

Abstract
Background: MicroRNAs have been identified to play important role in the development of human esophageal squamous carcinoma (ESCC). In this study, we examined the regulatory effects of microRNA-26b (miR-26b) on ESCC proliferation, cell-cycle transition and migration. Methods: Expressions of miR-26a and miR-2bb were analyzed in 8 ESCC cell lines, and 27 human ESCC tissues and paired adjacent non-tumor tissues. MiR-26a and miR-26b were either upregulated or downregulated in ESCC cell lines TE-1 and Kyse140 cells. Their effects on ESCC in vitro growth, cell-cycle transition and migration were analyzed by proliferation assay, cell-cycle assay and invasion assay, respectively. The association of miR-26b and its downstream target gene, tumor necrosis factor receptor-associated factor 5 (TRAF5), was analyzed by luciferase reporter assay and qRT-PCR. TRAF5 was downregulated in TE-1 and Kyse140 cells to analyze its direct effect on miR-26b downregulation induced ESCC inhibition. Results: MiR-26b expression was aberrantly upregulated in ESCC cell lines and human ESCC tissues, whereas miR-26a expression was unchanged. In TE-1 and Kyse140 cells, miR-26b downregulation had tumor-suppressive effect, whereas miR-26b downregulation or miR-26a upregulation/downregulation had no significant effect, on ESCC proliferation, cell-cycle transition and migration. TRAF5 is confirmed to be the downstream target of miR-26b in ESCC. SiRNA-mediated TRAF5 downregulation inversely regulated the inhibition of miR-26b downregulation on ESCC proliferation, cell-cycle transition and migration. Conclusion: our study demonstrates that miR-26b downregulation, through the inverse regulation on TRAF5, had tumor-suppressive effect on human ESCC.
Keywords: Esophageal squamous carcinoma, miR-26b, miR-26a, TRAF5, proliferation, migration
Introduction
Human esophageal cancer is one of the most malignant tumors in the world [1]. In the Unites States alone, yearly estimated number of new cases of esophageal cancer is more than 16,000, and yearly estimated number of esophageal cancer-related death is more than 150,000 [2]. There are two major sub-types of esophageal cancer, esophageal squamous carcinoma (ESCC) and esophageal adenocarcinoma (EAC). ESCC is the most dominant sub-type of esophageal cancer in developing countries, including China [3]. In recent decades, although great advances have been made in early diagnosis and personalized treatment, patients with ESCC or EAC still suffered from very poor prognosis, with estimated 5-year survival rate to be less than 25% [3,4]. Therefore, it is very important to unravel the underlying mechanisms of carcinogenesis and cancer development in human esophageal cancer.
MicroRNAs (miRNAs) are classes of small non-coding 18~22 nucleotides long small RNAs that bind the 3’ untranslated region (3’ UTR) of target genes to induce post-transcriptional gene repression or protein degradation [5]. Mounting evidences have demonstrated that miRNAs are important molecular regulators in human esophageal cancer, acting as either oncogenes or tumor-suppressors [6-8]. Recently, one of the tumor-associated miRNA, microRNA-26a (miR-26a) was shown to directly regulate cancer proliferation and cell cycle progression in EAC OE33 cells [9]. However, it is not clear whether miR-26a, or the other member of the miR-26 family, miR-26b, may functionally modulate ESCC, the other type of esophageal cancer.
Tumor necrosis factor receptor (TNFR)-associated factors (TRAFs), including TRAF1, 2, 3, 5, and 6, were molecular activators for TNFRs signaling pathway and interleukin-1 receptor-associated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway, to induce cell death or necrosis [10-12]. Among them, TRAF5 was shown to be involved in the regulation of Notch signaling pathway related NF-κB activation in brain cancer [13]. In human ESCC, TRAF5 was shown to be downregulated by the over-expression of transcription factor (paired box 2) PAX2, thus suggested to be correlated with Interleukin-5 induced cancer cell metastasis [14]. However, the exact function of TRAF5, or its association with epigenetic regulation of miRNA in ESCC, is largely unknown.
