids, the classic target organs of FGF23, the hormone binds FGF receptor (FGFR)/Klotho co-receptor complexes (20, 21) to stimulate phosphate excretion, inhibit PTH secretion, and decrease levels of active vitamin D (22–26). In patients with CKD, disordered phosphate metabolism occurs as a result of a defect in renal excretory capacity. Beginning in early stages of CKD, serum FGF23 levels rise progressively as kidney function declines (27), reaching levels 1000-fold above normal in advanced renal failure (28). While this massive increase in FGF23 helps to maintain normal serum phosphate levels (29), prospective human studies demonstrated a dose-dependent association between FGF23 levels, increased prevalence of cardiovascular disease and greater risk of mortality among CKD patients (30–34). A potential mechanism linking FGF23 to cardiovascular disease is its strong association with cardiac hypertrophy (30, 33, 35–38).
Several studies have shown that rodent models with elevated serum FGF23 levels develop cardiac hypertrophy (38–40). Since the experimental increase of FGF23 concentrations by injection of recombinant FGF23, application of a high phosphate diet, or induction of a CKD phenotype via renal ablation, adenine diet or Klotho deletion have many other systemic effects, it would be important to also conduct these studies in animals lacking FGF23 in order to determine a causative interrelation between FGF23 and cardiac hypertrophy. However, FGF23 knockout mice develop a severe aging phenotype including extensive vascular calcification and die within 12 weeks after birth (41). Moreover, prolonged FGF23 inhibition in rats by administration of an FGF23 blocking antibody induces cardiovascular injury (42). Therefore, in vivo studies in an FGF23 null state are not feasible.
Detailed analyses of the FGF23-induced signaling events have helped to circumvent this dilemma. FGF23 induces hypertrophic growth of cardiac myocytes in vitro (38, 43)**, and this effect requires the presence of FGFR4 (44)*. Moreover, FGF23 can activate FGFR4 in cardiac myocytes resulting in subsequent stimulation of the phospholipase Cγ (PLCγ) / calcineurin / nuclear target of activated T cells (NFAT) signaling pathway (44)*, which is a potent inducer of cardiac hypertrophy in response to other pathological stimuli (45). Global deletion of FGFR4 in mice on high phosphate diet and systemic blockade of FGFR4 or calcineurin in 5/6 nephrectomized rats prevent the development of cardiac hypertrophy (44, 46)*, indicating that the FGF23/FGFR4/PLCγ/calcineurin/NFAT signaling axis is a major inducer of cardiac hypertrophy in rodents with high serum FGF23 levels. Interestingly, these treatments in rats do not reduce blood pressure or improve kidney function, indicating that hypertension or other factors altered by kidney injury are not major drivers of cardiac hypertrophy in this particular animal model of CKD.
FGF23 elevations in mice with cell type-specific FGFR4 gene deletion will be necessary to confirm that FGFR4 in cardiac myocytes is required for the development of cardiac hypertrophy, which would also provide further evidence that indeed FGF23 can directly target cardiac myocytes in the rodent heart, as strongly suggested by the existing in vitro data derived from cardiac myocyte cultures (38, 43, 44)**, *. Constant infusions of FGF23 using osmotic minipumps are necessary to precisely characterize the cardiac effects of FGF23 in a time- and dose-dependent manner, and to determine if FGF23 elevations can eventually lead to heart failure. Furthermore, it needs to be investigated if FGF23, possibly after short or low-dose exposure, has beneficial effects on cardiac structure and function, as suggested by a study in isolated adult cardiac myocytes and ventricular muscle strips from mice reporting increased contractility following FGF23 treatment (43)**.
Combined, these experimental studies provide strong evidence that elevated FGF23 is a causal factor in the pathogenesis of uremic cardiomyopathy thereby repositioning FGF23 from biomarker of risk to mechanism of cardiac injury in CKD (Figure 2A). Human studies, although not capable of providing direct evidence for causality, support the existence of such an FGF23-driven pathomechanism in the heart. First, a recent retrospective study showed that in children with end-stage renal disease, FGFR4 expression levels and calcineurin/NFAT activity in the heart associate with cardiac hypertrophy (47)*. Second, lowering serum FGF23 levels in CKD patients using calcimimetics is associated with lower rates of cardiovascular events and death (48)**. Third, other human disorders with elevated serum FGF23, such as X-linked hypophosphatemia (XLH), are also associated with cardiac hypertrophy (49). Cardiac hypertrophy has been also detected in Hyp mice, an animal model of XLH (50)*. Fourth, serum FGF23 levels associate with cardiac hypertrophy in cardiovascular cohorts with no or only slightly impaired renal function (51–56). A recent study in a mouse model for myocardial infarct confirmed an association between primary cardiac injury and elevated serum FGF23 levels (57)*. It is possible that in this scenario an unknown endocrine heart-bone feedback mechanism elevates FGF23 synthesis and secretion in bone (56, 57)*.
Figure 2. Potential interplay between FGF23 and soluble Klotho in the regulation of cardiac remodeling in CKD.
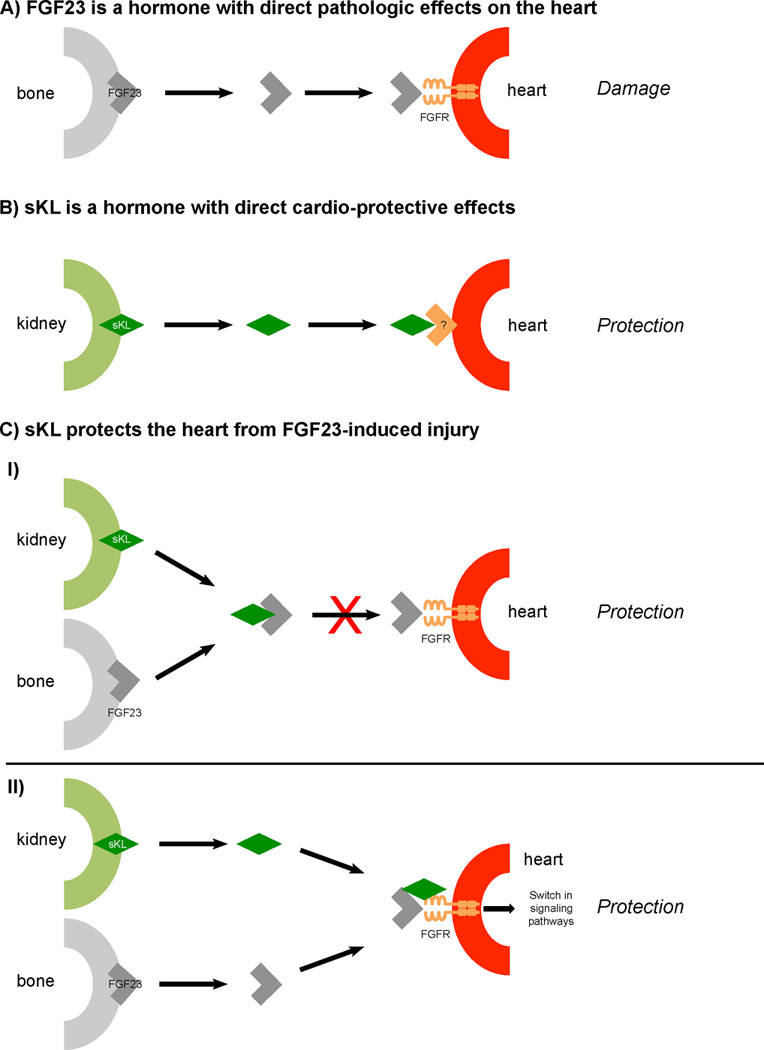
(A) FGF23 can directly target the myocardium via FGFR thereby inducing hypertrophic growth of cardiac myocytes and potentially further progressive damage including cardiac fibrosis, leading to impaired cardiac function. (B) Soluble Klotho (sKL) can directly target the heart via an unknown receptor thereby protecting the myocardium from pathologic stress stimuli that are associated with kidney injury, such as uremic toxins. (C.I) sKL can bind FGF23 in the circulation thereby blocking FGF23’s Klotho-independent pro-hypertrophic effects in the heart. (C.II) sKL can bind FGFR on cardiac cells, thereby modulating FGF23-induced signaling transduction in the myocardium. In the presence of elevated FGF23, sKL induces a switch from pro-hypertrophic calcineurin/NFAT signaling to different signaling branches with cardio-protective effects.
