

Abstract
Purpose
Cancers usually contain multiple unique tumor-specific antigens produced by single amino acid substitutions (AAS) and encoded by somatic non-synonymous single nucleotide substitutions. We determined whether adoptively transferred T cells can reject large, well-established solid tumors when engineered to express a single type of T cell receptor (TCR) that is specific for a single AAS.
Experimental Design
By exome and RNA sequencing of an UV-induced tumor, we identified an AAS in p68 (mp68), a co-activator of p53. This AAS seemed to be an ideal tumor-specific neoepitope because it is encoded by a trunk mutation in the primary autochthonous cancer and binds with highest affinity to the MHC. A high-avidity mp68-specific TCR was used to genetically engineer T cells as well as to generate TCR-transgenic mice for adoptive therapy.
Results
When the neoepitope was expressed at high levels and by all cancer cells, their direct recognition sufficed to destroy intra-tumor vessels and eradicate large, long-established solid tumors. When the neoepitope was targeted as autochthonous antigen, T cells caused cancer regression followed by escape of antigen-negative variants. Escape could be thwarted by expressing the antigen at increased levels in all cancer cells or by combining T cell therapy with local irradiation. Therapeutic efficacies of TCR-transduced and TCR-transgenic T cells were similar.
Conclusions
Gene therapy with a single TCR targeting a single AAS can eradicate large established cancer but a uniform expression and/or sufficient levels of the targeted neoepitope or additional therapy are required to overcome tumor escape.
Keywords: T cell receptor (TCR), tumor-specific single amino acid substitution (AAS), adoptive T cell therapy (ATT), tumor heterogeneity, tumor escape
Introduction
Cancers are thought to evolve from normal cells to a malignant cell population that often is highly diversified genetically and epigenetically. Most of this evolution seems to occur even before the tumor reaches a size that can be detected clinically. Genetic diversity results from mutations in genes that make the incipient cancer prone to accumulate additional mutations (e.g., mutations in p53 or possibly in p68, a co-activator of p53 (1)) and/or drive malignant growth. The diversity provides cancers with variants that can escape specific treatments (2). Mutations in cancers regularly include non-synonymous nucleotide substitutions that result in single amino acid substitutions (AAS). These somatic mutations are the basis of truly cancer-specific antigens (also referred to as neoantigens or neoepitopes) recognized by T cells (3).
This discovery two decades ago was soon confirmed in human cancer (4,5) and followed by the suggestion that tumor-specific T cell antigens recognized by T cells on a cancer may be predicted by analyzing its somatic mutations, an approach referred to as ‘reverse immunology’ (6). However, wide interest in targeting tumor-specific AAS only developed more recently (7,8) following the availability of high-throughput genomic analysis and computer algorithms predicting neoepitopes. Interest further intensified with the finding that the clinical efficacy of tumor-infiltrating T cells (TILs) (9) or immune checkpoint inhibitors (10,11) may correlate with endogenous T cell responses to neoepitopes. Consequently, stimulation of T cells with mutant peptides is being combined with immune checkpoint inhibitors (12) that can activate and/or rescue exhausted endogenous cancer-specific T cells (13). The success of these approaches, however, may be limited because they rely on stimulating tolerant T cells which revert to an ineffective state after transient activation (14).
To avoid these limitations, unbiased peripheral T cells of patients can be engineered to express antigen receptors of a chosen specificity. For example, T cells expressing anti-CD19 chimeric antibody receptors can eradicate advanced leukemia and lymphomas even though normal B cells are also ablated (15). Since targeting other non-mutant self-antigens on tumors by adoptive T cell transfer caused severe toxicity and only little efficacy (16), we engineered T cells to express TCRs recognizing a shared (or trunk) mutation to determine the conditions by which targeting a single cancer-specific point mutation eradicates a progressively growing, genetically heterogeneous cancer. To our knowledge this is the first study to show that under the right conditions, mutation-specific TCR gene therapy provides an effective, truly tumor-specific cancer treatment.
Material and Methods
Whole-exome and RNA sequencing
Genomic DNA and total RNA were extracted, DNA and RNAseq libraries were prepared, and sequenced by 150 bp paired-end reads on NextSeq 500 Desktop Sequencer or HiSeq2500 Sequencer (Illumina, San Diego, CA). Detailed information are provided in Supplementary Methods.
Read mapping and variant calling
Low-quality reads (more than 80% of bases were base quality less than 20) were excluded using FASTX toolkit (Gregory J. Hannon, Cold Spring Harbor Laboratory, NY). For whole-exome sequencing, sequence reads were mapped to the mouse reference genome mm10 using Burrows-Wheeler Aligner (BWA v0.7.10 (17)). Possible PCR duplicated reads were removed (Picard v1.91, Broad Institute, Cambridge, MA), and read pairs with a mapping quality <30 and mismatches more than 5% of read length were excluded. For RNAseq, sequence reads were mapped to the mouse reference genome mm10 using STAR (18), and possible PCR duplicated reads and reads with a mapping quality <30 were excluded. Somatic variants were called using a Fisher’s exact test-based method (19) and annotated using ANNOVAR (20). Detailed information on variant calling are provided in Supplementary Methods.
Neoepitope prediction
Binding affinity to H-2Kb and H-2Db was predicted for 8-, 9-, and 10-mer peptides containing non-synonymous variants and found to be expressed in Bulk (variant reads ≥1 in RNAseq). Amino acid sequences were analyzed by the NetMHC 3.4 server (21) and binding affinity is expressed as half maximum inhibitory concentration (IC50) of the respective neoepitope.
Cells
MC57 is a methylcholanthrene-induced C57BL/6-derived fibrosarcoma (provided by Pamela Ohashi (University of Toronto, Toronto, Ontario, Canada), with permission of Hans Hengartner (University Hospital Zurich, Zurich, Switzerland)). Its transfectants MC57-SIY and MC57-mp68 were generated in our laboratory and have been described (22,23). 8101 originated in a UV-treated C57BL/6 mouse and was generated in our laboratory (24). Fragments of the original 8101 tumor (1-2 mm in size) were frozen. Bulk is a primary tumor cell culture derived from approx. 20 fragments of the original 8101 tumor. The cell line Bulk-mp68 was generated using MFG-mp68-EGFP as described (23). All tumor cell lines, RMA-S cells (see reference in (24)) and Plat-E packaging cells (see reference in (25)) were maintained in Dulbecco's modified Eagles medium supplemented with 10% fetal calf serum (FCS, Gemini Bio-Products, West Sacramento, CA). Unless otherwise indicated, cell culture reagents were purchased from Invitrogen (Life Technologies, Carlsbad, CA). Before use, tumor cell lines were authenticated by sequencing and/or co-culture with antigen-specific T cells. RMA-S and Plat-E cells were shortly passaged after thawing of the initial frozen stock to generate master cell banks. Working batches were passaged no longer than 3 months and authenticated by cellular morphology.
