

Summary
We examined the contribution of endogenous cholinergic signaling to the acquisition and extinction of fear- related memory by optogenetic regulation of cholinergic input to the basal lateral amygdala (BLA). Stimulation of cholinergic terminal fields within the BLA in awake-behaving mice during training in a cued fear-conditioning paradigm slowed the extinction of learned fear as assayed by multi-day retention of extinction learning. Inhibition of cholinergic activity during training reduced the acquisition of learned fear behaviors. Circuit mechanisms underlying the behavioral effects of cholinergic signaling in the BLA were assessed by in vivo and ex vivo electrophysiological recording. Photo-stimulation of endogenous cholinergic input: (1) enhances firing of putative BLA principal neurons through activation of acetylcholine receptors (AChRs); (2) enhances glutamatergic synaptic transmission in the BLA and (3) induces LTP of cortical-amygdala circuits. These studies support an essential role of cholinergic modulation of BLA circuits in the inscription and retention of fear memories.
Introduction
Emotionally salient experiences can be indelibly inscribed in memory. Recall of either highly aversive or appetitive events engages distributed brain networks whose activity is tuned by various neuromodulators, including acetylcholine (ACh; (Hasselmo and Sarter 2011, Hermans et al. 2014, LeDoux 2012, Luchicchi et al. 2014, Picciotto et al. 2012). Cholinergic signaling has long been implicated in attending to and remembering emotionally potent experiences, but the precise role of ACh signaling in mnemonic processing of highly salient events has remained elusive (Dani and Bertrand 2007, Hasselmo and Sarter 2011, Luchicchi et al. 2014, Sarter et al. 2014). The major challenge to dissecting the role of endogenous ACh in the formation and retention of potent memories has been the inability to selectively and reversibly manipulate the activity of cholinergic inputs, which tend to be sparsely distributed, extensively arborized and intermingled with projections of other transmitter phenotypes (Wu et al. 2014).
Studies of fear in humans and in rodents have implicated the amygdala as an essential circuit component in the network that encodes the formation, recall, and extinction of fear memory (Adolphs 2013, Duvarci and Pare 2014, Herry and Johansen 2014, Zelikowsky et al. 2014). In particular, direct activation of principal neurons within the basal lateral amygdala (BLA) has been shown to be sufficient to associate sensory input with conditioned fear responses (Johansen et al. 2010, Tye et al. 2011). The strong similarities between the networks that are related to fear in humans with those in other species underscores the potential utility of dissecting the modulation of amygdala circuitry in animal models as a vital step to inform development of treatments for anxiety and fear disorders (Schiller and Delgado 2010).
In both humans and rodents, the BLA receives a particularly rich cholinergic projection arising primarily from the Nucleus Basalis (NBM) with some contribution from the horizontal limb of the diagonal band of Broca (Woolf 1991). The NBM cholinergic neurons and their projections to the BLA are interspersed with neurons and fibers of other transmitter phenotypes, preventing selective stimulation or inhibition by standard electrophysiological or pharmacological techniques. Even with selective immunoablation of basal forebrain cholinergic neurons, defining the role of ACh signaling in fear learning has remained a challenge (Baxter and Bucci 2013). In this study we used optogenetic techniques to specifically stimulate or inhibit of cholinergic inputs within the BLA, enabling us to assess the contribution of cholinergic signaling to cue conditioned fear behaviors in mice, and to test the role of cholinergic inputs in the modulation of cortical-BLA circuit plasticity in in vivo preparations and in acute, ex vivo brain slice preparations.
Results
Our first goal was to develop and validate methods for the selective activation of cholinergic neurons and their terminal fields in the BLA of awake-behaving mice, anesthetized mice, and in acute ex vivo slices of the BLA (Fig 1). We have used a set of excitatory (oChIEF); and inhibitory (eNpHR3.0) light sensitive probes engineered into adeno-associated viral (AAV) vectors. Both vectors are a flip excision switch design that requires Cre-mediated recombination for expression (Fig 1A). Viruses were injected into the basal forebrain nuclei of ChAT-Cre mice at one week post weaning (Fig 1B, top), resulting in robust expression of the labeled opto-probes in ChAT positive (cholinergic) neurons in the NBM and throughout the cholinergic terminal fields within the BLA 2–4 weeks post-infection (Fig 1C top).
FIGURE 1. Selective, optogenetic labeling of cholinergic NBM neurons and of their terminal field projections within the BLA.

A: Schematic of the viral vector (AAV9-DIO-oChIEF-tdTomato) used for optogenetic photo-stimulation experiments. The inverted open reading frame encodes oChIEF-tdTomato driven by the CAG promoter and flanked by loxP and lox2272 sites. In some experiments the vector was AAV9-Eflα-DIO-eNpHR3.0-eYFP.
B: Schematic of optogenetic labeling. Upper: AAV9-DIO-oChIEF-tdTomato was injected into the NBM of ChAT-Cre mice. Lower: To confirm the specificity of viral targeting to cholinergic neurons, the vector was injected into double transgenic mice expressing both Cre and a tauGFP fusion protein under the control of the ChAT promoter. Calibration 50 μm.
C: Confocal photomicrographs of single and double labeled NBM cell bodies and projections. Top left: oChIEF expressing NBM neurons (Calibration 50 μm). Top right: oChIEF labeled terminal fields in the basal lateral amygdala (BLA) but not the central amygdala (CeA) (Calibration 150 μm). The bottom images in C are double labeled by ChAT tau-eGFP (green) and oChIEF-tdTomato (red); left: NBM, right: NBM projections in the BLA (calibration 30 μm). The inset in the bottom right panel is a high power view of a ChAT+ NBM axon in the BLA showing the cytoskeletal staining of the tau-eGFP (green), surface labeling of the oChIEF-tdTomato (red) and the overlapping staining (yellow). Calibration 10 μm.
D: Quantification of oChIEF-tdTomato labeling of ChAT+ neurons. Left panel: number of NBM neurons per 100 μm thick coronal sections labeled by tau-eGFP (green), by oChIEF-tdTomato (red) and by both (yellow). Right panel: plot of the percentage of neurons in the NBM that expressed both tau-eGFP and oChIEF (n=6 mice). Data are shown as mean + SEM.
E: In vivo recording of oChIEF expressing NBM neurons. Left panels: Schematic of in vivo photo-stimulation and recording in NBM. Photo-stimulation was delivered at a rate of 1 to 25 Hz. Right panel: Plot of spike fidelity vs photo-stimulation frequency in vivo. Repeat photo-stimulation reliably triggered action potentials to each light pulse at frequencies of 1Hz- 20 Hz. (Calibration, top trace: 50 mV x 2 msec; bottom: 100 mV x 50 msec).
F: Ex vivo, whole cell patch clamp recording of oChIEF expressing cholinergic neurons in NBM. Left panel: Schematic of photo-stimulation and recording configuration in acute slice. Sample recordings of photo-stimulated action potentials at fast (above) and slow (below) time scales. Right panel: Photo-stimulation reliably triggered action potentials to each pulse up to 15 Hz. (Calibration: top: 30 mV x 2 msec; bottom: 10 mV x 100 msec).
G: Conductance response vs light intensity curve for photo-stimulation of oChIEF expressing NBM neurons. Top left: schematic of recording configuration in acute slice. Top Right: sample trace of light induced current in NBM cholinergic neurons (calibration 100 pA X 2 ms). Plot of the net charge elicited by 5 msec pulses of light at varying power levels. Maximal levels of net charge are elicited at 1 mW/mm2. Repeat pulse responses of consistent conductance are detected with light intensities of 1–7 mW/mm2. See Fig S1, S2 and S3.
Specificity and efficiency of viral vector expression in cholinergic neurons
To confirm specific viral expression in cholinergic neurons and their projections, we crossed ChAT-Cre mice with mice in which all the cholinergic neurons were genetically labeled with tauGFP, yielding a mouse line that expressed both Cre and tauGFP in all cholinergic neurons (Fig 1B, bottom). ChAT-Cre/tauGFP mice were injected with AAV9-DIO-oChIEF-tdTomato at 1 week post-weaning. Two weeks after gene delivery, we prepared coronal brain slices containing the NBM (bregma −0.4mm to −1.06mm) and quantified viral expression by counting the number of cells that were singly labeled with either the tauGFP or oChIEF-tdTomato and the number of cells that were doubly labeled with both fluorescent proteins (Figure 1C). Quantification of the total number of neurons per z stack projection that were singly labeled by either the tauGFP or oChIEF-tdTomato and the number of NBM neurons that were doubly labeled revealed an infection efficiency of ~50% (45.1 ± 1.6%, n= 6 animals). Viral targeting was specific for ChAT+ neurons and their projections: only 3% of the oChIEF-tdTomato positive NBM neurons lacked detectable tauGFP staining (Fig 1D). Likewise, high resolution imaging of the cholinergic projections within the BLA revealed that oChIEF-tdTomato labeling along axonal processes was localized immediately adjacent to, and overlapping with tauGFP (Fig 1C, bottom right; inset).