In contrast, other studies suggest that FGF23 does not directly harm the heart, but that rather other factors that correlate with FGF23 elevations in CKD cause cardiac hypertrophy (58–60). First, pharmacologic FGF23 blockade does not reduce cardiac hypertrophy in 5/6 nephrectomized rats (42). The outcome of this study seems to be contradictory to the observed anti-hypertrophic effects of FGF23 receptor blockade (38, 44)*. Although the respective studies used the same surgical rat model of CKD, they also exhibited important differences in other experimental parameters, such as normal versus high phosphate diet, start of pharmacological intervention at one hour versus 4 weeks post surgery, and analysis of cardiac alterations at 2 weeks versus 8 weeks after surgery. Therefore, it is impossible to directly compare the outcome of these studies. Furthermore as discussed above, FGF23 blockade by itself is detrimental and induces cardiovascular injury and decreases survival in rodents (41, 42). Second, although administration of the phosphate binder sevelamer in a mouse model of CKD lowered serum FGF23 levels and prevented progression of cardiac hypertrophy, a multiple regression analysis revealed that only serum phosphate, but not serum FGF23, independently correlated with left ventricular mass (61), indicating that phosphate’s calcifying effects on the vasculature rather than FGF23 effects on the heart are responsible for the observed cardiac hypertrophy in this particular animal model. Moreover in a human CKD study, sevelamer reduced serum FGF23 levels but did not significantly reduce cardiac hypertrophy or improve cardiac function (62). Third, by directly targeting distal tubules, FGF23 regulates renal sodium handling and elevates blood pressure, and FGF23’s hypertensive effect seems to be required for the development of cardiac hypertrophy in mice (50)*. Other studies have indicated that FGF23 might activate the renin-angiotensin system and elevate blood pressure (63), but detailed analyses are necessary to reveal the underlying molecular mechanism.
Overall, the molecular mechanisms by which FGF23 affects cardiac structure and function are not fully understood. As in human CKD studies, association analyses in animal models of CKD do not provide evidence to distinguish between direct versus indirect effects, and they will not help to provide novel insights into causality of events (39, 61). Instead, animal models such as gene knockout and knockin in mice should be used to study protein function in a tissue-specific manner in order to determine the contribution of a particular protein to a specific type of tissue injury, such as cardiac hypertrophy. In recent years, the reported number of cell types that are able to directly respond to FGF23 has been growing. Therefore, it is likely that besides its direct cardiac effects, systemic changes induced by FGF23, such as hypertension (50)* or inflammation (64), contribute to cardiac injury, suggesting that high serum FGF23 levels, as found in CKD, can hit the heart in several ways that might act synergistically.
Cardiac effects of Klotho
Klotho is a single-pass transmembrane protein that is expressed in several organs, including the kidney (20). Klotho acts as a co-receptor for FGF23 and increases the affinity of FGF23 for FGFR binding (21, 65, 66), thus mediating FGF23’s physiologic effects in the regulation of phosphate homeostasis. CKD is a state of Klotho deficiency (67–69), which can be due to a reduction in functional kidney mass and thereby a loss of Klotho expressing tubular epithelial cells. Furthermore, specific factors that are elevated in CKD can downregulate the expression of Klotho in the kidney, such as angiotensin II (AngII) (70–73), inflammatory cytokines (74–77) or phosphate (39). Vitamin D (78–80) and erythropoietin (81, 82) increase renal Klotho expression, and therefore vitamin D deficiency and anemia might contribute to a reduction in Klotho levels in CKD.
Mice lacking Klotho develop an aging and CKD-like phenotype including severe cardiovascular pathologies, such as vascular calcification as well as cardiac hypertrophy and fibrosis (20, 38, 39, 68). It is currently under debate if Klotho is expressed in the vasculature, and if a loss of membrane-bound Klotho in vascular smooth muscle cells and/or endothelial cells contributes to the calcification process (83, 84). Since Klotho is not expressed in the heart (20, 38, 74), a loss of membrane-bound Klotho cannot directly contribute to cardiac hypertrophy. Instead, cardiac effects of Klotho must be caused by changes in Klotho expression and/or function in its classic organs, like kidney or parathyroids, leading to secondary changes in levels of systemic factors that can directly target the heart, such as phosphate or FGF23.
The extracellular domain of Klotho can also exist as a circulating form (i.e. soluble Klotho, or sKL) that can be generated on mRNA level by alternative splicing or alternative transcriptional termination, or on protein level by proteolytic cleavage of transmembranous full-length Klotho (85–90). The kidney appears to be the major source of sKL (91, 92), and several different target cell types, such as endothelial cells and vascular smooth muscle cells, as well as a variety of cellular effects of sKL have been described. These functions seem to be exclusively beneficial and protective, including anti-oxidative, anti-apoptotic, anti-inflammatory, anti-fibrotic and anti-senescence effects (93–103). Several signal transduction pathways can be modulated by sKL thereby mediating its effects. These include the interference with the signaling of other paracrine and endocrine factors, such as insulin and insulin-like growth factor 1 (IGF1) (88, 104–106), Wnt (107, 108), AngII (39, 70, 97), transforming growth factor β (TGFβ) (39, 109), and erythropoietin (110), and the modulation of intracellular signaling mediators, such as PI3K (106), β-catenin (111), PKA (97), MAPK (95), NFκB (96, 102) and calpain (112, 113).
Based on the described findings, it has been postulated that sKL is a kidney-derived hormone with tissue-protective and anti-aging effects (Figure 2B). However, to date the sKL receptor has not been identified, and it remains unclear if sKL can directly activate signaling pathways and induce cellular effects in specific target cells. It has been shown that sKL can bind and inhibit TGFβ type II receptor (109) and FGFR1 (103), thereby competitively blocking fibrotic effects of TGFβ and FGF2, respectively. Furthermore, sKL suppresses the activation of insulin/IGF1 receptor (88) thereby blocking insulin and IGF1 signaling and contribute to anti-aging properties. However, it remains uncertain if sKL can directly bind these receptors. More detailed state-of-the-art binding analyses, including ELISA and surface plasmon resonance spectroscopy, are required to determine specificity and affinity of these potential ligand-receptor interactions. The identification of the sKL receptor is challenging, and the mechanism might not necessarily be based on sKL binding to one particular receptor protein. Instead, it may involve sKL’s activity as a deglycosylating enzyme (114, 115) that could affect several extracellular proteins or ectodomains of transmembrane proteins thereby regulating cellular functions. To date, the hunt for the sKL receptor(s) is still ongoing, and its success will be key in order to move the concept of sKL as a humoral factor forward.
It has been shown in animal modes and in patients with CKD that reduced renal expression of membrane-bound Klotho is accompanied by a decline in serum sKL levels (39, 68,92, 116, 117)*, *. Interestingly, recombinant sKL seems to be instable in uremic serum suggesting that decreased sKL production in the kidney may not be the sole reason for reduced serum sKL levels in CKD, and that increased sKL degradation in the circulation also contributes to sKL deficiency (116)*. Since in animal models of Klotho deficiency, restoration of Klotho expression and serum sKL levels ameliorates vascular calcification (68) and cardiac hypertrophy and fibrosis (39, 118, 119)*, it has been hypothesized that the reduction of sKL’s direct protective effects on the cardiovascular system causes injury (120, 121).
Several experimental studies indicate that sKL might directly interfere with cardiac hypertrophy and fibrosis. The induction of hypertrophic growth of cardiac myocytes in culture and of cardiac hypertrophy and fibrosis in rodents via β-adrenergic activation can be blocked by sKL administration (106, 122–124). In cultured cardiac myocytes, sKL inhibits AngII-, TGFβ- and indoxyl sulfate-induced hypertrophy (39, 119)*, and suppresses apoptosis that has been induced by hypoxia/reoxygenation or by β-adrenergic stimulation (124, 125). Furthermore, sKL attenuates AngII- and TGFβ-mediated activation of cultured cardiac fibroblasts (39). As in other tissues, cardiac sKL signaling seems to involve the blockade of IGF1, AngII, Wnt/β-catenin, PI3K, Akt and MAPK (39, 106, 124, 125), as well as a reduction in levels of reactive oxygen species (119, 125)*.