Generation and characterization of mp68-specific T cell clones, isolation of 1D9 TCR genes
T cell clones specific for mp68 (H-2Kb:SNFVAGI) were generated by immunizing C57BL/6 mice with Bulk tumor cells using procedures previously described (26) except that IL-2 (6 IU/ml) was used instead of T cell growth factor for expansion and cloning of the T cells. Specificity of established T cell cultures was analyzed by specific lysis of RMA-S cells loaded with mp68 (SNFVFAGI) or p68 wild-type peptide (SNFVSAGI, both peptides: 7.8×10−9 M, provided by Steven Meredith, University of Chicago, Chicago, IL) and Bulk tumor cells expressing the antigen at natural levels as described (24). 1D9 TCR sequences were determined by 5'-RACE-PCR following manufacturer's instructions (Life Technologies): TRAV1-CAVRSDTNAYKVIF-TRAJ30, TRBV19-CASSKRLSSYEQYF-TRBJ2-7. The 1D9 TCR was modified to form a second disulfide bond (27) and codon-optimized genes (GeneArt, Life Technologies) were integrated into MP71-PRE as described (MP71-1D9) (22).
Mice
C57BL/6 and Rag−/− (B6.129S7-Rag1tm1Mom) mice were purchased from the Jackson Laboratory. To generate a mouse that produces 1D9-transgenic T cells (1D9tg), the retrovirus vector plasmid encoding MP71-1D9 was injected into the nuclei of C57BL/6 zygotes as described (28). Transgenic mice backcrossed into the Rag−/− background solely expressed CD8+ 1D9tg T cells (1D9xRag−/−, B6.129S7-Rag1tm1Mom Tg(MP71-1D9)1Kshs). YFP×1D9×Rag−/− double-transgenic mice were generated as a pure source of fluorescently labeled 1D9tg T cells. YFP-transgenic Rag−/− mice have been described (29). OTIxRag−/− mice (B6.129S7-Rag1tm1Mom Tg(TcraTcrb)1100Mjb) were obtained by breeding Rag−/− and TCR OT-I-transgenic mice (provided by Matthew Mescher, University of Minnesota, Minneapolis, MN). Monospecific T cells of OT-IxRag−/− mice were used for the generation of 1D9td T cells. H-2KbDb−/−xRag−/− mice have been described (22). All animals were maintained at the University of Chicago facilities. The Institutional Animal Care and Use Committee at the University of Chicago approved all animal experiments, and all experiments were performed to conform to the relevant regulatory standards.
TCR gene transfer
Plat-E packaging cells were transiently transfected with MP71-1D9 or MP71-2C (22) by calcium phosphate precipitation. Virus supernatant was harvested and used for transduction of T cells isolated from OT-I-Rag−/− mice as detailed in Supplementary Methods.
Tumor challenge and treatment
Tumor cells were injected s.c. onto the shaved back of Rag−/− mice (1×107 Bulk, 2×106 MC57-mp68 or MC57-SIY). Tumor volumes were measured along 3 orthogonal axes every 3-4 days and tumor volume was calculated as abc/2. 1D9 T cells were administered i.v. on day 2 after transduction (transduced splenocytes of one OTIxRag−/− mouse per recipient, around 5×106 cells). Naive 1D9tg T cells were prepared from spleens of 1D9xRag−/− mice and administered i.v. without further culturing (splenocytes of one 1D9xRag−/− mouse per recipient, around 5×106 cells). Local radiation of tumors was done as described (30). Briefly, mice were locally irradiated using an x-ray generator (PCM 1000; Pantak) at a dose of 10 Gy. Mice were shielded with a lead cover leaving the subcutaneous tumors exposed through an opening on the side. Mice were irradiated when tumors reached the size of approx. 300 mm3 and T cells were transferred one day later.
Isolation of CD11b+ stromal cells
Tumors were isolated, single cell suspensions were generated by enzymatic digestion and CD11b+ cells were enriched using magnetic cell sorting as described (31). Sorted cells were used for co-cultures with 1D9td or 2Ctd T cells.
T cell analysis
To analyze antigen presentation by indicated tumor and stroma cells, co-cultures were performed with 1D9td and 2Ctd T cells as described (31). T cell activation was assessed by measuring IFN-γ content of 24 h co-culture supernatants by enzyme-linked immunosorbent assay (ELISA, Femto HS High Sensitivity, eBioscience), following the manufacturer's protocol. Cell-mediated lysis of indicated target cells was determined by standard 4.5 h chromium release assay as described (31).
Analysis of mp68 RNA
RNA was isolated from indicated tumors and PCR analysis to detect expression of the wildtype or mutant p68 allele was done as described (23).
Flow cytometry and antibodies
Erythrocytes in blood samples were lysed by ammonium chloride treatment. 1 μg Fc block (anti-mouse CD16/CD32, 2.4G2) was added to samples and cells were incubated with 50 μl PBS containing 1 μg of indicated anti-mouse antibodies: CD4 (GK1.5, allophycocyanin (APC), BioLegend, San Diego, CA), CD8 (53-6.7, PerCP, BioLegend), TCRvβ6 (RR4-7, fluorescein isothiocyanate (FITC) or APC, BD Biosciences (BD), Franklin Lakes, NJ) or H-2Kb:mp68 multimers (PE, NIH tetramer core facility, Bethesda, MD). Samples were washed in PBS and acquired using Calibur or LSR II flow cytometers (BD). Data analysis was performed using FlowJo software (TreeStar, Ashland, OR).
Longitudinal imaging
Implantation of windows, cancer cell injection, confocal microscopy and data analysis were done as described (29,32). Detailed information are provided in Supplementary Methods.