Fidelity of photo-stimulated suprathreshold firing in oChIEF expressing cholinergic NBM neurons
We next tested the properties of oChIEF expressing cholinergic NBM neurons in ChAT-Cre mice in both in vivo and in acute, ex vivo preparations. In vivo photo-activation of oChIEF expressing neurons within the NBM was elicited by 5 msec pulses of 473 nm laser light delivered at 1–25 Hz through a 200 μm diameter fiber-optic stereotactically positioned in the NBM. Extracellular recordings from the NBM reliably revealed one-to-one, photo-stimulation-locked action potential firing up to frequencies of ~20 Hz. Spike fidelity dropped off at higher stimulation frequencies (Fig 1E). Spike fidelity also dropped off at 20 Hz in whole cell patch clamp recordings from oChIEF expressing NBM neurons identified in acute slice (ex vivo) studies (Fig 1F). Ex vivo recordings in NBM demonstrated that tauGFP positive (“ChAT+”) neurons significantly differed from tauGFP- (“ChAT−”) in firing rate, threshold for firing and spike waveform and that the spike properties of oChIEF expressing, tauGFP+ neurons were identical to those of tauGFP+ NBM neurons that did not express oChIEF, suggesting that cholinergic NBM neurons were unaffected by expression of oChIEF-tdTomato (Fig S1).
In addition to assessing whether photo-stimulation effectively elicited depolarizing responses in the NBM, we further examined the relationship between the intensity of light exposure (mW/mm2) and the net change in conductance evoked in oChIEF expressing NBM neurons (Fig 1G). Light intensities of 0.7 mW/mm2 were sufficient to evoke maximal conductance responses. There was no further increase- or decrease- in light-evoked conductance with as much as 7 mW/mm2.
To assess potential experimental confounds of photo-stimulation per se, we performed a series of control studies using coherent wave length illumination in mice labeled with floxed viral GFP probes lacking the associated opsin channels and in ChAT-tauGFP mice (Fig S2). These illumination controls, in addition to studies with floxed opsin containing virus with mis-matched wavelength illumination, indicate that there were no confounding effects of illumination per se on behavioral measures, NBM or BLA excitability or cortical-BLA synaptic transmission in either in vivo or ex vivo studies (Fig S2).
Optogenetic manipulation of cholinergic tone in BLA alters fear learning and extinction behaviors
Because of the established literature relating the BLA to associative fear-conditioning (Gale et al. 2004, Myskiw et al. 2014) and the continuing debate as to the role of cholinergic signaling in fear and extinction learning behaviors (Knox and Keller 2015, Kutlu et al. 2016, Lalumiere and McGaugh 2005, Pidoplichko et al. 2013, Santini et al. 2012), we tested the effects of optogenetic manipulation of endogenous ACh signaling in the BLA using a classical associative, cue conditioning paradigm. To assess the contribution of cholinergic signaling in the BLA to fear learning and recall as well as to subsequent retention of extinction learning, we photo-activated oChIEF or eNpHR3.0 expressing cholinergic terminal fields in ChAT-Cre mice during the training period of the associative learning paradigm via implanted fiber optic cannulae targeted to the dorsal surface of the BLA (Fig 2).
FIGURE 2. Optogenetic manipulation of cholinergic signaling in BLA regulates fear and extinction learning Effects of activating cholinergic terminal fields in BLA during tone-shock fear-conditioning (A – C).

A: Training - Top: Schematic of training with optogenetic stimulation of oChIEF expressing cholinergic terminal fields in BLA. On the training day tone/shock pairs (green block/black line) were presented 3 times at 2 min intervals. At 27.5 s after each tone onset, mice in the opto group received 100 × 5 msec 473 nm optical pulses (20 Hz, 10 mW/mm2) to the dorsal surface of the BLA. Mice in control group were implanted with light guides but received no optical stimulation. Bottom: Plot of percent time spent freezing during training (opto-stimulation group: grey/blue boxes; control group: black boxes).
B: Recall - Top: Schematic of paradigm for test of recall. Twenty four hours after training mice were exposed to the same 30 s tone as on the training day and recall was measured as the time spent freezing. Bottom: There was no significant difference in recall between Control (Cont.) and oChIEF activated groups. Data are presented in box plot format to show the full range and distribution of the data; the boundaries of the box demarcate 75% of the data points, the whiskers delineate the 95% range and additional values outside of the 95% range are shown. The horizontal bar indicates the population median.
C: Extinction – Top: Schematic of paradigm for extinction training. Extinction learning involved exposing the mice to the same 30 s tone 10 times at pseudo-random intervals. Bottom: Retention of extinction learning was tested 24 hours after each extinction training session by measuring the time spent freezing in response to the first 30 s tone (mean ± SEM). Extinction training and test of recall were repeated daily for 7 days. Significant differences in freezing as a function of days of extinction learning were found between control and oChIEF groups at all days except on day 7.
Inhibition of cholinergic terminal fields: effects on recall and extinction (D – F)
D: Training – Top: Schematic of training paradigm with optogenetic inhibition of cholinergic terminal fields in BLA. On the training day, mice were exposed to one 30 s, 5 kHz, 80 dB tone (green block) co-terminating with a 2 s, 0.8 mA foot shock (black line). At the tone onset, mice in the Halo group (grey/yellow; n=22), received 2.5 min of continuous optical stimulation (12–15 mW/mm2, 590 nm, yellow block) of the dorsal surface of the BLA. Control mice (black, n = 18) received no optical stimulation. Bottom: Plot of time spent freezing over the 4 min of training with or without optogenetic inhibition. Prior to the cue x optogenetic pairing there was no significant difference in time spent freezing between groups. Immediately after the onset of optical stimulation, a highly significant difference in freezing was found (mean ±SEM; p < 0.001).
E: Recall - Top: Schematic of test of recall (as in 2B). Recall was measured as the time spent freezing to the tone. Bottom: There was a statistically significant difference in the extent of Recall between Control and Halo trained groups (p = 0.018).
F: Extinction - Top: Schematic of paradigm for extinction training. Extinction training and test of retention of extinction learning were performed as described above. Bottom: Overall there was no significant difference in the retention of extinction learning between groups.
G: Effect of intra-BLA infusion of cholinergic antagonists on fear conditioning and retention of extinction learning. To test the contribution of acetylcholine receptors to fear conditioning and retention of extinction learning we compared the behavior of mice with bi- lateral, intra BLA injections of saline alone (Vehicle Control) vs those injected with blocking concentrations of both nicotinic and muscarinic acetylcholine receptor antagonists (mecamylamine 20uM + atropine 1uM) vs those injected with blocking concentrations of atropine alone (1uM). Blocking concentrations of both nAChR and mAChR antagonists, but not mAChR antagonists alone, reduced recall (saline n=17, atropine & mecamylamine n=8, atropine n=9; one way ANOVA with Bonferroni correction: F(2,31)=6.935, p = 0.003 for group effect; saline recall vs atropine & mecamylamine recall, p=0.017; saline recall vs atropine alone recall, p=0.797; atropine alone recall vs atropine & mecamylamine recall, p=0.003). There was significantly greater retention of extinction learning in the group administered both atropine & mecamylamine as compared to vehicle control, while the degree of retention of extinction learning was unaffected by atropine (one way ANOVA on ranks; H=6.417, df=2, p=0.04; control vs mixed antagonists p<0.05).
H: Location of fiberoptic track terminations in fixed sections in oChIEF expressing (blue dots), or Halo–expressing mice (yellow dots). See Fig S2, Supplemental Video and Table S1.
We first tested whether altering the activity of cholinergic inputs in the BLA during the conditioned fear training per se would alter the extent of fear learning (Fig 2A, D). Control animals were either virally infected with fiber optic implants, but were not light exposed during training (Fig 2), or were infected with an AAV encoding GFP alone and were exposed to light during the training period (Fig S2). In addition, we compared the effects of a single tone-shock pairing with that of three tone-shock pairings (Table S1.). Post-hoc analyses across the 9 separate cohorts (87 mice; Table S1) revealed that all of the control condition mice were equivalent in their freezing behavior during the pre-training, training, and recall phases. The number of pairings did affect subsequent retention of extinction learning. For experiments using photo-stimulation we focused on the three tone-shock pairing to test the effect of increased cholinergic activation on the durability of fear learning, whereas for photo-inhibition of the cholinergic inputs we focused on the single pairing protocol, to minimize laser exposure times. Experimental animals were processed identically to controls except that these mice were exposed to photo-stimulation (oChIEF; 473 nm) or photo-inhibition (Halo; 590 nm) during the training period (Fig 2A, D respectively). Note that the photo treatments were only administered during the pairing protocol of the fear training; the test of the establishment of fear-learning and all assessments of the retention of extinction learning were conducted without any additional manipulations of cholinergic inputs by light (Fig 2A vs. B, C; Fig 2D vs. E, F).