Mechanistically, sKL seems to inhibit TRPC6, a calcium-permeable cation channel that is an important modulator of cardiac remodeling and an inducer of cardiac hypertrophy by stimulating calcium-mediated signaling including the calcineurin/NFAT pathway (126). Mice with cardiac-specific overexpression of TRPC6 spontaneously develop cardiac hypertrophy (126). Elevation of circulating sKL levels in these mice prevents the development of cardiac hypertrophy and prolongs survival (106). Furthermore, sKL inhibits TRPC6 cell surface expression and currents in cultured cardiac myocytes (106), suggesting that sKL can directly target cardiac myocytes and mediates its anti-hypertrophic and cardio-protective effects by reducing TRPC6 activity and thereby calcineurin/NFAT signaling.
To date it remains unclear if sKL can directly target the heart. Since all models of Klotho deficiency involve a reduction of both forms, membrane-bound Klotho and sKL, it is impossible to distinguish between sKL’s direct effects on the heart versus systemic changes mediated by membrane-bound Klotho. Blocking renal cleavage or release of sKL or specific inhibition of circulating sKL in healthy animals would help to clarify the situation. It has been shown that administration of inhibitors for α- and β-secretases, which modulate Klotho ectodomain shedding in the kidney (89, 90), cause a reduction in serum sKL levels in mice (92). However, potential cardiac effects of these inhibitors per se, or in the context of uremia have not been reported. Furthermore, in order to experimentally determine if sKL can indeed directly target cardiac myocytes, it is inevitable that the receptor of sKL is identified and deleted in a cell type-specific manner in the mouse heart, as discussed above for the cardiac FGF23 receptor.
Different from the described clinical studies showing strong associations between FGF23 and cardiac hypertrophy in CKD and non-CKD populations, human studies have failed to detect significant correlations between serum sKL levels, kidney function, cardiovascular disease and survival in patients with CKD (127, 128). Moreover, sKL levels do not associate with changes in cardiac mass or function or predict cardiovascular risk in cardiac patients with normal renal function (53, 129). The later finding is supported by experimental myocardial infarct models in rats and mice, showing that serum levels of FGF23 but not of sKL are elevated (57)*, indicating that at least in this particular type of primary cardiac injury FGF23 but not sKL might be causatively involved.
Of note, technical concerns hinder experimental and human studies and make data interpretation difficult. First, commercially available antibodies against Klotho, including the best-characterized antibody, termed KM2076 (130), seem to be unspecific and cross-react with other proteins (84, 116)*. Klotho ELISAs have provided inconstant results for serum sKL levels in CKD (116)*. A novel synthetic anti-Klotho antibody, termed sb106, has been shown to detect Klotho in tissue as well as sKL in serum and urine following an immunoprecipitation-immunoblot approach (92, 116)*. However, this antibody is currently not commercially available. Second, the sKL protein seems to be highly instable in blood, at least in the CKD milieu (92), and in urine in general, even when urine samples were frozen (131). Hence, prior analysis sKL degradation needs to be prevented to conserve sKL levels, but technical approaches to do so have not been described. Third, as sKL seems to be unstable, the generation of recombinant sKL for the treatment of cell cultures and for injections in animals is challenging. Since functional assays to monitor sKL activity are not available, it is currently impossible to determine if the applied sKL is indeed active, or if it underwent degradation most likely resulting in the loss or alteration of protein function.
Potential crosstalk of FGF23 and sKL in the regulation of cardiac remodeling
Overall, these exciting new studies provide strong support for the hypothesis that FGF23 and sKL can directly affect cardiac remodeling. Since FGF23 and Klotho act as a ligand-receptor pair that is a key component for the regulation of phosphate homeostasis, it would be somewhat surprising if the cardiac actions of both proteins would be uncoupled and independent from each other. However, it remains challenging to experimentally determine direct pathologic contributions of low sKL versus high FGF23. In all known scenarios of Klotho deficiency, such as in primary deficiency like Klotho knockout or hypomorphic mice, as well as in secondary deficiency like CKD, aging or phosphate loading, reduced serum sKL levels go hand in hand with elevated concentrations of circulating FGF23. Furthermore, the experimental increase in circulating sKL results in a reduction in serum FGF23 levels.
Since some of the important studies on cardiac sKL effects have not analyzed serum FGF23 levels (106, 119)*, and vise-versa studies focusing on direct cardiac actions of FGF23 have not analyzed sKL levels (38, 44)*, it remains difficult to compare both factors in regards to their direct causal involvement. Only a few experimental studies aimed to uncouple the reduction in sKL levels from the elevation of FGF23, followed by cardiac analyses. For example, it was reported that the normalization of serum levels of FGF23 and phosphate in CKD mice with heterozygous Klotho deletion does not abrogate cardiac hypertrophy (118)*, suggesting that Klotho prevents uremic cardiomyopathy by mechanisms that are independent of FGF23 and phosphate, most likely by direct effects of sKL on cardiac myocytes. However, in this key experiment the heart was only analyzed by determining the ratio of heart weight to body weight, a vague measurement for cardiac hypertrophy, and levels of sKL and renal Klotho were also not reported. In addition, association studies in mice have shown that higher serum FGF23 levels only correlate with cardiac hypertrophy and fibrosis in concert with reduced sKL levels (39), indicating that not high FGF23 but low sKL is the key contributor to uremic cardiomyopathy.
Can sKL act as a circulating receptor for FGF23?
The role of the full-length Klotho protein is to mediate FGF23 interactions with target cells by enhancing FGF23’s affinity for FGFR binding by over 20-fold (66). Based on in vitro analyses it has been questioned if sKL can efficiently bind FGF23, and it has been concluded that circulating sKL exert its effects independent of FGF23 (92). First, in the presence of sKL, FGF23 can still activate MAPK signaling in HEK293 cells (65). However, since dependent on the cell type and the presence of specific FGFR isoforms, FGF23 can stimulate different signal transduction pathways (132), the analysis of MAPK signaling in generic cell lines might miss the detection of potential inhibitory actions of sKL on FGF23 effects. Second, surface plasmon resonance spectroscopy revealed that sKL binds FGF23 only poorly in the absence of FGFR (133). However, since other factors such as serum proteins could affect the FGF23-sKL interaction, it needs to be experimentally determined if FGF23 and sKL can form a stable complex in the circulation. If true, such an interaction might most likely alter stability and half-life as well as biological availability and function of both proteins. Interestingly, at the recent annual Kidney Week of the American Society of Nephrology, exciting data has been presented using highly specific antibodies against FGF23 and sKL in co-immunoprecipitation studies indicating that indeed both proteins can interact with each other in the circulation (134)**. If some of these newly developed antibodies could specifically block or strengthen the interaction between FGF23 and sKL, they would provide a valuable tool that could be used to experimental determine the function of this interaction and to analyze the cardiac effects of both factors in a separate manner.
It is interesting to speculate that sKL can bind FGF23 thereby blocking Klotho-dependent and Klotho-independent effects in kidney and heart, respectively, and serving as a circulating decoy receptor for FGF23 (Figure 2C.I). However, experimental proof for such a hypothesis is still missing. A first step would be to determine, if sKL can block FGF23-induced FGFR4/PLCγ/calcineurin/NFAT signaling and hypertrophic growth in cultured cardiac myocytes. Interestingly, it has been shown that in myocardial tissue of CKD patients declining sKL protein levels in combination with elevated FGF23 production correlate with cardiac hypertrophy (47)*. This clinical finding indicates that a reduction in sKL’s inhibitory actions towards FGF23’s pro-hypertrophic effects might contribute to uremic cardiomyopathy, thereby supporting the hypothesis that sKL acts as a decoy receptor for FGF23.
Can sKL bind FGFRs and act as a circulating co-receptor for FGF23?
It has been shown in vitro that sKL can bind FGFRs (65) and increase the affinity of FGF23 for FGFR binding (133). Therefore it is possible that sKL acts as a circulating FGF23 co-receptor that can interact with membrane-bound FGFRs, thereby facilitating FGF23 binding to cell types that per se do not express Klotho and mediating temporary responsiveness to FGF23. Such a mechanism has been reported in fibroblast and myoblast cell lines where the combined treatment with FGF23 and sKL activated FGFR1 and MAPK signaling leading to increased cell survival (135). Furthermore, the transmembrane form of Klotho has been shown to modulate downstream signaling events (44, 136)*. Therefore, in cell types that can respond to FGF23 in a Klotho-independent manner via FGFR4, such as cardiac myocytes, sKL binding might cause a switch in signaling pathways leading to different cellular effects of FGF23 (Figure 2C.II).