Results
‘Reverse immunology’ identifies a mutant epitope of p68 as neoantigen with high MHC affinity and broad expression in a genetically heterogeneous cancer
The 8101 tumor was induced by repeated exposure of a normal C57BL/6 mouse to UV light (24). The tumor on the back of this mouse was excised along with heart and lung tissue to generate a strain of normal fibroblasts (HLF) (24). Approximately 20 fragments of the autochthonous tumor 8101 were minced to generate an uncloned culture of 8101 cancer cells (23), herein after called “Bulk” (Fig. 1A). This culture was only minimally expanded in vitro to provide cancer cells for sequencing and tumor induction in vivo. Additional 20 fragments were kept for individual analysis to capture the spatial heterogeneity of mutations in the autochthonous tumor and to properly select a mutation as target for adoptive T cell therapy (ATT) that will likely be present in all parts of the tumor (Fig. 1A).
Fig. 1. The neoepitope mp68, predicted by ‘reverse immunology’, is found throughout the genetically diverse tumor 8101 and is recognized by high-avidity T cell clones.

A, UV-irradiation caused the development of an autochthonous tumor (8101). The tumor was excised and 20 individual tumor fragments were adapted to culture (Bulk) or analyzed separately using whole exome sequencing. Heart-lung fibroblasts (HLF) were generated as autologous tissue control. B, Identification of suitable neoepitopes as therapeutic targets by ‘reverse immunology’. Venn diagram (from left to right): Number of mutations detected in Bulk after whole exome and RNA sequencing. Number of neoepitopes (8-, 9-, or 10-mer peptides) found to be expressed and predicted to bind to H-2Kb or -Db with affinities of ≤500 nM or ≤50 nM (NetMHC 3.4). C, Binding affinity to MHC-I (IC50) and RNA expression level of neoepitopes identifies mp68 (SNFVFAGI, red) as highly expressed antigen with highest MHC affinity. D, Phylogenetic representation of somatic mutational frequency in the 8101 tumor identifies mp68 as trunk mutation. Green represents the trunk mutation p68S551F that was found in all fragments. Branches shown in blue lack the mutation p53S238A. Numbers on the top of each branching indicate unique mutations in 20 individual fragments and the Bulk tumor cell culture of 8101. E, F, The T cell clone 1D9 efficiently lyses Bulk tumor cells and is specific for the mp68 neoepitope. Specific lysis of (E) RMA-S cells loaded with 7.8 pM SNFVFAGI peptide or (F) Bulk tumor cells by mp68-specific T cell clones. The T cell clone 1D9 is highlighted in red and was used for subsequent TCR isolation.
Whole exome next-generation sequencing of Bulk identified 3,710 mutations (average coverage depths: 146x) using HLF DNA as baseline representing normal tissue from the host (Fig. 1B, analysis pipeline see: Supplementary Fig. S1 and detailed data in Supplementary Table S1). RNAseq showed that 33% of these mutations (total number: 1,207) were expressed in Bulk (Fig. 1B). Since mutant peptide epitopes with the highest affinity to MHC-I (IC50: ≤4 nM) appear to be required for eradication of large established cancer by adoptively transferred T cells (31), we first wanted to determine which of the 1,207 mutations would be predicted as preferred epitope. We made use of an open access computer algorithm (NetMHC 3.4 (21)) to predict mutant peptides of Bulk that bind to MHC-I. A total of 1,106 potential neoepitopes could be determined when screening the expressed mutated genes for peptides (octa-, nona-, or decamers) that harbor a mutation and bind to H-2Kb or H-2Db (Fig. 1B, IC50: ≤500 nM). Only 194 of these mutant peptides were found to bind either one of the MHC molecules with higher affinity (Fig. 1B, IC50: ≤50 nM). Only 15 mutations (0.4% of all mutations detected in Bulk) resulted in neoepitopes that had an IC50 of ≤4 nM. Among those, highest RNA expression was found for the mutant gene of the DEAD box helicase p68 encoding a serine to phenylalanine exchange at position 551 (Fig. 1C, in red, mp68 in the following).
The above results suggested that mp68 should be an excellent target for adoptively transferred T cells to eradicate established 8101 tumors. To prevent treatment failure, it is essential that the vast majority of cancer cells express the targeted antigen. To this end, we sequenced the DNA of 20 tumor fragments that were randomly selected from the original tumor. Each fragment contained 3,900±561 mutations (mean±SD, average coverage depths: 81x (±10), Supplementary Table S2). Mutations of the fragments clustered as illustrated in Fig. 1D, demonstrating differences in clonotypic composition (analysis pipeline see: Supplementary Fig. S2). This spatial diversification is confirmed by a large number of mutations uniquely present in individual fragments (numbers given at the top of each branching). Twenty five percent of all mutations (total number: 1,792) were shared by all fragments and represent the ‘mutational trunk’. One of these mutations was p68S551F (mp68) described above. The average read frequency of the mutant allele (30±8% (mean±SD)) suggested that the mp68 gene was represented appropriately for expression. Another mutation, p53S238A, was previously used to mark the common ancestry of the 8101 tumor (33) and was detected in all but 2 of the tumor fragments (Fig. 1D).
The mp68 epitope, the immunodominant rejection antigen of the 8101 tumor, induces high-avidity T cell clones
As reported above, algorithm analysis of exome and RNAseq data selected mp68 as highest ranking neoepitope. This is precisely the same neoepitope we had previously found to be the immunodominant rejection antigen in 8101 by direct immunology (24). To obtain T cells recognizing this tumor-specific antigen, we generated a number of mp68-specific T cell clones from T cells of tumor-free mice that had been immunized with the uncloned 8101 cancer cells. Tumor-reactive T cells were expanded by repeated stimulation in vitro with irradiated 8101 tumor cells and subsequently cloned by limiting dilution. Fig. 1E shows that several clones achieved half maximal lysis of RMA-S cells at low peptide concentration (7.8 pM SNFVFAGI), while these T cells were not reactive against the corresponding p68 wild-type peptide (SNFVSAGI, specific lysis <10%, data not shown). These high avidities are in agreement with the previously determined EC50 of another mp68-specific T cell clone (24). Decisive for selecting 1D9 T cells as source of the TCR was the high reactivity against Bulk tumor cells expressing mp68 at natural levels (Fig. 1F). Thus, we determined the 1D9 TCRα- and β-chain sequences and cloned them into a retrovirus vector (Supplementary Fig. S3A) for transduction of CD8+ T cells (1D9td). The vector was also used to generate a 1D9 TCR-transgenic mouse (1D9tg, 1D9xRag−/− mice, Supplementary Fig. S3B) for comparison because many previous studies used TCR-transgenic T cells for adoptive therapy to target surrogate or mutated antigens on tumors (34). However, only TCR-transduced T cells are realistic for patient therapy. As recipient T cells for transduction, we used CD8+ T cells from OT-IxRag−/− mice, which only express the OT-I-TCR specific for the ovalbumin peptide SIINFEKL, an irrelevant target for our model. Transduction efficacy was measured using TCRvβ6-specific antibodies (representative staining is shown in Supplementary Fig. S3C). The specific lysis of Bulk tumor cells by 1D9td T cells (Fig. 2A) was comparable to the lysis determined for the original 1D9 T cell clone (Fig. 1F).