For activation of cholinergic inputs during training (Fig 2A; Table S1) oChIEF-expressing cholinergic fibers in the BLA were photo–activated concurrently with each tone/shock pairing during the training period. Over the course of the training protocol, the freezing behavior of both the control mice (no light, n=15) and the blue light exposed mice (n=18) showed a significant increase with time [F(17, 594) = 5.54, p< 0.0001]. However, there was no significant difference in the extent of freezing between the control and the photo-stimulated groups during the training period [F(1, 594) = 0.56, p = 0.45; Fig. 2A].
Twenty four hours post training we measured the extent of fear learning by assessing freezing behavior to the tone alone (Recall). In oChIEF expressing mice, the experimental (i.e. photo-stimulated during training) and the control mice displayed a similar, elevated freezing behavior in response to cue alone compared to freezing in response to the cue prior to training [control, p<0.001, U=84, oChIEF activated, p<0.001, U=78, Mann-Whitney Rank Sum test]. Comparison of the extent of recall in controls with those subject to optogenetic stimulation of oChIEF-expressing cholinergic inputs to the BLA during training revealed that photo-stimulation was without significant effect [Fig 2B; Recall following 3 tone-shock pairings, n= 2 cohorts, 15 controls vs. 18 photo-stimulated, Mann Whitney Rank sum; U=102.5, p = 0.247].
In contrast, subsequent examination of the time course of retention of extinction learning revealed that mice that had been exposed to photo-stimulation of the oChIEF expressing cholinergic terminal fields in the BLA during the initial training were significantly more resistant to extinction learning than controls (Fig 2C: Supp. Expt. Proc., Table S1). Control animals displayed significant reductions in the amount of time spent freezing to cue after only 1 day of extinction learning and returned to a new steady-state, baseline level of freezing within 3 days. In contrast, oChIEF expressing mice that had received photo-stimulation during the initial training period, showed a significantly greater response to the tone than the controls through 5 days of extinction training (Wilcoxon Rank Sum; oChIEF stimulated vs control; E1 p < 0.003; E2 p < 0.002; E3 p < 0.003; E5 p < 0.05 vs E7 p = 0.34). The difference in the profile of retention of extinction learning between groups was also significant as compared by two-way ANOVA [F(1, 227) = 26.784, p< 0.001; Fig 2C]. These results imply that increased endogenous cholinergic signaling in the BLA renders the fear-memory more resistant to extinction.
Photo-inhibition of cholinergic terminal fields within the BLA during training resulted in a strong and virtually immediate inhibition of the freezing response to the tone/shock pairing (Fig. 2D). This difference was highly significant [2-way ANOVA, F(1,181) = 51.8, p< 0.001]. Motor activity during periods in which there was no freezing did not differ between photo-inhibited and control mice (see Supplemental Video). Measures of recall of fear-learning 24 hours after training, again revealed significant increases in freezing in response to the cue in both control [n=18; p<0.001] and eNpHR3.0 expressing, photo-inhibited mice [n= 22; p<0.001]. However, comparison of the extent of recall revealed that the learned cue association was significantly decreased by photo-inhibition during training (Fig 2E; Mann-Whitney Rank Sum, U=226; p=0.018; n=18 controls vs 22 opto-inhibition; Supp. Video). Comparison of the profile of the retention of extinction learning in controls vs animals subjected to inhibition of cholinergic input during fear training did not reveal any difference (Fig 2F; two-way ANOVA [F(1, 188) = 0.836, p = 0.362]).
In sum, these data underscore the substantive and distinct effects of enhancement vs those of suppression of cholinergic signaling in the acquisition and extinction of fear conditioning behaviors. Cue conditioned fear-learning is robust 24 hours after training. Inhibition of cholinergic input within the BLA concomitant with cue-shock pairing was sufficient to significantly decrease the extent of learned fear behaviors. In contrast, photo-stimulation of cholinergic input within the BLA during training, did not alter the extent of recall, but significantly decreased the retention of the extinction of learned fear behavior.
In light of recent controversies about the role of cholinergic signaling per se, as opposed to other co-released neurotransmitters in striatum and cortex (Higley et al. 2011, Ren et al. 2011, Saunders et al. 2015) we directly tested the contribution of acetylcholine receptors (AChRs) in fear conditioning and retention of extinction learning. We examined the effects of intra-BLA infusion of vehicle vs. infusion of AChR blockers via chronically implanted cannulae during training, on recall and retention of extinction without further manipulation of the cholinergic signaling (Fig 2G; Table S1). The results of these experiments were (i) intra BLA infusion of vehicle alone did not alter the strength of fear conditioning or retention of extinction; (ii) intra BLA infusion of blocking concentrations of both nicotinic (nAChR) and muscarinic (mAChR) antagonists significantly depressed fear learning (ANOVA F(2,31)=6.935, p=0.003; control, n=17 vs. mixed antagonists, n=8, p<0.05) and enhanced the retention of extinction learning compared with vehicle control (one way ANOVA on ranks; H=6.417, df=2, p=0.04; control vs mixed antagonists p<0.05). In the 9 mice infused with blocking concentrations of mAChR antagonist alone, fear conditioning and extinction were not significantly different from the vehicle control, consistent with an important contribution of nAChR signaling. Post-hoc histological verification was used to confirm cannula placements and virus expression in the BLA (Fig 2H).
Excitation of BLA Neurons by Optogenetic Stimulation of Endogenous Cholinergic Input in vivo
We next sought to investigate the circuit level mechanisms that might underlie the behavioral effects of optogenetic stimulation or inhibition of cholinergic terminal fields in the BLA by combining photo-stimulation and electrophysiological recording in vivo (Fig 3). All in vivo experiments were conducted 3 to 4 weeks following injection of AAV-DIO-oChIEFtdTomato into the NBM/DB of ChAT-Cre mice. For a subset of these studies we also employed broad spectrum antagonists of acetylcholine receptors to complement the studies on the effects of Halorhodopsin and to assess if the effects of activating or inhibiting cholinergic terminal fields result from release of acetylcholine per se. In all in vivo recording studies animals were anesthetized and optical fibers, coupled to extracellular recording electrodes, were stereotactically positioned within the BLA to allow optogenetic stimulation of local cholinergic terminal fields and simultaneous collection of extracellular single unit recordings from BLA principal neurons. Single units were identified by a combination of template waveform matching and cluster analysis of primary components (Fig 3 and Fig S4). A subset of studies utilized an “injectrode” configuration to permit concurrent, local delivery of AChR antagonists.
FIGURE 3. Enhanced firing of BLA principal neurons elicited by photo-activation of oChIEF expressed in cholinergic terminal fields in vivo.

A: Top: Schematic of the recording configuration allowing optical stimulation of cholinergic projection fibers in the BLA with simultaneous extracellular recording. The inset shows sample waveforms obtained from a regular spiking unit that was included in analysis (black) and a fast spiking unit whose cellular identity was ambiguous and was therefore excluded from analysis (grey). Calibration: 500 mV X 0.2 msec. Middle and bottom: Representative traces from one of the 38 regular spiking neurons and the corresponding peri-stimulus histogram illustrating the modulation of spontaneous spike frequency after delivery of 100 pulses of 473 nm light (indicated by vertical blue bars; Spike amplitude: 200mV, optical stimulation duration: 5 s).
B: Population (n=19) frequency vs peri-stimulus time plots recorded before, during and after photo-activation of cholinergic terminal fields in the BLA during infusion of artificial CSF (control), of both AChR antagonists (MEC + ATR at 10μM and 500 nM, respectively) and after antagonist washout (Recovery). During the control period opto-stimulation evoked a statistically significant increase in firing frequency (from 0.71 Hz to 0.99 Hz). In the presence of MEC+ATR the baseline firing frequency was suppressed by more than 50% (0.32 Hz). Subsequent photo-stimulation in the presence of these AChR antagonists did not change unit activity. Both the baseline BLA firing rate and the excitatory response to photo-stimulation recovered after antagonist washout. Insets show sample waveforms of a representative unit before, during and after drug administration. Dotted lines show the average firing frequency obtained during the 20 s before and after optical stimulation. The black line represents the instantaneous mean frequency, while the shaded areas represent standard error of the mean (Calibration 500 mv X 0.2 msec).
C: Statistical analysis of firing rates obtained pre (black) and post (blue) photo-stimulation of cholinergic terminal fields within the BLA shows a significant excitatory response to opto-stimulation during the control/vehicle period and after saline wash out, but not in the presence of AChR antagonists (control n = 19, MEC+ATR n = 14, Recovery n = 14, within condition paired t-tests * p < 0.05, data shown as mean + SEM).
D: Cumulative spike count before and after photo-stimulation of cholinergic terminal fields (n=38 units) shows an approximately 400 msec pause in firing immediately after the onset of optical stimulation of cholinergic signaling (indicated by the blue bars) prior to the increase in firing rate seen in all 38 units. Inset: cumulative spike count over a narrower time range, before and during onset of photo stimulation.
E: Frequency vs peri-stimulus time plots of recordings obtained before, during and after photo-activation of cholinergic terminal fields in the BLA under control conditions, or in the presence of mecamylamine (20 μM). Blue bars represent the timing of the opto-stimulation. Dotted lines show the average firing frequency obtained during the 80 s before and after optical stimulation. The black line represents the instantaneous mean frequency, while the shaded areas represent standard error of the mean. See also Fig S2, S3 and S4.