In this study, we explored the expression pattern of miR-26 family, including miR-26a and miR-26b in ESCC cell lines and ESCC tumor tissues. We then used synthetic oligonucleotides to either upregulate or downregulate miR-26a and miR-26b in ESCC cell line TE-1 and Kyse140 cells, to analyze their functional mechanisms in ESCC proliferation, cycle-cycle transition and migration. Furthermore, we examined the hypothesis that TRAF5 gene is the downstream target of miR-26b in ESCC, and analyzed the effect of TRAF5 downregulation on miR-26b mediated mechanistic regulations on ESCC development. Our results would undoubtedly help to further the understanding on the miRNA-associated molecular mechanisms in ESCC.
Materials and methods
Ethic statements
In this study, all experiments were performed in accordance with the principles outlined in the Declaration of Helsinki, and local and federal government agencies. All experimental protocols were approved by the Clinical Research & Ethic Committees at Union Hospital & Central Hospital of Tongji Medical College of Huazhong University of Science and Technology in Wuhan, Hunan Province, China. All patients signed consent forms before enrollment in the study.
ESCC cell lines and human ESCC tumor samples
A normal human esophageal squamous epithelial cell line (ESEC) was established in our laboratory from biopsy samples of non-tumorous esophageal tissues using the method described previously [15]. The esophageal squamous carcinoma (ESCC) cell lines, TE-1, TE-2, Kyse30, Kyse140 and Eca-109 were obtained from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Kyse30, Kyse410, Kyse510 were purchased from Deutsche Sammlung von Mikroorganismen und Zelkulturen GmbH (DSMZ, Braunschweig, Germany). All cells were maintained in RPMI 1640 medium (Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS, Invitrogen, USA), 100 μg/ml streptomycin and 100 units/ml penicillin (Sigma-Aldrich, USA) and 0.2 mM non-essential amino acid (Invitrogen, USA) in a humidified tissue culture chamber with 5% CO2 at 37°C.
Human tumor samples of ESCC (T), and their adjacent non-tumorous esophageal tissues (ANT) were obtained from biopsies of twenty-seven patients in the Department of Cancer Chemotherapy and Radiotherapy at Tangshan Gongren Hospital, Tangshan Clinical Medical College of Hebei Medial University in Tangshan, Hebei Province, China. After surgical retrieval, all tissues were immediately snap frozen in liquid nitrogen and stored at -80°C until further experiments.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from ESCC cell lines or ESCC tumor samples using TRIzol kit (Invitrogen, USA), followed by RNeasy Mini Kit (Qiagen USA) according to the manufacturers’ instructions. 1000 ng RNA of each sample was reverse-transcribed into cDNA using an iScript cDNA synthesis kit (Bio-Rad, USA) according to the manufacturer’s instruction. To quantify gene expression levels of miR-26a and miR-26b, quantitative real-time PCR (qRT-PCR) was performed using a TaqMan miRNA PCR kit (Applied Biosystems, USA) according to the manufacturer’s instruction. U6 snRNA was applied as control template. To quantify mRNA levels of TRAF5, a Brilliant III Fast QPCR Kit (Agilent Technologies, Canada) was performed. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was applied as control template. Gene expressions were measured as fold-changes and compared using the 2-ΔΔCt method as described previously [16].
Controlled MicroRNA-26 expression assay
The synthetic miR-26a mimic (miR26a-m), miR-26a inhibitor (miR26a-I), miR-26b mimic (miR26b-m) and miR-26b inhibitor (miR26b-I) oligonucleotides, as well as control mimic (Ctrl-m) and control inhibitor (Ctrl-m) oligonucleotides were purchased from BiboBio (RiboBio, Guangzhou, China). In order to upregulate or downregulate miR-26a/miR-26b in ESCC cell lines, transfection of synthetic miRNA oligonucleotides was performed using a Lipofectamine 2000 reagent (Invitrogen, USA) according to the manufacturer’s instruction. 48 h later, transfection efficacy was examined by quantitative real time PCR.
Proliferation assay
Cancer proliferation of ESCC cell lines, TE-1 and Kyse140 cells, was measured by a CellTiter 96 Aqueous One Solution cell proliferation assay (Promega, USA) according to the manufacturer’s protocol. Briefly, TE-1 and Kyse140 cells were plated in 96-well plates (500 cells/well). The cell proliferation solution (20 μl/well) was added into culture for 1 h at 24, 48, 72 and 96 hours after plating. The optical density (O.D.) at absorbance of 570 nm was then measured and compared between different experimental conditions.