Fig. 2. Mutant-specific TCR gene therapy causes regression of primary 8101 tumors with the subsequent escape of antigen-negative variants.
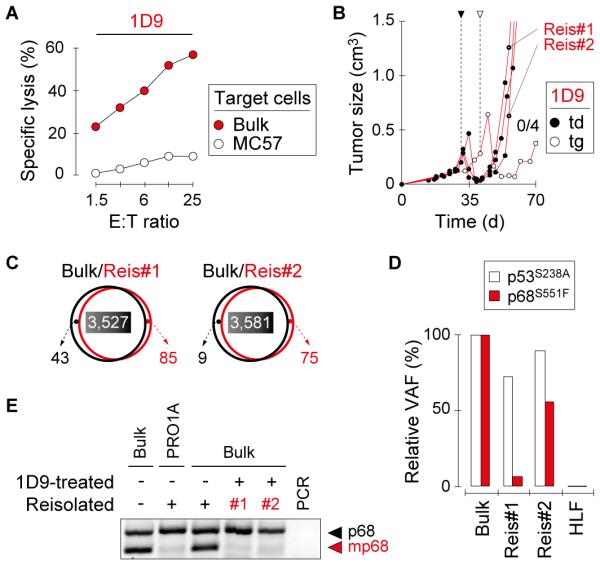
A, 1D9-engineered T cells efficiently lyse Bulk tumor cells. Specific lysis of Bulk and control MC57 tumor cells was analyzed in vitro using 1D9td T cells One representative experiment of three is shown. B, T cell therapy targeting mp68 causes regression of Bulk tumors that eventually escape. Mice with established Bulk tumors were treated with 1D9tg or 1D9td T cells. T cells were injected on day 29 (td) or 39 (tg), as indicated. Data are compiled from 3 independent experiments. Bulk tumor reisolates are indicated (Reis#1, Reis#2). C, Bulk tumor reisolates have similar mutational profiles compared to the parental tumor. DNA of Reis#1 and Reis#2 was analyzed by whole exome sequencing and compared to original Bulk tumor cells. The total number of mutations in each population is indicated. The numbers of shared mutations have colored background. D, Bulk tumor reisolates show diminished variant allele frequency (VAF) of mp68. The VAF of p53S238A and mp68 in Reis#1 and Reis#2 is shown in relation to the VAF detected in Bulk. E, Bulk tumor reisolates do not express the mp68 gene. RT-PCR was used to identify expression of the mutant allele of p68 in Reis#1 and Reis#2. Bulk tumor cells and a reisolated progressor variant of 8101 that escaped an immunocompetent normal host and lost the mp68 gene (PRO1A (33)) were used as controls.
mp68-specific TCR gene therapy causes regression of long-established primary 8101 tumors followed by escape of antigen-negative variants
After characterization of mp68 as an ideal target for ATT (high binding affinity to MHC-I, expressed in all regions of the primary tumor, and recognized by a TCR derived from a high-avidity T cell clone), we next assessed whether Bulk tumors could be rejected by mp68-specific TCR gene therapy. Thus, Rag−/− mice bearing Bulk tumors were treated by adoptive transfer of ~5×106 1D9 T cells. ATT led to initial regression of Bulk tumors, but all tumors eventually relapsed (Fig. 2B). The efficacy of 1D9tg and 1D9td T cells was similar. Whole exome sequencing of tumor reisolates (Reis#1, Reis#2) revealed a mutational pattern which was almost identical to the original Bulk tumor (Fig. 2C). However, the variant allelic frequency (VAF) of mp68 seemed to be reduced (Fig. 2D, Supplementary Table S1). This reduction was apparently related to the escape from mp68-directed T cell therapy as the proportion of the p53S238A mutation remained rather unchanged. Furthermore, Reis#1 and Reis#2 failed to express the mp68 RNA (Fig. 2E). RNA derived from in vitro-cultured Bulk tumor cells, an mp68-negative 8101 variant (PRO1A (33)) and a Bulk tumor reisolate from a mouse not treated with 1D9 T cells were used as controls. These results demonstrated that primary tumors escape mp68-specific ATT by loss of mp68 expression which is consistent with previous studies (23).
TCR gene therapy targeting mp68 is sufficient to eradicate established MC57 tumors transfected to express high amounts of the neoepitope
The failure of eradicating 8101 tumors by 1D9 T cell therapy raised the question whether 8101 was a cancer exceptionally difficult to treat or whether the destructive force of just CD8+ T cells grafted with a single type of TCR targeting a single tumor-specific mutant peptide was insufficient to achieve eradication of any large solid tumor. We therefore made use of the MC57 tumor model in which we had previously eradicated large solid tumors by adoptive transfer of CD8+ TCR-transgenic T cells targeting surrogate antigens (31,35). MC57 cancer cells were transduced with a retroviral vector to express the mp68 peptide coupled to GFP. This allowed us to express high levels of the targeted antigen in >99.9% of MC57-mp68 cancer cells (Supplementary Fig. S4). 1D9td T cells recognized MC57-mp68 but not mock-transfected MC57 cells in standard cytotoxicity assays (Fig. 3A). Remarkably, a single adoptive transfer of ~5×106 1D9 CD8+ T cells eradicated an established solid MC57-mp68 tumor in almost all of the Rag−/− mice (8/9 rejected, Fig. 3B, left). 1D9tg T cells were similarly effective as 1D9td T cells. The growth of MC57-mp68 was not affected by transfer of T cells transduced with a TCR of unrelated specificity (2C TCR, Mock, Fig. 3B, right). These experiments proved that T cells modified to express TCRs with a single specificity for a single mutant antigen can destroy very large and long-established solid cancers.