CONCLUSION
FGF23 appears to function as a circulating factor that can directly target cardiac myocytes and induce cardiac hypertrophy independently of other stress factors (38, 43, 44)**, *. Furthermore, Klotho deficiency predisposes the heart to stress-induced cardiac hypertrophy (106, 119)*. Thus, high FGF23 and low sKL may synergistically contribute to uremic cardiomyopathy by molecular mechanisms that are not completely understood. The theory that Klotho deficiency is deleterious whereas Klotho sufficiency is protective against pathologic effects of FGF23 might also explain other organ damage associated with CKD, as recently proposed for the endothelium (99)*. As CKD is a stage of both, low sKL and high FGF23, and both trends aggravate with declining renal function, such a pathomechanism would serve as a plausible explanation for the dramatic increase in severity and extent of secondary organ injury, such as cardiac hypertrophy and fibrosis, as kidney injury progresses (Figure 3).
Figure 3. Potential synergistic contributions of elevated FGF23 and soluble Klotho deficiency to uremic cardiomyopathy.
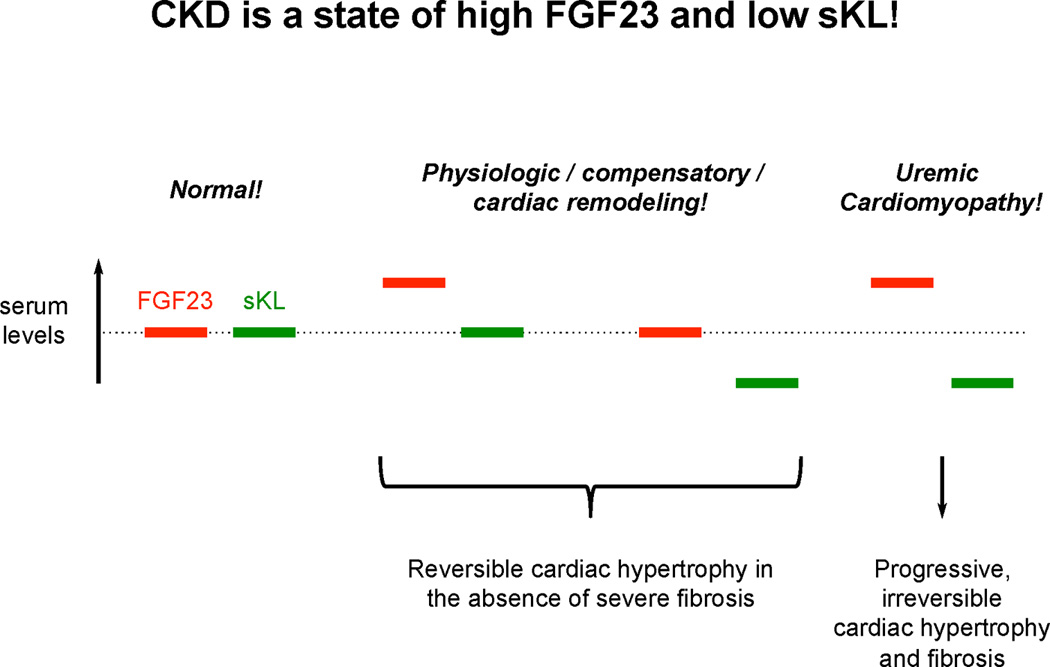
FGF23 and soluble Klotho (sKL) are part of an endocrine system that regulates cardiac remodeling, where FGF23 acts as an inducer and sKL as an inhibitor of cardiac hypertrophy. Under physiologic conditions, minor changes in the FGF23 and sKL balance can contribute to cardiac remodeling that might have adaptive or compensatory functions. In CKD, both factors are constantly out of balance leading to cardiac injury.
KEY POINTS.
Elevated serum levels of FGF23 and reduced serum levels of soluble Klotho are major contributors to uremic cardiomyopathy.
Cardiac effects of FGF23 and soluble Klotho may include blood pressure-dependent and blood pressure-independent mechanisms.
FGF23 can directly induce cardiac hypertrophy by targeting cardiac myocytes via FGFR4.
Soluble Klotho may directly target the heart via an unknown receptor thereby protecting the myocardium from pathologic stress stimuli that are associated with kidney injury, such as uremic toxins.
Soluble Klotho might block FGF23-induced cardiac hypertrophy via an unknown mechanism.
Acknowledgments
None
FINANCIAL SUPPORT AND SPONSORSHIP
This work was supported by the Katz Family Drug Discovery Center of the University of Miami Miller School of Medicine.
CONFLICT OF INTEREST
Dr. Faul has served as a consultant for Ultragenyx, has received research support from U3 Pharma GmbH and Roche, and is currently receiving a grant (R01HL128714) from the National Institutes of Health and a basic science award (1-16-IBS-087) from the American Diabetes Association. Dr. Grabner has been supported by a research fellowship from the Deutsche Forschungsgemeinschaft and a young investigator award from Roche, and he is currently receiving a Postdoctoral Fellowship (15POST22730025) from the American Heart Association.
REFERENCES
- 1.London GM, Pannier B, Guerin AP, Blacher J, Marchais SJ, Darne B, Metivier F, Adda H, Safar ME. Alterations of left ventricular hypertrophy in and survival of patients receiving hemodialysis: follow-up of an interventional study. J Am Soc Nephrol. 2001;12:2759–2767. doi: 10.1681/ASN.V12122759. [DOI] [PubMed] [Google Scholar]
- 2.Di Lullo L, Gorini A, Russo D, Santoboni A, Ronco C. Left Ventricular Hypertrophy in Chronic Kidney Disease Patients: From Pathophysiology to Treatment. Cardiorenal Med. 2015;5:254–266. doi: 10.1159/000435838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Middleton RJ, Parfrey PS, Foley RN. Left ventricular hypertrophy in the renal patient. J Am Soc Nephrol. 2001;12:1079–1084. doi: 10.1681/ASN.V1251079. [DOI] [PubMed] [Google Scholar]
- 4.Shlipak MG, Fried LF, Cushman M, Manolio TA, Peterson D, Stehman-Breen C, Bleyer A, Newman A, Siscovick D, Psaty B. Cardiovascular mortality risk in chronic kidney disease: comparison of traditional and novel risk factors. JAMA. 2005;293:1737–1745. doi: 10.1001/jama.293.14.1737. [DOI] [PubMed] [Google Scholar]
- 5.Zoccali C, Benedetto FA, Mallamaci F, Tripepi G, Giacone G, Cataliotti A, Seminara G, Stancanelli B, Malatino LS. Prognostic value of echocardiographic indicators of left ventricular systolic function in asymptomatic dialysis patients. J Am Soc Nephrol. 2004;15:1029–1037. doi: 10.1097/01.asn.0000117977.14912.91. [DOI] [PubMed] [Google Scholar]
- 6.Appel LJ, Wright JT, Jr, Greene T, Agodoa LY, Astor BC, Bakris GL, Cleveland WH, Charleston J, Contreras G, Faulkner ML, et al. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med. 2010;363:918–929. doi: 10.1056/NEJMoa0910975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA. 2011;305:2532–2539. doi: 10.1001/jama.2011.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, Wanner C, Krane V, Cass A, Craig J, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet. 2012;377:2181–2192. doi: 10.1016/S0140-6736(11)60739-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowrie EG, Huang WH, Lew NL. Death risk predictors among peritoneal dialysis and hemodialysis patients: a preliminary comparison. Am J Kidney Dis. 1995;26:220–228. doi: 10.1016/0272-6386(95)90177-9. [DOI] [PubMed] [Google Scholar]
- 10.Zager PG, Nikolic J, Brown RH, Campbell MA, Hunt WC, Peterson D, Van Stone J, Levey A, Meyer KB, Klag MJ, et al. "U" curve association of blood pressure and mortality in hemodialysis patients. Medical Directors of Dialysis Clinic, Inc. Kidney Int. 1998;54:561–569. doi: 10.1046/j.1523-1755.1998.00005.x. [DOI] [PubMed] [Google Scholar]
- 11.Winchester JF, Audia PF. Extracorporeal strategies for the removal of middle molecules. Semin Dial. 2006;19:110–114. doi: 10.1111/j.1525-139X.2006.00135.x. [DOI] [PubMed] [Google Scholar]
- 12.Lekawanvijit S, Kompa AR, Wang BH, Kelly DJ, Krum H. Cardiorenal syndrome: the emerging role of protein-bound uremic toxins. Circ Res. 2012;111:1470–1483. doi: 10.1161/CIRCRESAHA.112.278457. [DOI] [PubMed] [Google Scholar]
- 13.Shane E, Mancini D, Aaronson K, Silverberg SJ, Seibel MJ, Addesso V, McMahon DJ. Bone mass, vitamin D deficiency, and hyperparathyroidism in congestive heart failure. Am J Med. 1997;103:197–207. doi: 10.1016/s0002-9343(97)00142-3. [DOI] [PubMed] [Google Scholar]
- 14.Zittermann A, Schleithoff SS, Tenderich G, Berthold HK, Korfer R, Stehle P. Low vitamin D status: a contributing factor in the pathogenesis of congestive heart failure? J Am Coll Cardiol. 2003;41:105–112. doi: 10.1016/s0735-1097(02)02624-4. [DOI] [PubMed] [Google Scholar]
- 15.Pilz S, Tomaschitz A, Marz W, Drechsler C, Ritz E, Zittermann A, Cavalier E, Pieber TR, Lappe JM, Grant WB, et al. Vitamin D, cardiovascular disease and mortality. Clin Endocrinol (Oxf) 2011;75:575–584. doi: 10.1111/j.1365-2265.2011.04147.x. [DOI] [PubMed] [Google Scholar]
- 16.Schierbeck LL, Jensen TS, Bang U, Jensen G, Kober L, Jensen JE. Parathyroid hormone and vitamin D--markers for cardiovascular and all cause mortality in heart failure. Eur J Heart Fail. 2011;13:626–632. doi: 10.1093/eurjhf/hfr016. [DOI] [PubMed] [Google Scholar]
- 17.Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004;15:2208–2218. doi: 10.1097/01.ASN.0000133041.27682.A2. [DOI] [PubMed] [Google Scholar]