Cell cycle assay
ESCC cell cycle was examined by flow cytometry. Briefly, TE-1 and Kyse140 cells were quickly fixed in 70% ethanol, then co-cultured with 50 μg/ml propidium iodide (Thermo Fisher Scientific, USA) in phosphate-buffered saline (PBS, Sigma-Aldrich, USA) for 30 min at 37°C. DNA contents were analyzed using a FACSCaliburTM flow cytometry (Thermo Fisher Scientific, USA). The obtained histograms were analyzed by a Multicycle AV software (Phoenix Flow System, USA) to determine the cell population (in %) at G1, S or G2/M stages.
Invasion assay
ESCC invasion was examined by an in vitro scratch assay described previously [17]. Briefly, TE-1 and Kyse140 cells were maintained to ~80% confluence in 6-well plates. A sterile cell scrapper was used to create a linear wound across the well, followed by 3 times wash to remove floating cell debris. 24 h later, transmit-light images were obtained. The travelling distances by cells invading into the wound area were measured, and the relative invasion (in %) was characterized by normalizing the travelling distances under various experimental conditions against the travelling distance under control condition.
Luciferase reporter assay
The binding of human miR-26b on human TRAF5 gene was examined by a luciferase reporter assay. Briefly, 3’ UTR of human TRAF5 gene was cloned into a pmiR-REPORT vector (RiboBio. Guangzhou, China) to create a luciferase reporter, Lu-TRAF5. Putative binding domain by human miR-26b on TRAF5 3’ UTR was point-mutated, and also cloned into pmiR-REPORT vector to create a mutant luciferase reporter, Lu-TRAF5-mutated. In HEK293T cells, 50 ng/mL Lu-TRAF5 or Lu-TRAF5-mutated were co-transfected with 2500 p-mol Ctrl-m or miR26b-m for 48 h. Relative luciferase activities were measured by a dual-luciferase reporter assay (Promega, USA) and normalized to the luciferase activity under the control condition with Lu-TRAF5 and Ctrl-m co-transfection.
TRAF5 knockdown assay
The knockdown of TRAF5 mRNA in ESCC cell lines, TE-1 and Kyse140, were achieved through small-interfering RNA (siRNA) technology. The human TRAF5-specific siRNA (TRAF5-siRNA) and a control scrambled-siRNA (S-siRNA) were purchased from RiboBio (Guangzhou, China). The transfection of siRNA (50 nM) was performed using a GeneSilencer® reagent (Genlantis, San Diego, CA, USA) according to the manufacturer’s instruction. 24 h later, transfection efficacy was examined by quantitative real time PCR.
Statistical analysis
In our work, all experiments were biologically repeated for at least three times. Data are shown as mean ± SEM. Statistical comparisons were made by Student’s t-tests. An unpaired two-tailed P value < 0.05 was termed to be significantly different.
Results
MiR-26b but not miR-26a is upregulated in ESCC
In a recent study, it was shown that miR-26a played important role in regulating esophageal adenocarcinoma cells [9]. That leads us to wonder whether miR-26 family, including miR-26a and miR-26b, may also have functional roles in the other type of esophageal carcinoma, esophageal squamous carcinoma (ESCC).
We firstly examined the expressions of miR-26a and miR-26b in eight ESCC cell lines. As compared to the expressions of miR-26a and miR-26b in a normal human esophageal squamous epithelial cell line (ESEC), Qrt-PCR analysis demonstrated that miR-26a expression was un-changed (Figure 1A, ΔP > 0.05), whereas miR-26b expression was significantly upregulated in all of the 8 ESCC cell lines (Figure 1B, *P < 0.05).
Figure 1.