Fig. 3. Mutant-specific TCR gene therapy eradicates MC57 tumors expressing the mp68 neoepitope.

A, T cells expressing the 1D9 TCR lyse MC57 tumor cells stably transfected with the mp68 neoepitope. Specific lysis of indicated target cells was analyzed in vitro using T cells expressing the 1D9 TCR (T cells from OT-IxRag−/− mice transduced with the 1D9 TCR (1D9td) or the original anti-mp68 1D9 T cell clone). One representative experiment of two is shown. B, TCR gene therapy targeting mp68 causes rejection of MC57-mp68 tumors. H-2Kb-positive Rag−/− mice with established MC57-mp68 tumors were treated with 1D9td or 1D9tg T cells (left panel) or with T cells transduced with an irrelevant TCR (Mock, right panel). T cells were injected between day 15 and 19 as indicated by the arrow heads. C, TCR gene therapy can reject MC57-mp68 tumors in absence of stromal cross-presentation. H-2Kb-negative Rag−/− mice bearing established MC57-mp68 were treated as in (B). Data in (B) and (C) were compiled from 7 independent experiments. D, Stromal cross-presentation of mp68 induces high levels of cytokine release by 1D9td T cells. CD11b+ stromal cells were isolated from untreated MC57-mp68 and MC57-SIY tumors. Enriched stromal cells and cancer cells of the respective lines were co-cultured with 1D9td or 2Ctd T cells. IFN-γ content of supernatants was determined by ELISA. One representative experiment of two is shown.
Cross-presentation by the tumor stroma is not required for cancer eradication when the mp68 neoepitope is expressed uniformly and at high levels
Given the importance of stromal destruction for preventing relapse from T cell therapy in the MC57 and other experimental models (36), we asked whether recognition of mp68 on stromal cells by 1D9 T cells was required for eradication of MC57-mp68. Surprisingly, large MC57-mp68 tumors growing in Rag−/− mice devoid of MHC-I expression (H-2Kb−/−/Db−/−) were rejected by 1D9 T cells (4/4, Fig. 3C), indicating that 1D9 T cells received sufficient stimulation directly from MC57-mp68 tumor cells in absence of stromal antigen cross-presentation. However, large amounts of IFN-γ were released when stromal cells (mostly macrophages) from MC57-mp68 tumors were used as stimulators (Fig. 3D). The levels of IFN-γ were comparable to those stimulated by stromal cells from tumors expressing the SIY surrogate tumor antigen which is consistent with our previous studies (31).
Vessel destruction and bystander killing occur in the absence of stromal cross-presentation even though cross-presentation accelerates cancer cell destruction
To further investigate whether stromal cross-presentation of mp68 contributed to stromal destruction, we made use of longitudinal confocal microscopy (32) to follow the course of tumor destruction. MHC-I-positive and -negative Rag−/− mice with ~2 week-old MC57-mp68 tumors were treated with 1D9 T cells (Fig. 4A). In both situations, T cells rapidly vanished from the circulation after i.v. injection, but appeared in the tumor usually 2-3 days after transfer (designated as Day 0 in Fig. 4A). T cells appeared to be arrested in their movement in close vicinity of tumor vessels. Within the following 24 h, the blood flow completely ceased with or without cross-presentation (evidenced by immobile DiD-labeled erythrocytes (round) as compared to erythrocytes dashing through the tumor vessels (lines) on Day 0). The extravasation of DiD-labeled erythrocytes indicated that the tumor-endothelial barrier was destroyed. This evidence of vascular leakage was the microscopic correlate to the dark-red discoloration of the tumor observed after successful TCR gene therapy. Nevertheless, destruction of cancer cells (indicated by loss of green fluorescence observed in viable cancer cells) was clearly accelerated when 1D9 T cells could recognize cross-presented mp68 on CD11b+ tumor stromal cells (Day 1 in both Fig. 4A and Fig. 4B-C). This finding correlated with the ~10x higher IFN-γ release of T cells incubated with mp68 cross-presenting stromal cells if compared to stimulation with cancer cells (Fig. 3D). As control for the experiment, 1D9 T cells were transferred into a mouse bearing a MC57 tumor expressing an irrelevant antigen (SIYRYYGL coupled to GFP). Interestingly, 1D9 T cells did neither show homeostatic expansion (Supplementary Fig. S5) nor infiltration in the tumor (Supplementary Fig. S6). In contrast, 1D9 T cells showed strong expansion in mice bearing MC57-mp68 tumors, irrespective of H-2Kb expression by the host (Supplementary Fig. S5). Together, these data indicate that direct recognition of highly and uniformly expressed mp68 sufficed for the infused 1D9 T cells to localize to the solid tumor, find the cancer cells as stimulators to proliferate and to release sufficient amounts of cytokines to destroy the tumor vasculature and eradicate the cancer. Nevertheless, when the neoantigen was not cross-presented, cancer cell destruction was significantly delayed. Thus, stromal cross-presentation could become a determining factor for eradication of antigen-loss variants when the target antigen is not uniformly expressed by all cancer cells. This is suggested by previous experiments of adoptive therapy of tumors expressing surrogate antigens after transfection rather than transduction and containing significant numbers of antigen-negative cancer cells (see references in (36)).
Fig. 4. Stromal cross-presentation of mp68 accelerates elimination of cancer cells in established tumors whereas direct presentation by cancer cells suffices for tumor vessel destruction.

A, Longitudinal confocal microscopy imaging of cancer cell and tumor vessel destruction following adoptive T cell transfer. Cross-presentation of mp68 by the tumor stroma cause rapid destruction of cancer cells by 1D9 T cells entering the tumor. The left panel shows the longitudinal imaging of MC57-mp68 tumors in a H-2Kb-positive and the right panel shows a H-2Kb-negative Rag−/− mouse following adoptive transfer of 1D9 T cells of YFP×1D9×Rag−/− mice. Day 0 is the time when the first 1D9 T cell was detected in the skinfold window (see magnification, red). Viability of tumor tissue was analyzed by monitoring GFP expression (cancer cells, green) and blood flow (see bottom magnification, DiD-stained erythrocytes, purple). Data are representative for 3 independent experiments. B, Quantification of the timing of cancer cell and vascular viability in tumors with or without cross-presentation of mp68 by the tumor stroma shown in (A). Areas on day 0 were defined as 100%. C, Quantification of cancer cell destruction in tumors with or without cross-presentation of mp68 by the tumor stroma. GFP and DiD signals were compared to calculate the delay of cancer cell destruction after collapse of blood flow. Mean values (± SD) obtained from individual mice are shown (p=0.036, Wilcoxon Rank Sum Test). Data were pooled from 3 independent experiments.