- 18.Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone. 2004;35:455–462. doi: 10.1016/j.bone.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118:3820–3828. doi: 10.1172/JCI36479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 21.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 22.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 23.Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun. 2004;314:409–414. doi: 10.1016/j.bbrc.2003.12.102. [DOI] [PubMed] [Google Scholar]
- 24.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto K, Fukushima N. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280:2543–2549. doi: 10.1074/jbc.M408903200. [DOI] [PubMed] [Google Scholar]
- 26.Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, Miyamoto K, Fukushima N. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J Biol Chem. 2003;278:2206–2211. doi: 10.1074/jbc.M207872200. [DOI] [PubMed] [Google Scholar]
- 27.Isakova T, Gutierrez OM, Wolf M. A blueprint for randomized trials targeting phosphorus metabolism in chronic kidney disease. Kidney Int. 2009;76:705–716. doi: 10.1038/ki.2009.246. [DOI] [PubMed] [Google Scholar]
- 28.Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–1378. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, Yamashita T, Fukumoto S, Shimada T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010;78:975–980. doi: 10.1038/ki.2010.313. [DOI] [PubMed] [Google Scholar]
- 30.Gutierrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, Sarwar A, Hoffmann U, Coglianese E, Christenson R, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119:2545–2552. doi: 10.1161/CIRCULATIONAHA.108.844506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seiler S, Reichart B, Roth D, Seibert E, Fliser D, Heine GH. FGF-23 and future cardiovascular events in patients with chronic kidney disease before initiation of dialysis treatment. Nephrol Dial Transplant. 2010;25:3983–3989. doi: 10.1093/ndt/gfq309. [DOI] [PubMed] [Google Scholar]
- 32.Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Juppner H, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutierrez OM, Steigerwalt S, He J, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305:2432–2439. doi: 10.1001/jama.2011.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kendrick J, Cheung AK, Kaufman JS, Greene T, Roberts WL, Smits G, Chonchol M. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol. 2010;22:1913–1922. doi: 10.1681/ASN.2010121224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu HJ, Wu MS. Fibroblast growth factor 23: a possible cause of left ventricular hypertrophy in hemodialysis patients. Am J Med Sci. 2009;337:116–122. doi: 10.1097/MAJ.0b013e3181815498. [DOI] [PubMed] [Google Scholar]
- 36.Kirkpantur A, Balci M, Gurbuz OA, Afsar B, Canbakan B, Akdemir R, Ayli MD. Serum fibroblast growth factor-23 (FGF-23) levels are independently associated with left ventricular mass and myocardial performance index in maintenance haemodialysis patients. Nephrol Dial Transplant. 2011;26:1346–1354. doi: 10.1093/ndt/gfq539. [DOI] [PubMed] [Google Scholar]
- 37.Seeherunvong W, Abitbol CL, Chandar J, Rusconi P, Zilleruelo GE, Freundlich M. Fibroblast growth factor 23 and left ventricular hypertrophy in children on dialysis. Pediatr Nephrol. 2012;27:2129–2136. doi: 10.1007/s00467-012-2224-7. [DOI] [PubMed] [Google Scholar]
- 38.Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Aguillon-Prada R, Lincoln J, Hare JM, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, Shelton J, Amaral AP, Faul C, Taniguchi M, et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2014050465. This study showed that higher phosphate and lower Klotho correlate with more cardiac hypertrophy and fibrosis in models of uremic cardiomyopathy. In contrast, higher FGF23 is only associated with increased cardiac injury in concert with lower serum Klotho levels, indicating that reduced levels of soluble klotho but not high FGF23 is the main driver of uremic cardiomyopathy.
- 40.Shobeiri N, Pang J, Adams MA, Holden RM. Cardiovascular disease in an adenine-induced model of chronic kidney disease: the temporal link between vascular calcification and haemodynamic consequences. J Hypertens. 2013;31:160–168. doi: 10.1097/HJH.0b013e32835b15bb. [DOI] [PubMed] [Google Scholar]
- 41.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, Renshaw L, Hawkins N, Wang W, Chen C, et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest. 2012;122:2543–2553. doi: 10.1172/JCI61405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Touchberry CD, Green TM, Tchikrizov V, Mannix JE, Mao TF, Carney BW, Girgis M, Vincent RJ, Wetmore LA, Dawn B, et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am J Physiol Endocrinol Metab. 2013;304:E863–E873. doi: 10.1152/ajpendo.00596.2012. This is the first study to show that FGF23 increases cardiac contractility in vitro, thereby indicating that FGF23 might have beneficial effects on the heart.
- 44. Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, Li J, Shehadeh LA, Hare JM, David V, et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015 doi: 10.1016/j.cmet.2015.09.002. This experimental study shows that FGFR4 is requried for FGF23-induced cardiac hypertorphy. Since in the heart FGFR4 is only expressed on cardiac myocytes, this study is the first to indicate that FGF23 can directly target cardiac myocytes. Furthermore, it suggests that FGFR4 blockade might serve as a novel therapeutic approach to tackle uremic cardiomyopathy.
- 45.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 46.Di Marco GS, Reuter S, Kentrup D, Ting L, Grabner A, Jacobi AM, Pavenstadt H, Baba HA, Tiemann K, Brand M. Cardioprotective effect of calcineurin inhibition in an animal model of renal disease. Eur Heart J. 2011;32:1935–1945. doi: 10.1093/eurheartj/ehq436. [DOI] [PubMed] [Google Scholar]
- 47. Leifheit-Nestler M, Grosse Siemer R, Flasbart K, Richter B, Kirchhoff F, Ziegler WH, Klintschar M, Becker JU, Erbersdobler A, Aufricht C, et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol Dial Transplant. 2015 doi: 10.1093/ndt/gfv421. This study shows that the hypertrophic FGF23/FGFR4/calcineurin/NFAT signaling axis is activated in the mycodarium of patients with end-stage renal disease, indicating that this pathway might contribute to uremic cardiomyopathy in humans. Furthermore, it shows that the uremic heart produces FGF23 and accumulates soluble Klotho, suggesting the existence of a pro-hypertrophic paracrine FGF23 mechanism that might be inhibited by circulating Klotho.
- 48. Moe SM, Chertow GM, Parfrey PS, Kubo Y, Block GA, Correa-Rotter R, Drueke TB, Herzog CA, London GM, Mahaffey KW, et al. Cinacalcet, Fibroblast Growth Factor-23, and Cardiovascular Disease in Hemodialysis: The Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) Trial. Circulation. 2015;132:27–39. doi: 10.1161/CIRCULATIONAHA.114.013876. This clincial study provides the first evidence that lowering FGF23 levels might improve clinical outcome in dialysis patients. It provides proof-of-principal that lowering serum FGF23 levels can improve cardiac remodeling in CKD, and thereby it supports the hypothesis that FGF23 can directly induce cardiac hypertrophy.