The data presented in Fig 3 are from a total of 50 units recorded in animals with oChIEF expression in the cholinergic terminal fields within the BLA and with optical fiber and electrode/injectrode placement confirmed post hoc. Early studies examined the parameters for photo-stimulation and found that responses were optimized at 100 pulses (Fig S3). Figure 3A illustrates the experimental configuration for stimulation and recording (top) and shows a representative raw data trace of spike activity in one of the 38 regular spiking BLA neurons studied (middle). The inset of two spike waveforms shown in the top panel of Figure 3A illustrates the profile of a “regular spiking” BLA neuron (black) and a “fast spiking” neuron (grey). Regular spiking BLA neurons- which have longer duration action potentials and lower firing rate than the briefer and faster firing neurons- comprise >75% of the isolated units and are considered putative BLA principal neurons (Duvarci and Pare 2014). The relatively rare, fast firing neurons are thought to be BLA interneurons and were not included in this study. Below the raw data trace of this sample regular spiking neuron is the corresponding peri-stimulus time histogram (PSTH) of activity recorded for 20 seconds before and after photo-stimulation of oChIEF expressed in cholinergic inputs (5 sec, 20 Hz, 473nm). Photo-stimulation of cholinergic input to the BLA (indicated by the blue bars) elicited a robust increase in firing rate in this BLA unit, from 0.5 Hz before photo-stimulation to rates ranging from 2 to 6.5 Hz (average = 2.6 Hz) for the 20 seconds following photo-stimulation (Fig 3A, bottom and Fig S3).
We next tested whether the observed increased firing of putative BLA principal neurons was dependent on the activation of AChRs. We first tested the efficacy of a mixture of broad spectrum muscarinic and nicotinic AChR antagonists delivered locally to the recording site. Pooled data for 22 regular spiking BLA single units that were stably recorded through the control period and during and after intra-BLA infusion with the AChR blockers are shown in the merged PSTHs (Fig 3B, Fig S4). The insets in Fig 3B show examples of spike waveforms before, during and after recovery from drug treatment. Control and recovery conditions included intra BLA infusion with ACSF. The consistency of these waveforms suggests that it is the same unit recorded under all 3 conditions. Additional evaluation of the stability of recordings throughout these manipulations was obtained by principal component analyses (Fig S4). All 22 of these regular spiking units exhibited a statistically significant increase in firing rate in response to photo-stimulation of cholinergic input under control conditions (vehicle control: Fig 3B, top and Figure 3C left; R2=0.147; p≤0.05). Injectrode delivery of both atropine and mecamylamine within the BLA resulted in a profound, but reversible, decrease in baseline firing, as well as an ablation of the photo-stimulated increase in spiking activity of all of the units studied. These findings are consistent with AChR- mediated effects of photo-stimulation (Fig 3B, C middle panel). In addition, the notable decrease in baseline firing rate following delivery of the AChR antagonist cocktail (but prior to photo-stimulation) is consistent with control of baseline activity of putative BLA principal neurons, at least in part, by ongoing activation of AChRs. Both baseline activity and photo-stimulated increases in BLA firing recovered to pre-antagonist levels following a 30–60 min wash out of the antagonist cocktail, and with infusion of the vehicle control (Fig 3B,C last panel). Following drug wash out, the average firing rate in the vehicle control returned to ~0.7 Hz; the average firing with vehicle, post-photo-stimulation was increased to 1.0 Hz (R2=0.106; p ≤ 0.05; Fig 3C, right).
A subset of experiments testing the effects of the nicotinic AChR antagonist, mecamylamine (MEC), alone revealed a qualitatively similar profile to the mixed AChR antagonist cocktail. BLA unit baseline firing was decreased, the photo-stimulated firing rate increase was blocked, and both phenomena were fully recovered following washout of mecamylamine. As such, we conclude that nicotinic AChRs contribute to both the baseline firing and the observed increase in BLA firing elicited by optogenetic stimulation of cholinergic inputs to the BLA (Fig 3E). These data are consistent with our findings on effects of AChR antagonists on fear conditioning and retention of extinction learning (Fig 2G).
To examine the temporal relationship between changes in firing of regular spiking BLA neurons and photo-stimulation of the cholinergic terminal fields in BLA in vivo, we plotted cumulative spike counts for all 38 regular firing units and examined the population data obtained for the 2000 msec immediately before and after the onset of the photo-stimulation (Fig 3D; 20 Hz photo-stimulation indicated by vertical blue lines). This analysis revealed interesting complexity in the temporal pattern of activity following photo-stimulation of cholinergic terminal fields. The first 1–8 flashes elicited an immediate pause in firing that lasted ~400 msec (Fig 3D; inset). This brief pause was followed by a sustained increase in firing frequency that remained elevated for up to 30 minutes following termination of the photo-stimulation.
Photo-stimulation of cholinergic input in acute slice (ex-vivo) preparations
We next sought to examine the synaptic mechanisms by which ACh exerts circuit level effects in the BLA by combining photo-stimulation of cholinergic terminal fields with whole cell patch clamp recording from putative BLA principal neurons in ex vivo (acute slice) preparations. All ex vivo recordings were from adult mice of the same age as those tested in both the behavioral and in vivo recording studies. First, we tested the effect of photo-activation of oChIEF expressing cholinergic input on firing rates of putative BLA principal neurons (n=21). The typical firing rate of these neurons at rest potential (Vm= −55–60 mV) was extremely low (<0.1–0.5Hz; Fig 4Bi). Current injection to depolarize the BLA neurons increases the baseline spike rate to 1 Hz (Fig 4Bii). Under both of these recording condition, 10 Hz photo-stimulation of cholinergic terminal fields elicited a robust increase in firing rate that persisted above baseline rates for 5–35 minutes (Fig 4C).
FIGURE 4. Enhanced firing of BLA neurons elicited by photo-activation of oChIEF expressed in cholinergic terminal fields ex vivo.

A: Schematic of configuration for photo-stimulation and recording in acute slice preparation.
B: Current clamp recordings from two regular spiking BLA neurons before, during and after 100 flashes of 473 nm light (10 Hz x 10s; 5 msec duration). (i) Representative recording at resting membrane potential = −60mV (n=9 neurons). The spontaneous action potential frequency at rest potential (<0.1 Hz) increased by 5–10 fold after photo-stimulation. One of 9 neurons tested showed a small hyperpolarization prior to subsequent increased firing. (ii) Representative recordings with current injection to bring membrane potential to ~−50mV (n=12 neurons). Nine of 12 neurons showed a 5mV hyperpolarization in response to blue light that was followed by increased firing rate.
C: Population firing frequency responses to photo-stimulation in regular spiking BLA neurons at rest potential. Left: box and scatter plots of firing frequency data prior to blue light stimulation (Cont.), at the peak firing frequency and the mean firing frequency during the 10 min following photo-stimulation. There was a statistically significant increase in both the peak and the mean firing frequency (Paired Wilcoxon Signed Rank Test, Peak, p = 0.031, n=6; Mean, p=0.031, n = 6). Right: Representation of the duration of elevated activity following photo-stimulation of cholinergic inputs to BLA (n=6).
D: Firing frequency ± photo-stimulation in sequential recordings from putative principal BLA neurons treated with muscarinic and/or nicotinic AChR antagonists. Sequential recordings show robust increase in firing rate with the first round of photo-stimulation (p<0.05, n=3). Photo-stimulation in the presence of atropine (0.5 μM) elicited a significant increase in firing (p<0.05, n=3). Following washout of atropine and wash in of mecamylamine (10 μM), control baseline firing rates were restored. Subsequent photo-stimulation in the presence of mecamylamine was without significant effect. Recovery of photo-stimulated increase in firing was seen after washout of mecamylamine (p<0.05; n=3) Right: Opto-stimulation with both mecamylamine and atropine in the bath did not increase the firing rate. All data are presented normalized to initial control firing rate. See also Fig S2.
The timing of detectable changes in membrane potential relative to the onset of photo-stimulation appeared to depend on the prior state of the recorded neuron. In 9 of 12 neurons in which the membrane potential was set to −50mV by current injection, photo-stimulation elicited a membrane hyperpolarization within ~80 msec of the first pulse (Fig 4Bii). Without current injection, small (2–4 mV) depolarizations could be detected in 4 of 9 neurons immediately after the first pulse (i.e. within the time required to fully open the shutter, ~8ms). In all but one of the neurons examined at their resting membrane potential (i.e. in the absence of current injection), membrane hyperpolarization was not detected. All 21 neurons studied by ex vivo current clamp recording displayed increased firing rate within 1–10 seconds of the onset of photo-stimulation of the cholinergic terminal fields within the BLA, as seen in the in vivo recordings. This increase in firing rate post photo-stimulation of the cholinergic inputs to BLA was seen whether or not it was preceded by a transient hyperpolarization.