Established 8101 tumors are eradicated by TCR gene therapy when all cancer cells express mp68 antigen at high levels
Antigen cross-presentation accelerated tumor destruction and has facilitated tumor rejection in other models (35), we assessed whether the escape of 8101 tumors by outgrowth of antigen-loss variants correlated with lack of stromal cross-presentation of mp68. Indeed, CD11b+ stromal cells isolated from Bulk tumors stimulated far less IFN-γ release by 1D9 T cells than CD11b+ stromal cells isolated from MC57-mp68 (Fig. 5A (left) compared with Fig. 3D). Thus, the level of mp68 in Bulk tumor cells may not have provided sufficient antigen for direct or cross-presentation to stimulate sufficient cytokine release to cause stromal destruction and antigen-negative variants could escape (Fig. 2B). We then generated Bulk tumor cells that were transduced to express higher levels of mp68 antigen (Bulk-mp68, Supplementary Fig. S4B). Bulk-mp68 tumor cells were killed effectively in vitro (Fig. 5B) and stimulated ~20x higher IFN-γ release by 1D9 T cells (Fig. 5A, right) by direct antigen presentation of the Bulk-mp68 cancer cells or stroma cells derived from Bulk-mp68 tumors. Most importantly, 1D9 TCR gene therapy eradicated 4 of 5 Bulk-mp68 tumors established for over a month and at least 1 cm in diameter (Fig. 5C). Thus, resistance of primary tumors to 1D9 treatment was overcome by increasing the level of mp68 antigen that is naturally expressed by Bulk tumor cells. However, because the antigen was uniformly expressed on the transduced Bulk tumor cells, there were probably no variants that could require stromal presentation to be eradicated. In any case, proper expression of the mp68 antigen allowed eradication of large Bulk tumors by adoptive transfer of peripheral T cells engineered to express a single type of TCR and specific for a single AAS.
Fig. 5. Escape of primary cancers from mp68-specific T cell therapy is thwarted by uniform and high expression of antigen or when T cell therapy follows local irradiation.

A, Bulk-mp68 cancer cells or stromal cells isolated from Bulk-mp68 tumors induce release of high levels of IFN-γ by 1D9td T cells. CD11b+ stromal cells were isolated from Bulk tumors either unmodified or over-expressing mp68. Enriched stromal cells and cancer cells of the respective lines were co-cultured with 1D9td or 2Ctd T cells. IFN-γ content of supernatants was determined by ELISA. One representative experiment of two is shown. (B) Bulk tumor cells modified to express high levels of mp68 are recognized by 1D9-transduced T cells. Specific lysis of Bulk tumor cells over-expressing mp68 (Bulk-mp68) was analyzed in vitro using 1D9td T cells. T cells transduced with the 2C TCR were used as control. One representative experiment of two is shown. C, TCR gene therapy causes rejection of Bulk tumors overexpressing mp68. Mice with established Bulk-mp68 tumors were treated with 1D9tg T cells. T cells were injected between day 41 and 81 when tumors were established; timescale indicates time post T cell transfer. Data are compiled from 3 independent experiments. D, Irradiation prevents escape of parental Bulk tumors after TCR gene therapy. Growth of established Bulk tumors in Rag−/− mice after 1D9 T cell therapy combined with local radiation (1D9tg (n=5), 1D9td (n=3)). Control mice received only 1D9 T cells (1D9tg (n=2), 1D9td (n=1)) or radiation (n=3). Mice were treated between day 28 and 40 when tumors were established. Data are compiled from 4 independent experiments (p=0.01, Log-rank-test for progression-free survival of animals receiving either 1D9 T cells alone or in combination with local irradiation).
Local tumor irradiation followed by adoptive T cell transfer reduces relapse of 8101 tumors expressing the autochthonous mp68 antigen
We have previously shown that chemotherapeutic drugs or local irradiation can synergize with ATT to achieve eradication of cancers that fail to be rejected because they express too low levels of the targeted antigen (30). The efficacy of the combined treatment correlated with antigen loading onto the stroma from cancer cells damaged or stressed by the chemo- or radiotherapy. We followed the same experimental design (30) to improve the effectiveness of mutation-specific TCR gene therapy of heterogeneous tumors expressing the original autochthonous mp68 neoepitope. Indeed, the combination with local irradiation of Bulk tumors significantly improved therapeutic outcome after adoptive transfer of ~5×106 1D9 T cells (rejection in 4/8 mice, (Fig. 5D)) while treatment of Bulk tumors with either 1D9 T cells or local irradiation alone was ineffective.
Discussion
Most human cancers have been present for years and the smallest lesions when first detected are usually at least ~1 cm in diameter harboring ~109 cancer cells. These two facts give guidance for shaping preclinical models. First, the size of the targeted cancer cell population determines the likelihood of escape and tumors in mice must have a size that provides them with a realistic chance of escaping. Escape remains the biggest threat to any cancer therapy including ATT (37). The time of establishment of experimental tumors is the second critical variable. Tumor transplantation invariably induces acute inflammation that greatly helps the host (or experimentalist) to eradicate transplanted tumors at early stages (i.e. during the first two weeks after transplantation) (38). When a tumor is targeted a month after transplantation, therapeutic procedures often become ineffective even when tumors remain rather small (39). Therefore, it is important that TCR gene therapy described in this study eradicated tumors that were not only large (~1 cm in diameter) but had also been established for a month or longer.
Our studies of the 8101 tumor demonstrate that ‘reverse immunology’, the sequencing of a tumor’s genetic information and the computational prediction of neoepitopes, predicts the neoantigen mp68 we had previously identified as immunodominant rejection antigen of 8101. While our study showed consistency between direct and ‘reverse immunology’, tracing of suitable neoepitopes without guidance by specific T cells can be misleading. For example, predicting the binding of cysteine-containing peptides to MHC is problematic (40) and some neoepitopes predicted to bind may be destroyed by proteasomal cleavage (25). Predicting neoepitopes in cancer by combining sequencing with mass spectometry analysis of eluted peptides (41) or multimer staining of specific T cells (12) could improve reliability.