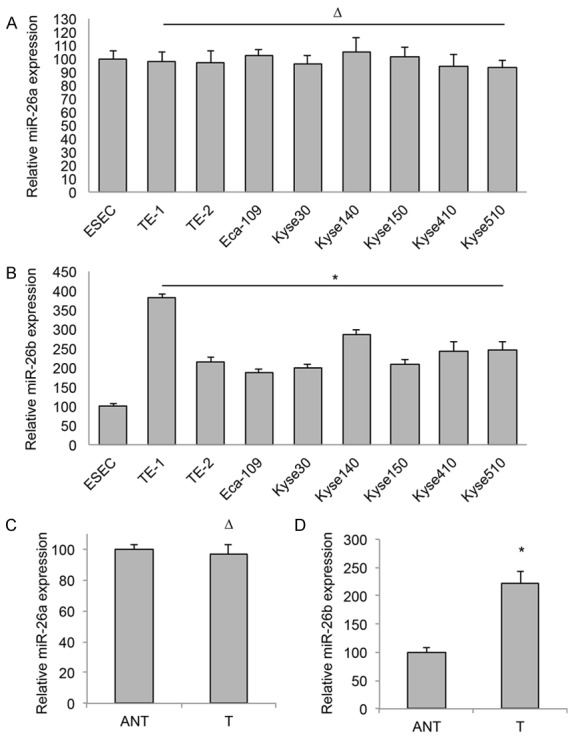
MiR-26b is upregulated in ESCC. Gene expression levels of miR-26a (A) and miR-26b (B) were examined by qRT-PCR in eight ESCC cell lines, TE-1, TE-2, Eca-109, Kyse30, Kyse140, Kyse150, Kyse410 and Kyse510. The expression levels of miR-26a and miR-26b were then normalized to the expression level of miR-26a or miR-26b in a normal human esophageal squamous epithelial cell line (ESEC) (ΔP > 0.05, *P < 0.05). Expression levels of miR-26a (C) and miR-26b (D) were also examined by qRT-PCR in 27 human ESCC tumor (T) tissues and paired adjacent non-tumor (ANT) tissues (ΔP > 0.05, *P < 0.05).
We then examined 27 clinical samples of paired human ESCC tumor (T) tissues and their adjacent non-tumor (ANT) tissues. Analysis of qRT-PCR showed that the averaged expression level of miR-26a was about the same between T and ANT tissues (Figure 1C, ΔP > 0.05), whereas the averaged expression level of miR-26b was significantly upregulated in T tissues than in ANT tissues (Figure 1D, *P < 0.05). These results suggest that miR-26b, not miR-26a is upregulated in ESCC, and it may have functional role in the development of human ESCC.
MiR-26b downregulation inhibited proliferation and induced cell-cycle arrest in ESCC
As we discovered that miR-26b was aberrantly upregulated in both ESCC cell lines and clinical samples, we hypothesized that it had biological functions in ESCC. In order to examine it, we transfected two ESCC cell lines, TE-1 and Kyse140 cells with synthetic miR-26b mimics (miR26b-m) or miR-26b inhibitor (miR26b-I). Analysis of qRT-PCR showed that, as compared to control miRNAs (Ctrl-m & Ctrl-I), miR26b-m significantly upregulated and miR26b-I significantly downregulated gene expression of miR-26b in both TE-1 and Kyse140 cells (Figure 2A, *P < 0.05).
Figure 2.
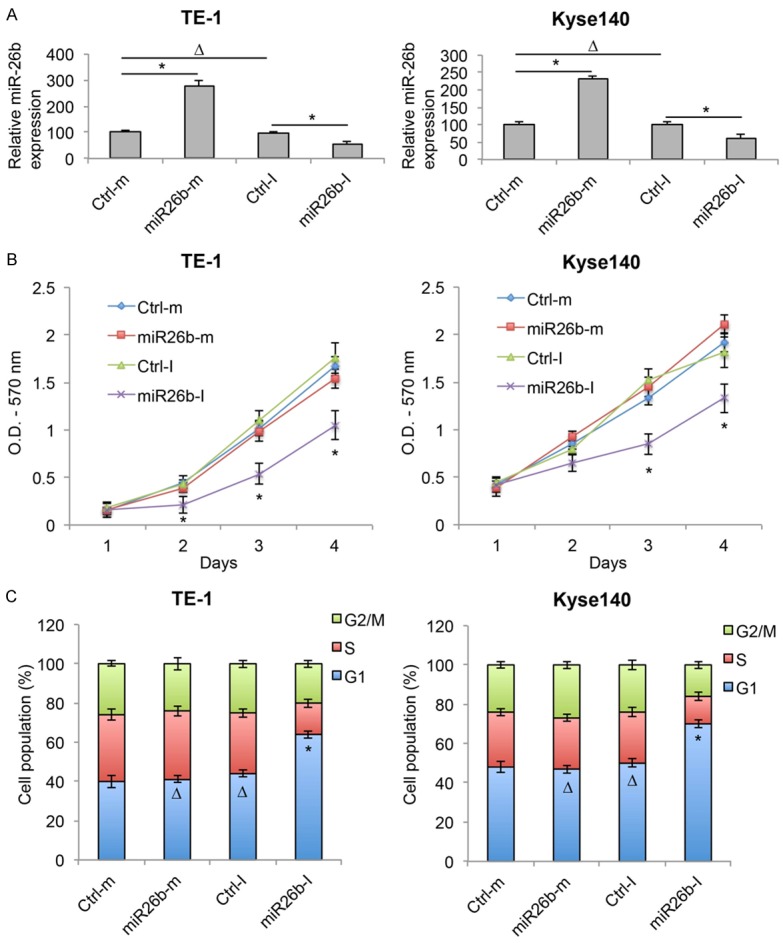
MiR-26b downregulation inhibited proliferation and cell cycle transition in ESCC. A. ESCC cell lines TE-1 and Kyse140 were transfected with miR-26b mimics (miR26b-m) or inhibitor (miR26b-I), as well as control mimic miRNA (Ctrl-m) and control inhibitor miRNA (Ctrl-I). The transfection efficiencies were verified by qRT-PCR (ΔP > 0.05, *P < 0.05). B. After transfection, in vitro growth of TE-1 and Kyse140 cells were examined for 4 consecutive days using a proliferation assay (*P < 0.05). C. Post-transfection cell cycle transition was examined by a cell-cycle assay. Cell populations at G1, S and G2/M stages were calculated (ΔP > 0.05, *P < 0.05).