To further assess the ACh receptor(s) that mediate the photo-stimulation induced increase in firing frequency we repeated this experiment in the presence of mAChR and nAChR antagonists. Fig 4D presents data from sequential recordings in a subset of neurons that met recording criteria throughout the time required to test all 4 conditions (i.e. control ± photo-stimulation, mAChR antagonist alone ± photo-stimulation, nAChR antagonist alone ± photo-stimulation and recovery/post control ± photo-stimulation). The initial response to photo-stimulation was a robust increase in firing (~4x control). After the firing rate had returned to baseline or near baseline levels (10–30 min) we tested the mAChR antagonist atropine. Atropine (0.5 μM) did not block the increase in action potential firing elicited by photo-stimulation. In some experiments, application of muscarinic antagonists appeared to somewhat increase baseline firing frequency and augment the excitatory effects of photo-stimulation (Fig 4D). The nAChR antagonist mecamylamine, significantly inhibited the increased firing frequency of BLA principal neurons that was elicited by photo-stimulation (Fig 4D). The photo-stimulated response completely recovered following washout of all antagonists (Fig 4D). Overall, these sequential recordings (Fig 4D) and a separate set of population recordings with both muscarinic and nicotinic blockers (Fig 4D right; Mec + Atrop ± photo-stimulation, n = 11; Wilcoxon Rank Sum, p=0.12), indicate that both mAChRs and nAChRs appear to contribute to the changes in BLA excitability elicited by photo-activation of cholinergic terminal fields. Thus, endogenous ACh released by stimulation of the NBM projections to the BLA - with the same pattern as used in our behavioral and in vivo recording studies – increased BLA pyramidal neuron excitability by activating AChRs. In view of the effects of AChR antagonists on baseline firing, ongoing AChR activity may influence resting activity ex vivo as it does in vivo.
Endogenous ACh signaling modulates glutamatergic synaptic transmission in BLA
The observed increase in supra-threshold activity following photo-stimulation of oChIEF-expressing cholinergic terminal fields in BLA could arise by direct and/or indirect actions of ACh. We probed the synaptic mechanisms that contribute to the enhanced firing of BLA neurons seen following optogenetic stimulation by assessing whether photo-stimulation of endogenous ACh inputs elicited changes in glutamatergic transmission in the BLA. We examined TTX resistant, glutamatergic EPSCs (Fig 5A,B) in putative BLA principal neurons. Voltage clamp recordings collected in the presence of both TTX and bicuculline (to block Na+ channel and GABA-A receptor mediated changes in excitability, respectively) revealed a baseline rate of glutamatergic transmission (EPSCs) at ~2 Hz. Following a 10 Hz burst of photo-stimulation of the cholinergic inputs to the BLA, the TTX-resistant, glutamatergic EPSC frequency increased significantly without changing amplitude, consistent with an increase in release probability (Fig 5A,B: TTX-resistant EPSC frequency: p < 0.01, n=11, Paired Wilcoxon Signed Rank test; amplitude: p = 0.2, n = 11, Paired Wilcoxon Signed Rank test). Assessment of cumulative data also indicated a significant decrease in inter-event intervals without a significant shift in the cumulative distribution of TTX-resistant EPSC amplitudes (Fig 5B, Kolmogorov-Smirnov test, ISI p < 0.01; amplitude p = 0.9; n=11). The basic AChR pharmacology of photo-stimulated changes in TTX-resistant EPSCs matched that seen in our studies of photo-stimulated changes in BLA firing. Photo-stimulation of oChIEF expressing cholinergic input significantly increased the TTX resistant EPSC frequency in the presence or absence of atropine (0.5μM atropine: pre vs post photo-stimulation: 0.97 Hz vs 1.27 Hz, n=10, p=0.002), whereas mecamylamine abolished the facilitation of TTX-resistant EPSCs induced by photo-stimulation of cholinergic terminal fields (pre vs post photo-stimulation: 0.96 Hz vs 0.84 Hz, n=7, p=0.8).
FIGURE 5. Photo-activation of cholinergic terminal fields in BLA modulates glutamatergic synaptic transmission.

A: Left: Schematic of configuration for voltage clamp recording of TTX resistant glutamatergic EPSCs in the BLA. Right: Sample recordings of TTX resistant glutamatergic EPSCs before (black) and after (blue) a 10 Hz burst of 473 nm light flashes (calibration: 10 pA x 200 msec).
B: Population data of TTX resistant glutamatergic EPSCs before (black) and after (blue) photo-stimulation, presented both as cumulative plots of inter-event intervals (left) and amplitude (right) and as box plots (insets) of the population data. Stimulation of cholinergic terminal fields in the BLA elicited a significant left shift in the inter-event interval cumulative probability curve (p < 0.01, n = 11), as well as a significant increase in TTX resistant glutamatergic EPSCs frequency (p < 0.01, n = 11). There was no statistically significant effect on TTX resistant glutamatergic EPSC amplitude.
C: Effect of optical stimulation on paired-pulse facilitation of evoked excitatory synaptic transmission (eEPSCs). Left: Schematic of configuration for voltage clamp recording with electrical stimulation of cortical inputs to the BLA. Right: An inter-stimulus interval of 50 msec revealed modest potentiation of the second response compared with the first (P2 vs P1) before (black trace) and after (blue trace) a 10 Hz burst of 473 nm light flashes. The amplitudes of both the first and second eEPSC were increased following photo-activation of cholinergic terminal fields (calibration: 50 pA x 20 msec).
D: Box and scatter plots of population data of paired pulse eEPSCs ± photo-activation of cholinergic input to BLA (n=8). There was a statistically significant increase in the amplitudes of both the first (p=0.014) and second (p=0.014) response to paired pulse stimulation when comparing before (black) and after (blue) photo-activation of oChIEF expressed in ChAT+ terminal fields in the BLA. See also Fig S2.
The TTX-resistant, glutamatergic EPSC activity assessed above arises from all glutamatergic inputs to the BLA principal neurons. We next asked whether photo-stimulation of oChIEF expressing cholinergic terminal fields in BLA would alter the profile of stimulus-evoked glutamatergic transmission (eEPSCs; Fig 5C, D). Cortical afferents to the BLA in the external capsule (EC) were electrically stimulated (Fig 5C). Prior studies testing paired-pulse stimulation of cortical-BLA synapses over a 20–200 ms inter-stimulus interval (ISI) revealed robust paired-pulse facilitation in young (pre weaning) mice in response to a 50 ms ISI, consistent with low release probability synapses (paired pulse ratio ~ 1.5; Jiang et al., 2008). Using the same stimulation protocol in the 2–3 month old mice studied here, a more modest paired pulse facilitation was seen (Fig 5C, D). Following photo-stimulation (10 Hz), the amplitudes of both the first (Paired Wilcoxon Signed Rank test, p< 0.05, n=8) and second (p< 0.05, n=8) paired-pulse eEPSCs were significantly increased, consistent with photo-stimulation eliciting increased glutamate release and/or increased post synaptic responsiveness (Fig 5C & D). The paired-pulse ratio was not different following photo-stimulation of cholinergic input (pre vs post photo-stimulation: 1.28 ± 0.17 vs. 1.08 ± 0.07 p = 0.29, n=8, Wilcoxon signed rank), although the trend toward a decrease is consistent with the increase in release probability revealed by the analysis of TTX-resistant glutamatergic EPSCs (Fig 5A,B).
Photo-stimulation of cholinergic input enhances plasticity of cortical-BLA excitatory transmission
Because of the established literature linking synaptic plasticity to learning and memory and prior studies demonstrating nAChR potentiation of glutamatergic transmission in regions related to mnemonic processing (Dani and Bertrand 2007, Halff et al. 2014, Zhang et al. 2010, Zhong et al. 2013), we tested whether endogenous ACh signaling in the BLA might exert long lasting changes in cortical-BLA synaptic transmission. The studies presented in Figures 6 & 7 examine possible synaptic plasticity correlates for the observed, persistent changes in fear-related behaviors following manipulation of endogenous ACh signaling in the BLA in awake behaving mice (Fig 2).
FIGURE 6. Stimulation of cholinergic inputs to BLA enhances plasticity induced by θ burst stimulation of cortical afferents.

A: Left: Schematic of configuration for voltage clamp recording from BLA principal neurons. Right: sample recordings of single electrical evoked responses of cortico-BLA synapses before θ burst (4 × 50 Hz bursts, 200 ms inter-burst intervals) (black, control), post θ burst alone (red), and post θ burst plus photo-stimulation (blue). θ burst alone briefly increased the eEPSC amplitude and success rate. After θ burst plus optogenetic stimulation, both the amplitude and the rate of successful evoked responses to 0.1 Hz stimulation of the EC were significantly increased.