We suggest that the clinically relevant situation is likely to be mirrored by tumors composed of cancer cells expressing the tumor-specific mutations at natural levels, resembled by Bulk used in this study. Even though we targeted an immunodominant rejection antigen expressed in every analyzed part of the original tumor, previously unnoticed antigen-negative variants or cancer cells that lost antigen expression foiled cancer eradication by T cell therapy. We targeted a mutation in p68, a coactivator of p53 (1), which probably played a role as driver in the formation of the cancer but sustained expression was not required for tumor maintenance. Nevertheless, mp68 provided a subnanomolar affinity neoepitope expressed in comparison with other neoepitopes of the tumor at comparatively high levels. Also, mp68 was found in all analyzed regions of the cancer, which suggested that this neoepitope was not affected by usual spatial tumor heterogeneity. Better targets would obviously be driver mutations essential for cancer cell function and survival and expressed at higher levels so that antigen escape does not occur (3,42-44). However, such ideal targets may be rare. Given the remarkable genetic and phenotypic diversity of cancers (45), we therefore believe that escape by antigen-negative variants will likely remain an important problem in targeting tumors, experimentally or clinically.
We propose several complementary solutions to solve the problem of escape from TCR gene therapy. First, targeting multiple independent neoepitopes on the same cancer could reduce the chance for therapy-induced selection of escape variants (26,46). For example, the 8101 primary cancer contained another neoepitope that was retained by mp68-loss variants and was recognized by a different cytolytic T cell clone (24). Secondly, tumor-specific CD4+ T cells facilitate the accumulation of CD8+ T cells in the tumor microenvironment (47) and also cooperate with CD8+ T cells during the effector phase in bystander killing of cancer variants thereby preventing tumor escape (48). Thus, a properly chosen combination of two mutation-specific TCRs, one restricted to MHC-I and the other to MHC-II is likely to be a fruitful approach. Certainly, focusing on highly expressed mutant proteins that are cross-presented by the tumor stroma and thereby sensitize the stroma for destruction will greatly increase the killing of antigen-negative variants as bystanders (30,35). Finally, TCR gene therapy could clinically be combined with adjuvant therapy, like chemotherapy or irradiation as used in this study. For example stereotactic irradiation can be given to many sites of tumor growth in patients. Even though such treatments may fail to kill a significant number of tumor cells, irradiation may increase trafficking of T cells to irradiated tumors (49). Furthermore, chemo- or radiotherapy can increase the amount of antigen released from cancer cells (30) as can be shown when a high affinity TCR probe is available for measuring the amount of peptide:MHC on stromal cells. This seems to be of particular importance when targeting neoepitopes whose expression levels do not support sufficient cross-presentation and stimulation of T cells. Synergy has also recently been reported between radiotherapy and checkpoint blockade (50). Together, mutation-specific TCR gene therapy could benefit from various additional clinical treatment.
Supplementary Material
Translational relevance.
Neoantigens encoded by somatic tumor-specific mutations are preferred targets for T cells. However, studies using adoptive transfer of tumor-infiltrating lymphocytes or checkpoint inhibitors rely on activating T cells that might be tolerant. Higher efficacy could be achieved when patients’ unbiased peripheral T cells are adoptively transferred after they are engineered to express mutation-specific TCRs. In a reductionist approach, we show eradication of large solid tumors after adoptive transfer of peripheral T cells engineered to express a single type of TCR and specific for a single AAS. Targeting the autochthonous neoepitope in the primary cancer caused regression, but relapse followed because of too many antigen-negative variants or low antigen expression levels on cancer cells. Rejection of the primary tumor was achieved by uniform and/or increased expression of the antigen or in combination with local irradiation. These results give important guidance for designing mutation-specific TCR gene therapy in the clinic.
Acknowledgements
We thank Kordelia Hummel for excellent technical support and David Binder for help with the radiation experiments. We thank Vytas Bindokas for guidance in the longitudinal imaging studies, Linda Degenstein at the University of Chicago Transgenics/ES Cell Technology Mouse Core Facility as well as the University of Chicago Flow Cytometry Core Facility. We also thank Thomas Blankenstein and Michael Spiotto for providing suggestions regarding the manuscript. This work was supported by NIH grants R01-CA22677 and R01-CA37156, the Cancer Center at the University of Chicago, a Collaborative Research Grant of the Berlin Institute of Health and the Einstein-Stiftung Berlin.
Financial support:
NIH grants R01-CA22677 (H.S., Y.N., W.U.) and R01-CA37156 (H.S.), Collaborative Research Grant of the Berlin Institute of Health (H.S., W.U.), Einstein-Stiftung Berlin (H.S.)
Footnotes
Disclosure:
The authors have no conflict of interests.