After TE-1 and Kyse140 cells were transfected with synthetic miRNAs, a cell proliferation assay showed that ESCC in vitro growth was significantly reduced by miR26b-I, but unaffected by miR26b-m (Figure 2B, *P < 0.05). In addition, a cell-cycle assay demonstrated that significant amount of ESCC cells were arrested at G1 stage by miR26b-I, whereas cell-cycle transition was un-affected by miR26b-m (Figure 2C, *P < 0.05, ΔP > 0.05). These results suggest that miR-26b downregulation had tumor-suppressive effects, whereas miR-26b upregulation had no effect, on cancer proliferation and cell cycle transition in ESCC.
MiR-26b downregulation inhibited cancer migration in ESCC
After TE-1 and Kyse140 cells were transfected with synthetic miRNAs, we also examined ESCC migration capability through an in vitro invasion assay. At the onset of assay, a cell scraper created a linear wound area on the monolayers of ESCC cells. 24 h post wound-creation, the effects of synthetic miRNA transfections on ESCC cells invading into the wound areas were examined. By comparing the transmit-light images of TE-1 cells at the onset (0 h) and the end (24 h) of invasion assay, we noticed that markedly less cells invaded into the wound area with miR26b-I transfection (Figure 3A). Quantitative measurement indicated that miR26b-I significantly reduced, where miR26b-m had little effect on, the invasion capability of TE-1 cells (Figure 3B, *P < 0.05, ΔP > 0.05). Images of transfected Kyse140 cells in invasion assay showed similar result as TE-1 cells, with miR26b-I hindering the cells from invading into the wound area (Figure 3C). Quantitative measurement also showed that invasion capability of Kyse140 cells was significantly reduced by miR26b-I, but not by miR26b-m (Figure 3D, *P < 0.05, ΔP > 0.05). These results suggest that miR-26b downregulation had tumor-suppressive effect on cancer migration in ESCC.
Figure 3.