B: Plot of eEPSCs obtained from a representative BLA principal neuron in response to 0.1 Hz stimulation before (black), after θ burst stimulation alone (red), and after optogenetic stimulation paired with θ burst stimulation (blue). Electrical stimulations of the external capsule that did not elicit a post synaptic response (i.e. failures) are indicated by amplitudes of 0 pA. The number of failures was briefly reduced after θ burst stimulation alone. Thirty mins following θ burst stimulation + photo-stimulation of the cholinergic terminal fields, failures were eliminated (i.e. eEPSC success probability approached 100%). The amplitude of eEPSCs briefly increased after θ burst stimulation alone whereas enhancement of eEPSC amplitude was sustained for the duration of the recording after pairing of θ burst stimulation and photo- stimulation.
C: Box and scatter plot of population data of eEPSCs amplitude (upper) and eEPSC success probability (Ps; lower) before patterned stimulation (control, shown in black), after θ burst stimulation (red), and after θ burst plus opto stimulation (blue). Top: θ burst stimulation alone induced a significant but brief increase in amplitude (p < 0.03, n = 11). Paired θ burst stimulation plus optogenetic stimulation also significantly increased the population eEPSC amplitude (p < 0.001, n = 11). The increase induced by θ burst stimulation plus photo-stimulation was significantly greater than the increase induced by θ burst stimulation alone. Bottom: Success rate was not increased by θ burst alone. A significant increase was seen after paired stimulation (p = 0.007, n = 8). See also Fig S2 and S5.
FIGURE 7. Photo-stimulation of cholinergic terminal fields in the BLA is sufficient to elicit LTP of cortical-BLA glutamatergic synaptic transmission.
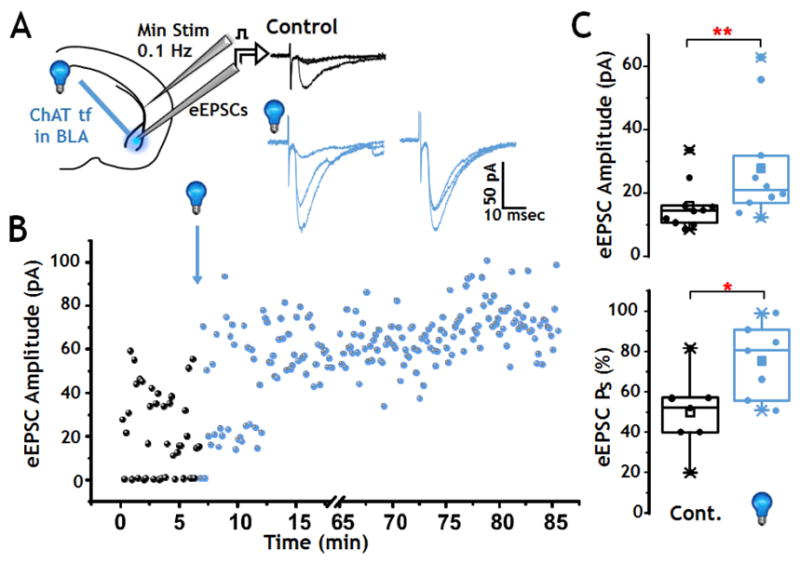
A: Left: Schematic of configuration for voltage clamp recording in BLA. Right: Sample recordings of single evoked responses of cortico-BLA synapses before photo-stimulation (black) and after photo-stimulation (blue) of cholinergic inputs. After photo-stimulation, both the EPSC amplitude and success rate increased.
B: Plot of 0.1 Hz evoked EPSCs obtained from a representative BLA principal neuron before and after photo-stimulation of oChIEF expressing cholinergic terminal fields in the BLA (blue). The number of failures was significantly reduced and the eEPSC amplitude was increased after photo-stimulation.
C: Population analyses (box and scatter plots) of changes in eEPSC amplitude (Top) and success probability (Bottom) obtained following photo-stimulation of cholinergic input to the BLA. Photo-stimulation significantly increased the amplitude of eEPSCs (p = 0.005, n = 10) and success rate (p = 0.02, n = 7).
Theta burst stimulation (θ) of cortical-BLA circuits reliably elicits a transient potentiation of glutamatergic synaptic transmission. Pairing θ stimulation with exposure to nicotine lowers the threshold for activation of long term potentiation (Jiang et al. 2013, Jiang and Role 2008, Mansvelder and McGehee 2000). To examine whether photo-activation of oChIEF expressing cholinergic inputs in the BLA altered the profile of glutamatergic synaptic transmission before and after θ stimulation of cortical- BLA synapses, we evoked transmission with “minimal” stimulation of the external capsule (EC), i.e. at a stimulation strength of sufficiently low intensity to evoke a steady state response rate of ~50% successes at 0.1 Hz stimulation (Fig 6A, control; Fig 6B first 8 minutes). A single θ stimulation of the EC typically elicited a brief potentiation reflected in a transient increase in the evoked response success rate and/or an increase in eEPSC amplitude that returned to baseline within 3–5 minutes (Fig 6A, B “θ alone”).
Pairing the θ stimulation with 10 Hz photo-stimulation of oChIEF expressing cholinergic inputs elicited a sustained potentiation of glutamatergic synaptic transmission, with a significant increase in both the stimulus-evoked response success rate and in the eEPSC amplitude. Cortical-BLA transmission remained elevated for the duration of the recording (53 min, Fig 6B). Pooled data from 11 experiments in which θ stimulation alone elicited brief potentiation of minimally evoked eEPSCs shows that pairing of θ stimulation of cortical-BLA inputs with optogenetic activation of cholinergic inputs significantly enhanced cortical-BLA glutamate release probability and eEPSC amplitude compared with either control or with θ stimulation alone (Fig 6C; post θ + photo-stimulation vs control: amplitude, p<0.01, success probability, Ps, p<0.01. Post θ + photo-stimulation vs. post θ alone: amplitude, p < 0.01, Ps, p < 0.05; Paired Wilcoxon Signed Rank test). Repeat θ did not result in sustained potentiation of cortical-BLA synapses (Fig S6). Thus, pairing of θ stimulation of cortical inputs with photo-stimulation of oChIEF expressing cholinergic inputs to the BLA was sufficient to convert short term enhancement of cortical-BLA glutamatergic transmission into a long lasting potentiation by increasing both the probability of evoked glutamate release and the magnitude of evoked post synaptic responses in cortical-BLA circuits.
To assess whether cholinergic stimulation per se might be sufficient to induce plasticity of cortical-BLA circuits, we examined the ability of patterned photo-stimulation of cholinergic inputs to strengthen evoked excitatory cortical-BLA transmission in the absence of any pairing protocol (Fig 7A). We found that a burst of 100 (10 bursts at 10 Hz) flashes of 473 nm light could elicit long lasting potentiation of glutamatergic transmission evoked by minimal-stimulation of the cortical input (LTP ≥ 35–78 min; Fig 7A, B). The LTP was reflected both in statistically significant increases in the probability of evoked responses to minimal intensity stimulation [Paired Wilcoxon Rank Sum; p<0.05, n=7] and an increase in the amplitude of the post synaptic (glutamatergic) eEPSCs (Paired Wilcoxon Signed Rank test; p<0.01, n=10; Fig 7C). Control studies demonstrate that 10 Hz x 10 s exposure to 473 nm light in a ChATtau-GFP (non-opsin) expressing mouse does not affect synaptic transmission over >70 minutes (Fig S2D). Photo-stimulation of oChIEF expressing cholinergic inputs in the presence of AChR antagonists had no effect on synaptic plasticity (n=19). Taken together these studies suggest that photo-stimulated release of endogenous ACh can not only lower the threshold for synaptic plasticity in a paired stimulation protocol but that activation of cholinergic inputs alone is sufficient to elicit LTP of glutamatergic transmission in cortical-BLA circuits.
Discussion
Our key findings are: (a) a single period of patterned stimulation or inhibition of cholinergic terminal fields in the BLA during conditioned fear training was sufficient to alter fear learning and the retention of extinction learning; (b) endogenous cholinergic signaling is required for normal fear learning and for the retention of extinction; (c) endogenous cholinergic signaling modulates the excitability of putative BLA principal neurons via interaction with both muscarinic and nicotinic AChRs; ACh signaling within the BLA regulates firing rates in a similar manner in vivo and ex vivo, and (d) endogenous ACh modulates the threshold for LTP ex vivo. The fact that stimulation of cholinergic terminal fields in the BLA is sufficient to induce LTP at cortical-BLA synapses and to drive persistent increases in BLA neuronal spiking both in vivo and ex vivo supports our proposal that the manipulation of cholinergic tone within the BLA regulates cue-conditioned fear learning and retention of fear extinction behaviors by mechanisms that involve ACh signaling per se (Fig 8).
Figure 8. Proposed mechanism for effect of BLA cholinergic tone on conditioned fear learning and retention of extinction learning behaviors.