- 49.Nehgme R, Fahey JT, Smith C, Carpenter TO. Cardiovascular abnormalities in patients with X-linked hypophosphatemia. J Clin Endocrinol Metab. 1997;82:2450–2454. doi: 10.1210/jcem.82.8.4181. [DOI] [PubMed] [Google Scholar]
- 50. Andrukhova O, Slavic S, Smorodchenko A, Zeitz U, Shalhoub V, Lanske B, Pohl EE, Erben RG. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol Med. 2014;6:744–759. doi: 10.1002/emmm.201303716. This experimental study shows that FGF23 executes its cardiac effects by inducing higher renal tubular reabsorption of Na+ resulting in elevated blood pressure.
- 51.Seiler S, Cremers B, Rebling NM, Hornof F, Jeken J, Kersting S, Steimle C, Ege P, Fehrenz M, Rogacev KS, et al. The phosphatonin fibroblast growth factor 23 links calcium-phosphate metabolism with left-ventricular dysfunction and atrial fibrillation. Eur Heart J. 2011;32:2688–2696. doi: 10.1093/eurheartj/ehr215. [DOI] [PubMed] [Google Scholar]
- 52.Isakova T, Houston J, Santacruz L, Schiavenato E, Somarriba G, Harmon WG, Lipshultz SE, Miller TL, Rusconi PG. Associations between fibroblast growth factor 23 and cardiac characteristics in pediatric heart failure. Pediatr Nephrol. 2013;28:2035–2042. doi: 10.1007/s00467-013-2515-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shibata K, Fujita S, Morita H, Okamoto Y, Sohmiya K, Hoshiga M, Ishizaka N. Association between circulating fibroblast growth factor 23, alpha-Klotho, and the left ventricular ejection fraction and left ventricular mass in cardiology inpatients. PLoS One. 2013;8:e73184. doi: 10.1371/journal.pone.0073184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imazu M, Takahama H, Asanuma H, Funada A, Sugano Y, Ohara T, Hasegawa T, Asakura M, Kanzaki H, Anzai T, et al. Pathophysiological impact of serum fibroblast growth factor 23 in patients with nonischemic cardiac disease and early chronic kidney disease. Am J Physiol Heart Circ Physiol. 2014;307:H1504–H1511. doi: 10.1152/ajpheart.00331.2014. [DOI] [PubMed] [Google Scholar]
- 55.Wohlfahrt P, Melenovsky V, Kotrc M, Benes J, Jabor A, Franekova J, Lemaire S, Kautzner J, Jarolim P. Association of Fibroblast Growth Factor-23 Levels and Angiotensin-Converting Enzyme Inhibition in Chronic Systolic Heart Failure. JACC Heart Fail. 2015;3:829–839. doi: 10.1016/j.jchf.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 56.Andersen IA, Huntley BK, Sandberg SS, Heublein DM, Burnett JC., Jr Elevation of circulating but not myocardial FGF23 in human acute decompensated heart failure. Nephrol Dial Transplant. 2015 doi: 10.1093/ndt/gfv398. [DOI] [PubMed] [Google Scholar]
- 57. Andrukhova O, Slavic S, Odorfer KI, Erben RG. Experimental Myocardial Infarction Upregulates Circulating Fibroblast Growth Factor-23. J Bone Miner Res. 2015;30:1831–1839. doi: 10.1002/jbmr.2527. This is the first study showing that experimental myocardial infarct augments cardiac and circulating levels of FGF23.
- 58.Agarwal I, Ide N, Ix JH, Kestenbaum B, Lanske B, Schiller NB, Whooley MA, Mukamal KJ. Fibroblast growth factor-23 and cardiac structure and function. J Am Heart Assoc. 2014;3:e000584. doi: 10.1161/JAHA.113.000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Unsal A, Kose Budak S, Koc Y, Basturk T, Sakaci T, Ahbap E, Sinangil A. Relationship of fibroblast growth factor 23 with left ventricle mass index and coronary calcificaton in chronic renal disease. Kidney Blood Press Res. 2012;36:55–64. doi: 10.1159/000339026. [DOI] [PubMed] [Google Scholar]
- 60.Sinha MD, Turner C, Booth CJ, Waller S, Rasmussen P, Goldsmith DJ, Simpson JM. Relationship of FGF23 to indexed left ventricular mass in children with non-dialysis stages of chronic kidney disease. Pediatr Nephrol. 2015;30:1843–1852. doi: 10.1007/s00467-015-3125-3. [DOI] [PubMed] [Google Scholar]
- 61.Maizel J, Six I, Dupont S, Secq E, Dehedin B, Barreto FC, Benchitrit J, Poirot S, Slama M, Tribouilloy C, et al. Effects of sevelamer treatment on cardiovascular abnormalities in mice with chronic renal failure. Kidney Int. 2013;84:491–500. doi: 10.1038/ki.2013.110. [DOI] [PubMed] [Google Scholar]
- 62.Chue CD, Townend JN, Moody WE, Zehnder D, Wall NA, Harper L, Edwards NC, Steeds RP, Ferro CJ. Cardiovascular effects of sevelamer in stage 3 CKD. J Am Soc Nephrol. 2013;24:842–852. doi: 10.1681/ASN.2012070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dai B, David V, Martin A, Huang J, Li H, Jiao Y, Gu W, Quarles LD. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLoS One. 2012;7:e44161. doi: 10.1371/journal.pone.0044161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han X, Li L, Yang J, King G, Xiao Z, Quarles LD. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2 D in macrophages. FEBS Lett. 2016;590:53–67. doi: 10.1002/1873-3468.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goetz R, Ohnishi M, Kir S, Kurosu H, Wang L, Pastor J, Ma J, Gai W, Kuro-o M, Razzaque MS, et al. Conversion of a paracrine fibroblast growth factor into an endocrine fibroblast growth factor. J Biol Chem. 2012;287:29134–29146. doi: 10.1074/jbc.M112.342980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pavik I, Jaeger P, Ebner L, Wagner CA, Petzold K, Spichtig D, Poster D, Wuthrich RP, Russmann S, Serra AL. Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: a sequence suggested from a cross-sectional study. Nephrol Dial Transplant. 2013;28:352–359. doi: 10.1093/ndt/gfs460. [DOI] [PubMed] [Google Scholar]
- 68.Hu MC, Shi M, Zhang J, Quinones H, Griffith C, Kuro-o M, Moe OW. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22:124–136. doi: 10.1681/ASN.2009121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kitagawa M, Sugiyama H, Morinaga H, Inoue T, Takiue K, Ogawa A, Yamanari T, Kikumoto Y, Uchida HA, Kitamura S, et al. A decreased level of serum soluble Klotho is an independent biomarker associated with arterial stiffness in patients with chronic kidney disease. PLoS One. 2013;8:e56695. doi: 10.1371/journal.pone.0056695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mitani H, Ishizaka N, Aizawa T, Ohno M, Usui S, Suzuki T, Amaki T, Mori I, Nakamura Y, Sato M, et al. In vivo klotho gene transfer ameliorates angiotensin II-induced renal damage. Hypertension. 2002;39:838–843. doi: 10.1161/01.hyp.0000013734.33441.ea. [DOI] [PubMed] [Google Scholar]
- 71.Zhou Q, Lin S, Tang R, Veeraragoo P, Peng W, Wu R. Role of Fosinopril and Valsartan on Klotho Gene Expression Induced by Angiotensin II in Rat Renal Tubular Epithelial Cells. Kidney Blood Press Res. 2010;33:186–192. doi: 10.1159/000316703. [DOI] [PubMed] [Google Scholar]