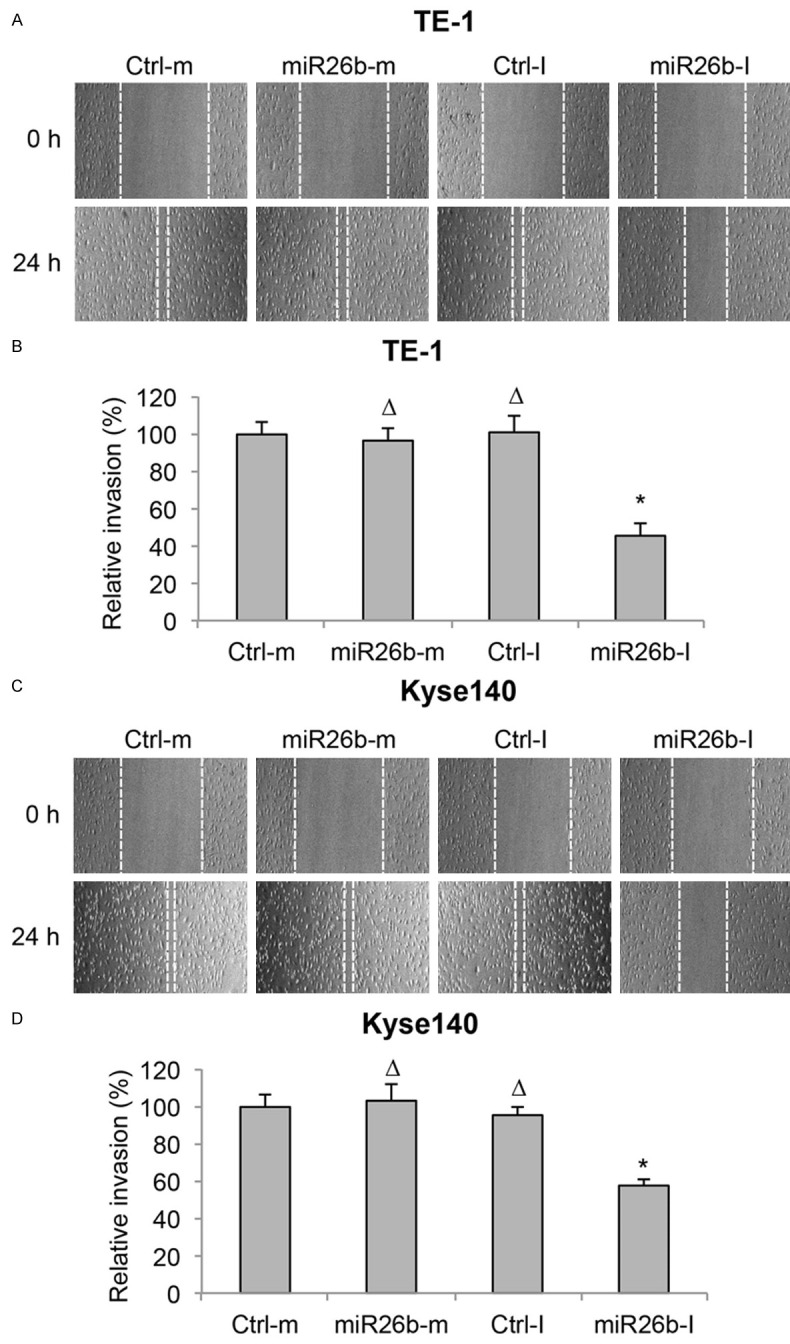
MiR-26b downregulation inhibited migration in ESCC. A. TE-1 cell lines were transfected with miR-26b mimics (miR26b-m) or inhibitor (miR26b-I), as well as control mimic miRNA (Ctrl-m) and control inhibitor miRNA (Ctrl-I). An in vitro invasion assay was performed by creating a linear wound area on cell monolayer and monitored for 24 h. B. Invasion capability in miRNA-transfected TE-1 cells was measured by comparing the travelling distances of TE-1 cells invaded into the wound areas (ΔP > 0.05, *P < 0.05). C, D. Kyse140 cells were treated the same way as TE-1 cells and the invasion capability was measured (ΔP > 0.05, *P < 0.05).
MiR-26a had no effect on cancer development in ESCC
As we discovered that miR-26b had significant functional role in regulating human ESCC, we wondered whether the other member of the miR-26 family, miR-26a, had any effects on ESCC development. To do so, we transfected TE-1 and Kyse140 cells with synthetic miR-26a mimics (miR26a-m) or inhibitor (miR26a-I), as well as control miRNAs (Ctrl-m & Ctrl-I). 48 h after transfection, analysis of qRT-PCR showed that, miR26a-m significantly upregulated and miR26a-I significantly downregulated gene expression of miR-26a in both TE-1 and Kyse140 cells (Figure 4A, *P < 0.05).
Figure 4.