References
- 1.Bates GJ, Nicol SM, Wilson BJ, Jacobs A-MF, Bourdon J-C, Wardrop J, et al. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J. 2005;24:543–53. doi: 10.1038/sj.emboj.7600550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 3.Monach PA, Meredith SC, Siegel CT, Schreiber H. A unique tumor antigen produced by a single amino acid substitution. Immunity. 1995;2:45–59. doi: 10.1016/1074-7613(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 4.Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–4. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 5.Coulie PG, Lehmann F, Lethé B, Herman J, Lurquin C, Andrawiss M, et al. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci USA. 1995;92:7976–80. doi: 10.1073/pnas.92.17.7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183:725–9. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68:889–92. doi: 10.1158/0008-5472.CAN-07-3095. [DOI] [PubMed] [Google Scholar]
- 8.Offringa R. Antigen choice in adoptive T-cell therapy of cancer. Curr Opin Immunol. 2009;21:190–9. doi: 10.1016/j.coi.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19:747–52. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31:e439–42. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–81. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–91. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schietinger A, Delrow JJ, Basom RS, Blattman JN, Greenberg PD. Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science. 2012;335:723–7. doi: 10.1126/science.1214277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. 2013;31:999–1008. doi: 10.1038/nbt.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30:2843–51. doi: 10.1093/bioinformatics/btu356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 20.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 2008;36:W509–12. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engels B, Chervin AS, Sant AJ, Kranz DM, Schreiber H. Long-term persistence of CD4+ but rapid disappearance of CD8+ T cells expressing an MHC class I-restricted TCR of nanomolar Affinity. Molecular Therapy. 2012;20:652–60. doi: 10.1038/mt.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber K, Arina A, Engels B, Spiotto MT, Sidney J, Sette A, et al. Spleen cells from young but not old immunized mice eradicate large established cancers. Clin Cancer Res. 2012;18:2526–33. doi: 10.1158/1078-0432.CCR-12-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dubey P, Hendrickson RC, Meredith SC, Siegel CT, Shabanowitz J, Skipper JC, et al. The immunodominant antigen of an ultraviolet-induced regressor tumor is generated by a somatic point mutation in the DEAD box helicase p68. J Exp Med. 1997;185:695–705. doi: 10.1084/jem.185.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Popovic J, Li L-P, Kloetzel PM, Leisegang M, Uckert W, Blankenstein T. The only proposed T-cell epitope derived from the TEL-AML1 translocation is not naturally processed. Blood. 2011;118:946–54. doi: 10.1182/blood-2010-12-325035. [DOI] [PubMed] [Google Scholar]
- 26.Ward PL, Koeppen H, Hurteau T, Schreiber H. Tumor antigens defined by cloned immunological probes are highly polymorphic and are not detected on autologous normal cells. J Exp Med. 1989;170:217–32. doi: 10.1084/jem.170.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ross SR, Solter D. Glucocorticoid regulation of mouse mammary tumor virus sequences in transgenic mice. Proc Natl Acad Sci USA. 1985;82:5880–4. doi: 10.1073/pnas.82.17.5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arina A, Schreiber K, Binder DC, Karrison TG, Liu RB, Schreiber H. Adoptively Transferred Immune T Cells Eradicate Established Tumors despite Cancer-Induced Immune Suppression. J Immunol. 2014;192:1286–93. doi: 10.4049/jimmunol.1202498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204:49–55. doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, et al. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell. 2013;23:516–26. doi: 10.1016/j.ccr.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schietinger A, Arina A, Liu RB, Wells S, Huang J, Engels B, et al. Longitudinal confocal microscopy imaging of solid tumor destruction following adoptive T cell transfer. Oncoimmunology. 2013;2:e26677. doi: 10.4161/onci.26677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schreiber K, Wu TH, Kast WM, Schreiber H. Tracking the common ancestry of antigenically distinct cancer variants. Clin Cancer Res. 2001;7:871s–875s. [PubMed] [Google Scholar]
- 34.Hanson HL, Donermeyer DL, Ikeda H, White JM, Shankaran V, Old LJ, et al. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 2000;13:265–76. doi: 10.1016/s1074-7613(00)00026-1. [DOI] [PubMed] [Google Scholar]
- 35.Spiotto MT, Rowley DA, Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med. 2004;10:294–8. doi: 10.1038/nm999. [DOI] [PubMed] [Google Scholar]
- 36.Engels B, Rowley DA, Schreiber H. Targeting stroma to treat cancers. Seminars in Cancer Biology. 2012;22:41–9. doi: 10.1016/j.semcancer.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bai X-F, Liu J, Li O, Zheng P, Liu Y. Antigenic drift as a mechanism for tumor evasion of destruction by cytolytic T lymphocytes. J Clin Invest. 2003;111:1487–96. doi: 10.1172/JCI17656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wen FT, Thisted RA, Rowley DA, Schreiber H. A systematic analysis of experimental immunotherapies on tumors differing in size and duration of growth. Oncoimmunology. 2012;1:172–8. doi: 10.4161/onci.1.2.18311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schreiber H. Cancer Immunology. In: Paul WE, editor. Fundamental Immunology. Seventh Lippincott Williams & Wilkins; Philadelphia: 2012. pp. 1200–34. [Google Scholar]
- 40.Parker KC, Shields M, DiBrino M, Brooks A, Coligan JE. Peptide binding to MHC class I molecules: implications for antigenic peptide prediction. Immunol Res. 1995;14:34–57. doi: 10.1007/BF02918496. [DOI] [PubMed] [Google Scholar]
- 41.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–6. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 42.Chiari R, Foury F, De Plaen E, Baurain JF, Thonnard J, Coulie PG. Two antigens recognized by autologous cytolytic T lymphocytes on a melanoma result from a single point mutation in an essential housekeeping gene. Cancer Res. 1999;59:5785–92. [PubMed] [Google Scholar]
- 43.Beck-Engeser GB, Monach PA, Mumberg D, Yang F, Wanderling S, Schreiber K, et al. Point mutation in essential genes with loss or mutation of the second allele: relevance to the retention of tumor-specific antigens. J Exp Med. 2001;194:285–300. doi: 10.1084/jem.194.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karanikas V, Colau D, Baurain JF, Chiari R, Thonnard J, Gutierrez-Roelens I, et al. High frequency of cytolytic T lymphocytes directed against a tumor-specific mutated antigen detectable with HLA tetramers in the blood of a lung carcinoma patient with long survival. Cancer Res. 2001;61:3718–24. [PubMed] [Google Scholar]
- 45.Tao Y, Ruan J, Yeh S-H, Lu X, Wang Y, Zhai W, et al. Rapid growth of a hepatocellular carcinoma and the driving mutations revealed by cell-population genetic analysis of whole-genome data. Proc Natl Acad Sci USA. 2011;108:12042–7. doi: 10.1073/pnas.1108715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wortzel RD, Philipps C, Schreiber H. Multiple tumour-specific antigens expressed on a single tumour cell. Nature. 1983;304:165–7. doi: 10.1038/304165a0. [DOI] [PubMed] [Google Scholar]
- 47.Wong SBJ, Bos R, Sherman LA. Tumor-specific CD4+ T cells render the tumor environment permissive for infiltration by low-avidity CD8+ T cells. J Immunol. 2008;180:3122–31. doi: 10.4049/jimmunol.180.5.3122. [DOI] [PubMed] [Google Scholar]
- 48.Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J Exp Med. 2010;207:2469–77. doi: 10.1084/jem.20092450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174:7516–23. doi: 10.4049/jimmunol.174.12.7516. [DOI] [PubMed] [Google Scholar]
- 50.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124:687–95. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.