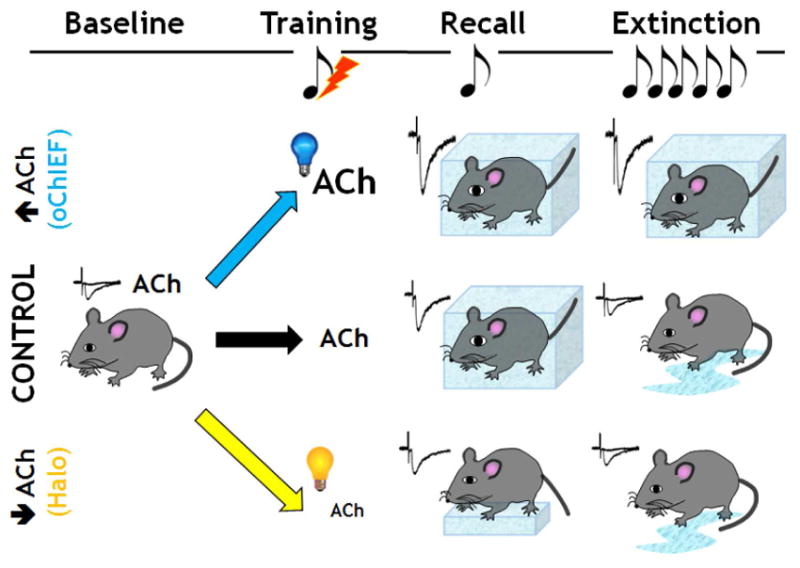
Under control conditions, fear conditioning induces freezing behavior measured in response to exposure to the conditioned stimulus (tone) (= recall). The freezing response decreases following several days of multiple tone exposures. Enhancing endogenous cholinergic signaling in the BLA during conditioned fear training did not alter freezing behavior assayed 24 hrs later, but decreased the response to the extinction sessions, consistent with the fear memory being more persistent over long periods of time. In contrast, reducing cholinergic signaling in the BLA during the initial training period reduced the freezing behavior during recall and led to greater retention of the extinction learning.
This study provides direct evidence for an essential role of cholinergic input to the amygdala in the formation and retention of one of the best studied emotionally salient memories: fear. Our findings underscore that ACh is more than just a modulator of fear memories: ACh is a requisite component of conditioned fear learning. We propose that the cholinergic regulation of fear learning derives, at least in part, from the effects of ACh on the excitability of, and on the glutamatergic transmission to, the principal neurons in the BLA. Our antagonist studies -in vivo and ex vivo - underscore that it is ACh per se, rather than other co-stored and/or co-released transmitters (Higley et al. 2011, Ren et al. 2011, Saunders et al. 2015), that is responsible for the observed effects on behavior and on BLA neuronal excitability.
The current in vivo electrophysiological studies also provide support to the notion that ACh can act rapidly and exert prolonged changes in network excitability (Chen et al. 2015, Eggermann et al. 2014, Luchicchi et al. 2014, Sarter et al. 2014). In the BLA brief photo-activation of cholinergic terminal fields elicited rapid changes in action potential firing (τdelay= 62 msec; Fig 3D) or in membrane potential (τdelay=10–100 msec) and slower but prolonged increases in firing rates of putative principal neurons (Fig 3, 4). In other in vivo recording studies in which we assessed the action potential firing elicited by photo-activation of the cholinergic neurons in the NBM per se and the consequent enhancement of BLA neuronal firing, the τdelay was about 6 times longer––but still consistent with the “rapid ACh transients” reported by Sarter et al. (Sarter et al. 2014).
We used acute slice electrophysiological recording to analyze the circuit and synaptic mechanisms that might underlie the sustained changes in learning and the enhanced BLA excitability following in vivo manipulation of cholinergic inputs to the BLA. Although less direct than in vivo assays, our ex vivo findings are also consistent with AChR-mediated potentiation of cortical-amygdala glutamatergic transmission that can persist for > an hour after the initial ACh signaling has occurred. Photo-stimulation of cholinergic projections enhances the probability of glutamate release, the magnitude of evoked EPSPs in cortical-BLA circuits and lowers the threshold for LTP. These modulatory effects of endogenous ACh are strikingly reminiscent of the effects of brief, low dosage exposure of cortical-BLA synapses to nicotine (Jiang et al. 2013, Jiang and Role 2008). Indeed, in all of our studies, nAChRs were an essential component of the observed effects of cholinergic stimulation. Inhibition of both nAChR and mAChR activity by intra-BLA infusion of antagonists during training inhibits conditioned fear learning and enhances extinction, whereas mAChR block alone does not. Likewise, nAChR antagonists blocked the increased excitability observed in response to photo-stimulation in vivo and blocking nAChRs alone significantly lowered the baseline firing rate of BLA neurons in vivo. These data are consistent with a role for ongoing activation of AChRs in setting the steady state level of BLA firing.
Both our in vivo and ex vivo studies show that the net effect of cholinergic input is to increase excitability. Recent studies have demonstrated that a subset of neurons within the amygdala is activated both during conditioned fear learning, and during recall. Direct stimulation of these BLA neurons can elicit fear behaviors, providing strong evidence that these neurons are part of a memory trace (Nonaka et al. 2014, Redondo et al. 2014). The likelihood that a neuron is recruited into the memory trace is related to the excitability of the neuron: electrophysiological studies demonstrate altered pre synaptic plasticity of cortical inputs to BLA neurons in the memory trace (Nonaka et al. 2014, Yiu et al. 2014). Our findings that endogenous acetylcholine modulates the excitability of principal neurons in the BLA, regulates cortical-BLA synaptic plasticity and influences the acquisition of fear related behaviors, argue strongly for a critical role of acetylcholine modulation in amygdala based learning and memory.
Our findings that the net effect of cholinergic input is to increase excitability contrast with the conclusions of a recent report of net inhibitory actions of photo-stimulation of cholinergic inputs to BLA (Unal et al. 2015). The dissimilarity of our results most likely derives from differences in recording paradigms and/or in the stimulation protocol for photo-activation of cholinergic inputs. We find that photo-stimulation of cholinergic inputs with current injection to depolarize the recorded BLA neurons, elicits a transient hyperpolarization, followed by the slower, longer lasting increases in BLA excitability that are seen in vivo and ex vivo in neurons at rest potential. The photo-stimulation paradigms are also somewhat different: we adopted the specific protocols for cholinergic terminal field stimulation in our in vivo and ex vivo studies based on what we had found to be effective in changing the associative fear learning of the animals (Table S1).
In all of our studies of photo-stimulation of cholinergic input to BLA principal neurons, we observed only three direct, light-evoked post synaptic responses (Fig S6). In these instances the recorded responses were fast, inward synaptic currents. The rarity of this result underscores that under our conditions, the predominant effect of photo-stimulation of cholinergic input in BLA is modulation of glutamatergic transmission rather than direct activation of post synaptic receptors.
Overall we find that stimulation of endogenous ACh release during exposure to conditioned fear training results in a more persistent memory of the learned fear. Equally important: interference with cholinergic input to the BLA during fear training ablates the formation of the memory and results in a rapid return to control behaviors following extinction training. In view of the key role of ACh in attention, cue detection, and mnemonic processing (Sarter et al. 2014) as well as the relationship between cholinergic degeneration and the progressive loss of short term memory in diseases such as Alzheimer’s disease, the use of cholinergic enhancement therapies for pro-cognitive treatments remains an area of intense study. Likewise, the more anecdotal but intriguing literature potentially linking nicotine exposure to the exacerbation of cued memory recall, as in post-traumatic stress disorder (Rasmusson et al. 2006), makes it tempting to speculate that manipulation of endogenous cholinergic signaling in specific terminal fields might provide novel treatment approaches to memory dysfunction.
Experimental procedures
Mouse use was approved by the Institutional Animal Care and Use Committee at Stony Brook University.
Viral delivery
ChAT-Cre mice (P21 to P30) were infected in the NBM with AAV9-CAG-DIO-oChIEF-tdTomato, AAV9-Ef1a-DIO-eNpHR3.0-eYFP or AAV9-CAG-DIO-eGFP. Viral stocks were obtained from the UNC vector core.
Behavioral Testing
Animals underwent cue associated fear conditioning and extinction training with either opto-stimulation or locally delivered AChR antagonists within the BLA. Details of the behavioral paradigms used are presented in the Supplemental Experimental Procedures and Table S1.
Electrophysiological recording
In vivo and ex vivo electrophysiological recordings were done in the BLA and in the NBM. The procedures used for electrophysiological recordings, opto-stimulation and drug delivery are provided in the Supplemental Procedures.
Statistics
Statistical analyses of electrophysiological data typically used non-parametric statistical tests including the Kolmogorov-Smirnov for cumulative distributions with n>1000, Mann-Whitney and Paired-Sample Wilcoxon Signed Rank Test with Origin 9.1. The median values are reported from the entire population tested. The ex vivo electrophysiological data are from 153 recordings from 62 mice. Unless otherwise noted n values reported are numbers of recordings. Behavioral data were analyzed by Mann-Whitney, one and two way ANOVA with Bonferroni correction for multiple comparisons.
Supplementary Material
Highlights.
Opto stimulation of ACh in BLA during cue-fear training makes memory more durable
Stimulating ACh input to BLA in vivo and ex vivo increases neuronal excitability
Stimulating ACh input to BLA can elicit LTP
All of the above effects are dependent on Acetylcholine receptors (AChRs)
Acknowledgments
Support: NS 022061, DP1-OD007014, K12GM102778 and F30MH105087. We thank Drs. Ge and Fontanini for expert advice and comments on the manuscript, and N. Joseph, C. Waddell and M. Myers for technical support.