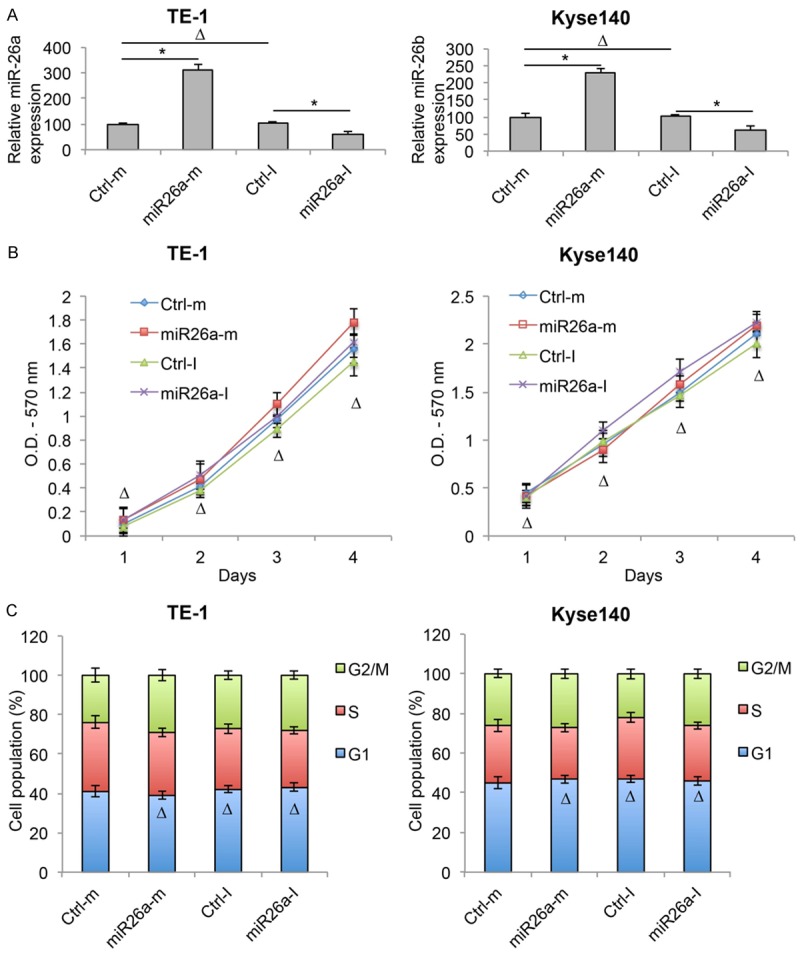
MiR-26a did not regulate proliferation or cell cycle in ESCC. A. TE-1 and Kyse140 were transfected with miR-26a mimics (miR26a-m) or inhibitor (miR26a-I), as well as control mimic miRNA (Ctrl-m) and control inhibitor miRNA (Ctrl-I). The transfection efficiencies were verified by qRT-PCR (ΔP > 0.05, *P < 0.05). B. After transfection, in vitro growth of TE-1 and Kyse140 cells were examined for 4 consecutive days using a proliferation assay (ΔP > 0.05). C. Post-transfection cell cycle transition was examined by a cell-cycle assay. Cell populations at G1, S and G2/M stages were calculated (*P < 0.05).
Post transfection, TE-1 and Kyse140 cells were examined by a cell proliferation assay. It showed that cancer cell growth was unaffected by either miR26a-m or miR26a-I (Figure 4B, ΔP > 0.05). A cell-cycle assay demonstrated that cell cycle transition was also unaffected by either miR26a-m or miR26a-I (Figure 4C, ΔP > 0.05).
In addition, after miR-26a was either upregulated or downregulated in TE-1 and Kyse140 cells, cancer cell migrations were examined by an in vitro invasion assay. It showed in both TE-1 and Kyse140 cells, neither miR26a-m nor miR26a-I had effect on ESCC migration (Figure 5A-D, ΔP > 0.05).
Figure 5.