- 72.Yoon HE, Ghee JY, Piao S, Song JH, Han DH, Kim S, Ohashi N, Kobori H, Kuro-o M, Yang CW. Angiotensin II blockade upregulates the expression of Klotho, the anti-ageing gene, in an experimental model of chronic cyclosporine nephropathy. Nephrol Dial Transplant. 2011;26:800–813. doi: 10.1093/ndt/gfq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saito K, Ishizaka N, Mitani H, Ohno M, Nagai R. Iron chelation and a free radical scavenger suppress angiotensin II-induced downregulation of klotho, an anti-aging gene, in rat. FEBS Lett. 2003;551:58–62. doi: 10.1016/s0014-5793(03)00894-9. [DOI] [PubMed] [Google Scholar]
- 74.Ohyama Y, Kurabayashi M, Masuda H, Nakamura T, Aihara Y, Kaname T, Suga T, Arai M, Aizawa H, Matsumura Y, et al. Molecular cloning of rat klotho cDNA: markedly decreased expression of klotho by acute inflammatory stress. Biochem Biophys Res Commun. 1998;251:920–925. doi: 10.1006/bbrc.1998.9576. [DOI] [PubMed] [Google Scholar]
- 75.Thurston RD, Larmonier CB, Majewski PM, Ramalingam R, Midura-Kiela M, Laubitz D, Vandewalle A, Besselsen DG, Muhlbauer M, Jobin C, et al. Tumor necrosis factor and interferon-gamma down-regulate Klotho in mice with colitis. Gastroenterology. 2010;138:1384–1394. 1394 e1381–1394 e1382. doi: 10.1053/j.gastro.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Radhakrishnan VM, Ramalingam R, Larmonier CB, Thurston RD, Laubitz D, Midura-Kiela MT, McFadden RM, Kuro OM, Kiela PR, Ghishan FK. Post-translational loss of renal TRPV5 calcium channel expression, Ca(2+) wasting, and bone loss in experimental colitis. Gastroenterology. 2013;145:613–624. doi: 10.1053/j.gastro.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moreno JA, Izquierdo MC, Sanchez-Nino MD, Suarez-Alvarez B, Lopez-Larrea C, Jakubowski A, Blanco J, Ramirez R, Selgas R, Ruiz-Ortega M, et al. The inflammatory cytokines TWEAK and TNFalpha reduce renal klotho expression through NFkappaB. J Am Soc Nephrol. 2011;22:1315–1325. doi: 10.1681/ASN.2010101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lau WL, Leaf EM, Hu MC, Takeno MM, Kuro-o M, Moe OW, Giachelli CM. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012;82:1261–1270. doi: 10.1038/ki.2012.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Forster RE, Jurutka PW, Hsieh JC, Haussler CA, Lowmiller CL, Kaneko I, Haussler MR, Kerr Whitfield G. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochem Biophys Res Commun. 2011;414:557–562. doi: 10.1016/j.bbrc.2011.09.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ritter CS, Zhang S, Delmez J, Finch JL, Slatopolsky E. Differential expression and regulation of Klotho by paricalcitol in the kidney, parathyroid, and aorta of uremic rats. Kidney Int. 2015;87:1141–1152. doi: 10.1038/ki.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sugiura H, Yoshida T, Mitobe M, Shiohira S, Nitta K, Tsuchiya K. Recombinant human erythropoietin mitigates reductions in renal klotho expression. Am J Nephrol. 2010;32:137–144. doi: 10.1159/000315864. [DOI] [PubMed] [Google Scholar]
- 82.Leone F, Lofaro D, Gigliotti P, Perri A, Vizza D, Toteda G, Lupinacci S, Armentano F, Papalia T, Bonofiglio R. Soluble Klotho levels in adult renal transplant recipients are modulated by recombinant human erythropoietin. J Nephrol. 2014;27:577–585. doi: 10.1007/s40620-014-0089-5. [DOI] [PubMed] [Google Scholar]
- 83.Faul C, Wolf M. Hunt for the culprit of cardiovascular injury in kidney disease. Cardiovasc Res. 2015;108:209–211. doi: 10.1093/cvr/cvv228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lewin E, Olgaard K. The vascular secret of Klotho. Kidney Int. 2015;87:1089–1091. doi: 10.1038/ki.2015.80. [DOI] [PubMed] [Google Scholar]
- 85.Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. 1998;242:626–630. doi: 10.1006/bbrc.1997.8019. [DOI] [PubMed] [Google Scholar]
- 86.Shiraki-Iida T, Aizawa H, Matsumura Y, Sekine S, Iida A, Anazawa H, Nagai R, Kuro-o M, Nabeshima Y. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. 1998;424:6–10. doi: 10.1016/s0014-5793(98)00127-6. [DOI] [PubMed] [Google Scholar]
- 87.Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, Fujimori T, Nabeshima Y. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004;565:143–147. doi: 10.1016/j.febslet.2004.03.090. [DOI] [PubMed] [Google Scholar]
- 88.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M, Kaether C. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. 2009;583:3221–3224. doi: 10.1016/j.febslet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A. 2007;104:19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lindberg K, Amin R, Moe OW, Hu MC, Erben RG, Ostman Wernerson A, Lanske B, Olauson H, Larsson TE. The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol. 2014;25:2169–2175. doi: 10.1681/ASN.2013111209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, Ravikumar P, Gillings N, Bian A, Sidhu SS, et al. Renal Production, Uptake, and Handling of Circulating alphaKlotho. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2014101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, Kurosu H, Miyoshi M, Ogawa Y, Castrillon DH, Rosenblatt KP, et al. Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem. 2005;280:38029–38034. doi: 10.1074/jbc.M509039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ikushima M, Rakugi H, Ishikawa K, Maekawa Y, Yamamoto K, Ohta J, Chihara Y, Kida I, Ogihara T. Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem Biophys Res Commun. 2006;339:827–832. doi: 10.1016/j.bbrc.2005.11.094. [DOI] [PubMed] [Google Scholar]
- 95.Maekawa Y, Ohishi M, Ikushima M, Yamamoto K, Yasuda O, Oguro R, Yamamoto-Hanasaki H, Tatara Y, Takeya Y, Rakugi H. Klotho protein diminishes endothelial apoptosis and senescence via a mitogen-activated kinase pathway. Geriatr Gerontol Int. 2011;11:510–516. doi: 10.1111/j.1447-0594.2011.00699.x. [DOI] [PubMed] [Google Scholar]
- 96.Buendia P, Carracedo J, Soriano S, Madueno JA, Ortiz A, Martin-Malo A, Aljama P, Ramirez R. Klotho Prevents NFkappaB Translocation and Protects Endothelial Cell From Senescence Induced by Uremia. J Gerontol A Biol Sci Med Sci. 2014 doi: 10.1093/gerona/glu170. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y, Kuro-o M, Sun Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell. 2012;11:410–417. doi: 10.1111/j.1474-9726.2012.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Banerjee S, Zhao Y, Sarkar PS, Rosenblatt KP, Tilton RG, Choudhary S. Klotho ameliorates chemically induced endoplasmic reticulum (ER) stress signaling. Cell Physiol Biochem. 2013;31:659–672. doi: 10.1159/000350085. [DOI] [PubMed] [Google Scholar]
- 99. Six I, Okazaki H, Gross P, Cagnard J, Boudot C, Maizel J, Drueke TB, Massy ZA. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS One. 2014;9:e93423. doi: 10.1371/journal.pone.0093423. This experimental study shows that soluble Klotho mitigates potentially noxious effects of FGF23 on the endothelium, thereby providing first evidence for the hypothesis that Klotho might act as a circulating inhibitor for FGF23.