Footnotes
Author Contributions
Conceptualization: LR, JL, DT; Methodology: LR, DT, LJ, SK, JL, GL; Formal analysis: LR, DT, LJ, SK, JL, SW, EB, GL; Investigation: LJ, SK, JL, SW EB, GL; Writing: LR, LJ, DT, SK, JL, SW, EB, GL.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adolphs R. The biology of fear. Curr Biol. 2013;23(2):R79–93. doi: 10.1016/j.cub.2012.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter MG, Bucci DJ. Selective immunotoxic lesions of basal forebrain cholinergic neurons: twenty years of research and new directions. Behav Neurosci. 2013;127(5):611–618. doi: 10.1037/a0033781. [DOI] [PubMed] [Google Scholar]
- Chen N, Sugihara H, Sur M. An acetylcholine-activated microcircuit drives temporal dynamics of cortical activity. Nat Neurosci. 2015;18(6):892–902. doi: 10.1038/nn.4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Pare D. Amygdala microcircuits controlling learned fear. Neuron. 2014;82(5):966–980. doi: 10.1016/j.neuron.2014.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Kremer Y, Crochet S, Petersen CC. Cholinergic signals in mouse barrel cortex during active whisker sensing. Cell Rep. 2014;9(5):1654–1660. doi: 10.1016/j.celrep.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR, Wiltgen B, Fanselow MS. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci. 2004;24(15):3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halff AW, Gomez-Varela D, John D, Berg DK. A novel mechanism for nicotinic potentiation of glutamatergic synapses. J Neurosci. 2014;34(6):2051–2064. doi: 10.1523/JNEUROSCI.2795-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Sarter M. Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology. 2011;36(1):52–73. doi: 10.1038/npp.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans EJ, Henckens MJ, Joels M, Fernandez G. Dynamic adaptation of large-scale brain networks in response to acute stressors. Trends Neurosci. 2014;37(6):304–314. doi: 10.1016/j.tins.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Herry C, Johansen JP. Encoding of fear learning and memory in distributed neuronal circuits. Nat Neurosci. 2014;17(12):1644–1654. doi: 10.1038/nn.3869. [DOI] [PubMed] [Google Scholar]
- Higley MJ, Gittis AH, Oldenburg IA, Balthasar N, Seal RP, Edwards RH, Lowell BB, Kreitzer AC, Sabatini BL. Cholinergic interneurons mediate fast VGluT3-dependent glutamatergic transmission in the striatum. PLoS One. 2011;6(4):e19155. doi: 10.1371/journal.pone.0019155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Emmetsberger J, Talmage DA, Role LW. Type III neuregulin 1 is required for multiple forms of excitatory synaptic plasticity of mouse cortico-amygdala circuits. J Neurosci. 2013;33(23):9655–9666. doi: 10.1523/JNEUROSCI.2888-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Role LW. Facilitation of cortico-amygdala synapses by nicotine: activity-dependent modulation of glutamatergic transmission. J Neurophysiol. 2008;99(4):1988–1999. doi: 10.1152/jn.00933.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Hamanaka H, Monfils MH, Behnia R, Deisseroth K, Blair HT, LeDoux JE. Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc Natl Acad Sci U S A. 2010;107(28):12692–12697. doi: 10.1073/pnas.1002418107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox D, Keller SM. Cholinergic neuronal lesions in the medial septum and vertical limb of the Diagonal Bands of Broca induce contextual fear memory generalization and impair acquisition of fear extinction. Hippocampus. 2015;22 doi: 10.1002/hipo.22553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, Holliday E, Gould TJ. High-affinity alpha4beta2 nicotinic receptors mediate the impairing effects of acute nicotine on contextual fear extinction. Neurobiol Learn Mem. 2016;128:17–22. doi: 10.1016/j.nlm.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalumiere RT, McGaugh JL. Memory enhancement induced by post-training intrabasolateral amygdala infusions of beta-adrenergic or muscarinic agonists requires activation of dopamine receptors: Involvement of right, but not left, basolateral amygdala. Learn Mem. 2005;12(5):527–532. doi: 10.1101/lm.97405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. Evolution of human emotion: a view through fear. Prog Brain Res. 2012;195:431–442. doi: 10.1016/B978-0-444-53860-4.00021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchicchi A, Bloem B, Viana JN, Mansvelder HD, Role LW. Illuminating the role of cholinergic signaling in circuits of attention and emotionally salient behaviors. Front Synaptic Neurosci. 2014;6:24. doi: 10.3389/fnsyn.2014.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27(2):349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Myskiw JC, Izquierdo I, Furini CR. Modulation of the extinction of fear learning. Brain Res Bull. 2014;105:61–69. doi: 10.1016/j.brainresbull.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Nonaka A, Toyoda T, Miura Y, Hitora-Imamura N, Naka M, Eguchi M, Yamaguchi S, Ikegaya Y, Matsuki N, Nomura H. Synaptic plasticity associated with a memory engram in the basolateral amygdala. J Neurosci. 2014;34(28):9305–9309. doi: 10.1523/JNEUROSCI.4233-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76(1):116–129. doi: 10.1016/j.neuron.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoplichko VI, Prager EM, Aroniadou-Anderjaska V, Braga MF. alpha7-Containing nicotinic acetylcholine receptors on interneurons of the basolateral amygdala and their role in the regulation of the network excitability. J Neurophysiol. 2013;110(10):2358–2369. doi: 10.1152/jn.01030.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson AM, Picciotto MR, Krishnan-Sarin S. Smoking as a complex but critical covariate in neurobiological studies of posttraumatic stress disorders: a review. J Psychopharmacol. 2006;20(5):693–707. doi: 10.1177/0269881106060193. [DOI] [PubMed] [Google Scholar]
- Redondo RL, Kim J, Arons AL, Ramirez S, Liu X, Tonegawa S. Bidirectional switch of the valence associated with a hippocampal contextual memory engram. Nature. 2014;513(7518):426–430. doi: 10.1038/nature13725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Qin C, Hu F, Tan J, Qiu L, Zhao S, Feng G, Luo M. Habenula “cholinergic” neurons co-release glutamate and acetylcholine and activate postsynaptic neurons via distinct transmission modes. Neuron. 2011;69(3):445–452. doi: 10.1016/j.neuron.2010.12.038. [DOI] [PubMed] [Google Scholar]
- Santini E, Sepulveda-Orengo M, Porter JT. Muscarinic receptors modulate the intrinsic excitability of infralimbic neurons and consolidation of fear extinction. Neuropsychopharmacology. 2012;37(9):2047–2056. doi: 10.1038/npp.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Lustig C, Howe WM, Gritton H, Berry AS. Deterministic functions of cortical acetylcholine. Eur J Neurosci. 2014;39(11):1912–1920. doi: 10.1111/ejn.12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Granger AJ, Sabatini BL. Corelease of acetylcholine and GABA from cholinergic forebrain neurons. Elife. 2015;4 doi: 10.7554/eLife.06412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller D, Delgado MR. Overlapping neural systems mediating extinction, reversal and regulation of fear. Trends Cogn Sci. 2010;14(6):268–276. doi: 10.1016/j.tics.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471(7338):358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal CT, Pare D, Zaborszky L. Impact of Basal forebrain cholinergic inputs on basolateral amygdala neurons. J Neurosci. 2015;35(2):853–863. doi: 10.1523/JNEUROSCI.2706-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37(6):475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- Wu H, Williams J, Nathans J. Complete morphologies of basal forebrain cholinergic neurons in the mouse. Elife. 2014;3:e02444. doi: 10.7554/eLife.02444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu AP, Mercaldo V, Yan C, Richards B, Rashid AJ, Hsiang HL, Pressey J, Mahadevan V, Tran MM, Kushner SA, Woodin MA, Frankland PW, Josselyn SA. Neurons are recruited to a memory trace based on relative neuronal excitability immediately before training. Neuron. 2014;83(3):722–735. doi: 10.1016/j.neuron.2014.07.017. [DOI] [PubMed] [Google Scholar]
- Zelikowsky M, Hersman S, Chawla MK, Barnes CA, Fanselow MS. Neuronal ensembles in amygdala, hippocampus, and prefrontal cortex track differential components of contextual fear. J Neurosci. 2014;34(25):8462–8466. doi: 10.1523/JNEUROSCI.3624-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang TA, Tang J, Pidoplichko VI, Dani JA. Addictive nicotine alters local circuit inhibition during the induction of in vivo hippocampal synaptic potentiation. J Neurosci. 2010;30(18):6443–6453. doi: 10.1523/JNEUROSCI.0458-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Talmage DA, Role LW. Nicotine elicits prolonged calcium signaling along ventral hippocampal axons. PLoS One. 2013;8(12):e82719. doi: 10.1371/journal.pone.0082719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.