- 100.Maekawa Y, Ishikawa K, Yasuda O, Oguro R, Hanasaki H, Kida I, Takemura Y, Ohishi M, Katsuya T, Rakugi H. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine. 2009;35:341–346. doi: 10.1007/s12020-009-9181-3. [DOI] [PubMed] [Google Scholar]
- 101.Zeng Y, Wang PH, Zhang M, Du JR. Aging-related renal injury and inflammation are associated with downregulation of Klotho and induction of RIG-I/NF-kappaB signaling pathway in senescence-accelerated mice. Aging Clin Exp Res. 2016;28:69–76. doi: 10.1007/s40520-015-0371-y. [DOI] [PubMed] [Google Scholar]
- 102.Zhao Y, Banerjee S, Dey N, LeJeune WS, Sarkar PS, Brobey R, Rosenblatt KP, Tilton RG, Choudhary S. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes. 2011;60:1907–1916. doi: 10.2337/db10-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guan X, Nie L, He T, Yang K, Xiao T, Wang S, Huang Y, Zhang J, Wang J, Sharma K, et al. Klotho suppresses renal tubulo-interstitial fibrosis by controlling basic fibroblast growth factor-2 signalling. J Pathol. 2014;234:560–572. doi: 10.1002/path.4420. [DOI] [PubMed] [Google Scholar]
- 104.Chateau MT, Araiz C, Descamps S, Galas S. Klotho interferes with a novel FGF-signalling pathway and insulin/Igf-like signalling to improve longevity and stress resistance in Caenorhabditis elegans. Aging (Albany NY) 2010;2:567–581. doi: 10.18632/aging.100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Abramovitz L, Rubinek T, Ligumsky H, Bose S, Barshack I, Avivi C, Kaufman B, Wolf I. KL1 internal repeat mediates klotho tumor suppressor activities and inhibits bFGF and IGF-I signaling in pancreatic cancer. Clin Cancer Res. 2011;17:4254–4266. doi: 10.1158/1078-0432.CCR-10-2749. [DOI] [PubMed] [Google Scholar]
- 106.Xie J, Cha SK, An SW, Kuro OM, Birnbaumer L, Huang CL. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun. 2012;3:1238. doi: 10.1038/ncomms2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–806. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- 108.Satoh M, Nagasu H, Morita Y, Yamaguchi TP, Kanwar YS, Kashihara N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am J Physiol Renal Physiol. 2012;303:F1641–F1651. doi: 10.1152/ajprenal.00460.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Doi S, Zou Y, Togao O, Pastor JV, John GB, Wang L, Shiizaki K, Gotschall R, Schiavi S, Yorioka N, et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. 2011;286:8655–8665. doi: 10.1074/jbc.M110.174037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hu MC, Shi M, Cho HJ, Zhang J, Pavlenco A, Liu S, Sidhu S, Huang LJ, Moe OW. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney Int. 2013;84:468–481. doi: 10.1038/ki.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhou L, Li Y, Zhou D, Tan RJ, Liu Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J Am Soc Nephrol. 2013;24:771–785. doi: 10.1681/ASN.2012080865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Manya H, Inomata M, Fujimori T, Dohmae N, Sato Y, Takio K, Nabeshima Y, Endo T. Klotho protein deficiency leads to overactivation of mu-calpain. J Biol Chem. 2002;277:35503–35508. doi: 10.1074/jbc.M206033200. [DOI] [PubMed] [Google Scholar]
- 113.Nabeshima Y, Washida M, Tamura M, Maeno A, Ohnishi M, Shiroishi T, Imura A, Razzaque MS, Nabeshima Y. Calpain 1 inhibitor BDA-410 ameliorates alpha-klotho-deficiency phenotypes resembling human aging-related syndromes. Sci Rep. 2014;4:5847. doi: 10.1038/srep05847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tohyama O, Imura A, Iwano A, Freund JN, Henrissat B, Fujimori T, Nabeshima Y. Klotho is a novel beta-glucuronidase capable of hydrolyzing steroid beta-glucuronides. J Biol Chem. 2004;279:9777–9784. doi: 10.1074/jbc.M312392200. [DOI] [PubMed] [Google Scholar]
- 115.Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24:3438–3450. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Barker SL, Pastor J, Carranza D, Quinones H, Griffith C, Goetz R, Mohammadi M, Ye J, Zhang J, Hu MC, et al. The demonstration of alphaKlotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrol Dial Transplant. 2015;30:223–233. doi: 10.1093/ndt/gfu291. This study reports the development of a novel anti-Klotho antibody and assay that can specifically detect Klotho in tissue as well as in serum and urine.
- 117.Sakan H, Nakatani K, Asai O, Imura A, Tanaka T, Yoshimoto S, Iwamoto N, Kurumatani N, Iwano M, Nabeshima Y, et al. Reduced renal alpha-Klotho expression in CKD patients and its effect on renal phosphate handling and vitamin D metabolism. PLoS One. 2014;9:e86301. doi: 10.1371/journal.pone.0086301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Xie J, Yoon J, An SW, Kuro OM, Huang CL. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2014040325. This is the first experimental study to support the hypothesis that Klotho prevents uremic cardiomyopathy by mechanisms that are independent of FGF23 and phosphate.
- 119. Yang K, Wang C, Nie L, Zhao X, Gu J, Guan X, Wang S, Xiao T, Xu X, He T, et al. Klotho Protects Against Indoxyl Sulphate-Induced Myocardial Hypertrophy. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2014060543. This animal study shows that elevation of soluble Klotho protects that heart from uremic toxin-induced cardiac hypertrophy, indicating that Klotho-deficiency in CKD results in reduced cardio-protection.
- 120.Fu H, Liu Y. Loss of Klotho in CKD Breaks One's Heart. J Am Soc Nephrol. 2015;26:2305–2307. doi: 10.1681/ASN.2015020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Floege J, Fliser D. Klotho Deficiency and the Cardiomyopathy of Advanced CKD. J Am Soc Nephrol. 2015;26:1229–1231. doi: 10.1681/ASN.2014090951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Song S, Si LY. Klotho ameliorated isoproterenol-induced pathological changes in cardiomyocytes via the regulation of oxidative stress. Life Sci. 2015;135:118–123. doi: 10.1016/j.lfs.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 123.Jia Z, Wei L, Liu Q, Zhu Z, Yang J, Yang X, Gan S, Chen W, Zhang L. Impact of transfection with recombinant adenovirus vector-mediated Klotho gene on myocardial remodeling in a rat model of heart failure. Zhonghua Xin Xue Guan Bing Za Zhi. 2015;43:219–226. [PubMed] [Google Scholar]
- 124.Song S, Gao P, Xiao H, Xu Y, Si LY. Klotho suppresses cardiomyocyte apoptosis in mice with stress-induced cardiac injury via downregulation of endoplasmic reticulum stress. PLoS One. 2013;8:e82968. doi: 10.1371/journal.pone.0082968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ai F, Chen M, Li W, Yang Y, Xu G, Gui F, Liu Z, Bai X, Chen Z. Protective role of Klotho on cardiomyocytes upon hypoxia/reoxygenation via downregulation of Akt and FOXO1 phosphorylation. Mol Med Rep. 2015;11:2013–2019. doi: 10.3892/mmr.2014.2899. [DOI] [PubMed] [Google Scholar]
- 126.Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Seiler S, Wen M, Roth HJ, Fehrenz M, Flugge F, Herath E, Weihrauch A, Fliser D, Heine GH. Plasma Klotho is not related to kidney function and does not predict adverse outcome in patients with chronic kidney disease. Kidney Int. 2013;83:121–128. doi: 10.1038/ki.2012.288. [DOI] [PubMed] [Google Scholar]
- 128.Buiten MS, de Bie MK, Bouma-de Krijger A, van Dam B, Dekker FW, Jukema JW, Rabelink TJ, Rotmans JI. Soluble Klotho is not independently associated with cardiovascular disease in a population of dialysis patients. BMC Nephrol. 2014;15:197. doi: 10.1186/1471-2369-15-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brandenburg VM, Kleber ME, Vervloet MG, Larsson TE, Tomaschitz A, Pilz S, Stojakovic T, Delgado G, Grammer TB, Marx N, et al. Soluble klotho and mortality: the Ludwigshafen Risk and Cardiovascular Health Study. Atherosclerosis. 2015;242:483–489. doi: 10.1016/j.atherosclerosis.2015.08.017. [DOI] [PubMed] [Google Scholar]
- 130.Kato Y, Arakawa E, Kinoshita S, Shirai A, Furuya A, Yamano K, Nakamura K, Iida A, Anazawa H, Koh N, et al. Establishment of the anti-Klotho monoclonal antibodies and detection of Klotho protein in kidneys. Biochem Biophys Res Commun. 2000;267:597–602. doi: 10.1006/bbrc.1999.2009. [DOI] [PubMed] [Google Scholar]
- 131.Adema AY, Vervloet MG, Blankenstein MA, Heijboer AC. alpha-Klotho is unstable in human urine. Kidney Int. 2015;88:1442–1444. doi: 10.1038/ki.2015.238. [DOI] [PubMed] [Google Scholar]
- 132.Kawai M. The FGF23/Klotho axis in the regulation of mineral and metabolic homeostasis. Horm Mol Biol Clin Investig. 2016 doi: 10.1515/hmbci-2015-0068. [DOI] [PubMed] [Google Scholar]
- 133.Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A. 2010;107:407–412. doi: 10.1073/pnas.0902006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sidhu S. American Society of Nephrology - Kidney Week. San Diego, CA: 2015. Circulating Klotho Levels: Best Way to Measure and How to Interpret Exisiting Data. This public presentation revealed for the first time that soluble Klotho and FGF23 can interact with each other in the circulation.
- 135.Smith RC, O'Bryan LM, Farrow EG, Summers LJ, Clinkenbeard EL, Roberts JL, Cass TA, Saha J, Broderick C, Ma YL, et al. Circulating alphaKlotho influences phosphate handling by controlling FGF23 production. J Clin Invest. 2012;122:4710–4715. doi: 10.1172/JCI64986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Olauson H, Lindberg K, Amin R, Sato T, Jia T, Goetz R, Mohammadi M, Andersson G, Lanske B, Larsson TE. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet. 2013;9:e1003975. doi: 10.1371/journal.pgen.1003975. [DOI] [PMC free article] [PubMed] [Google Scholar]