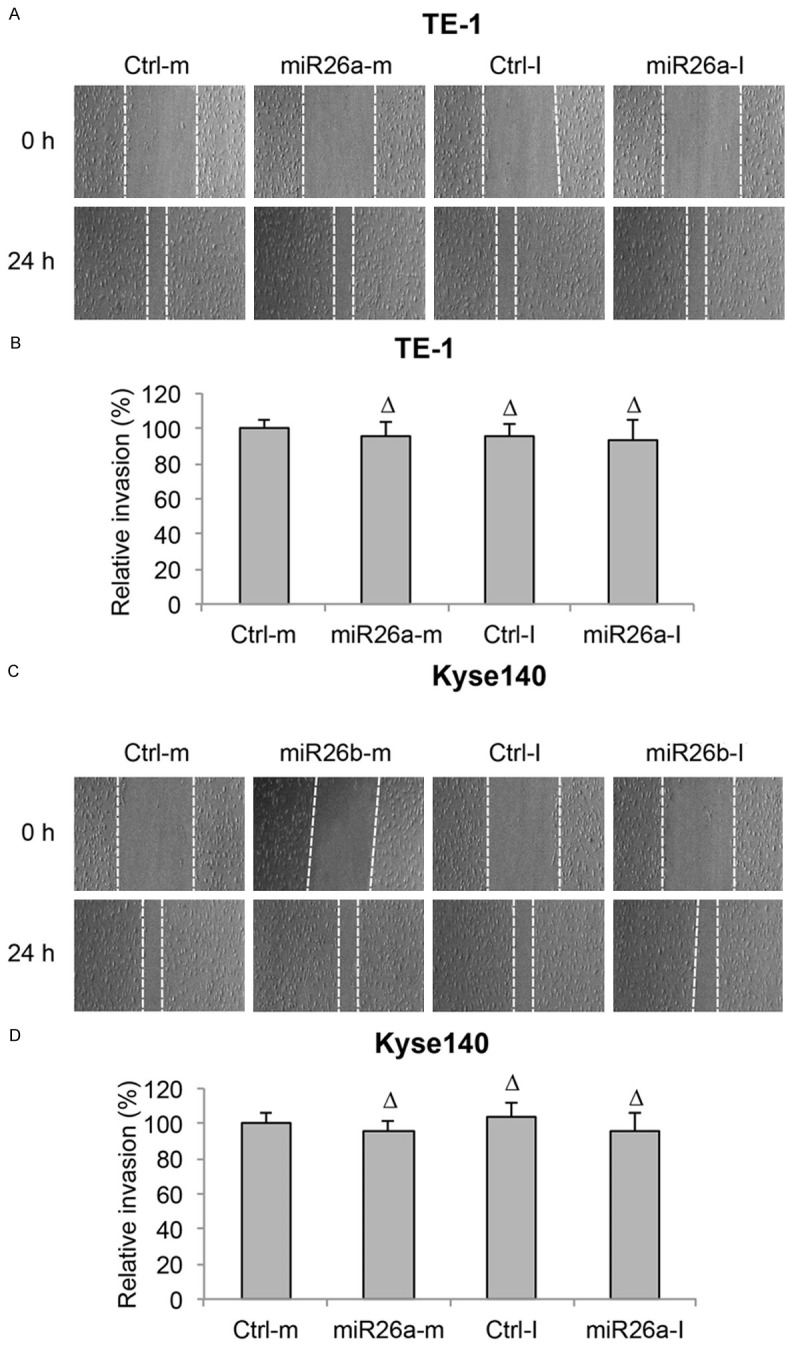
MiR-26a did not regulate migration in ESCC. A. TE-1 cell lines were transfected with miR-26a mimics (miR26a-m) or inhibitor (miR26a-I), as well as control mimic miRNA (Ctrl-m) and control inhibitor miRNA (Ctrl-I). An in vitro invasion assay was performed by creating a linear wound area on cell monolayer and monitored for 24 h. B. Invasion capability in miRNA-transfected TE-1 cells was measured by comparing the travelling distances of TE-1 cells invaded into the wound areas (ΔP > 0.05). C, D. Kyse140 cells were treated the same way as TE-1 cells and the invasion capability was measured (ΔP > 0.05).
Thus, these results suggest that miR-26a, either by upregulation or downregulation, had no effect on cancer proliferation, cell cycle progression or cancer migration in ESCC.
TRAF5 is inversely regulated by miR-26a in ESCC
As we discovered that miR-26b downregulation had tumor suppressive effects on ESCC development, we wondered what signaling pathway was regulated by miR-26b in ESCC. Using several public databases for miRNA target prediction, such as TargetScan (www.targetscan.org), we discovered that TRAF5 was a possible downstream target of human miR-26b (Figure 6A). We then conducted a dual-luciferase reporter assay in HEK293T cells. The analysis on relative luciferase activities showed that miR-26b did bind 3’ UTR of human TRAF5 gene, but not the mutated 3’ UTR of TRAF5 gene (Figure 6B, *P < 0.05, ΔP > 0.05).
Figure 6.
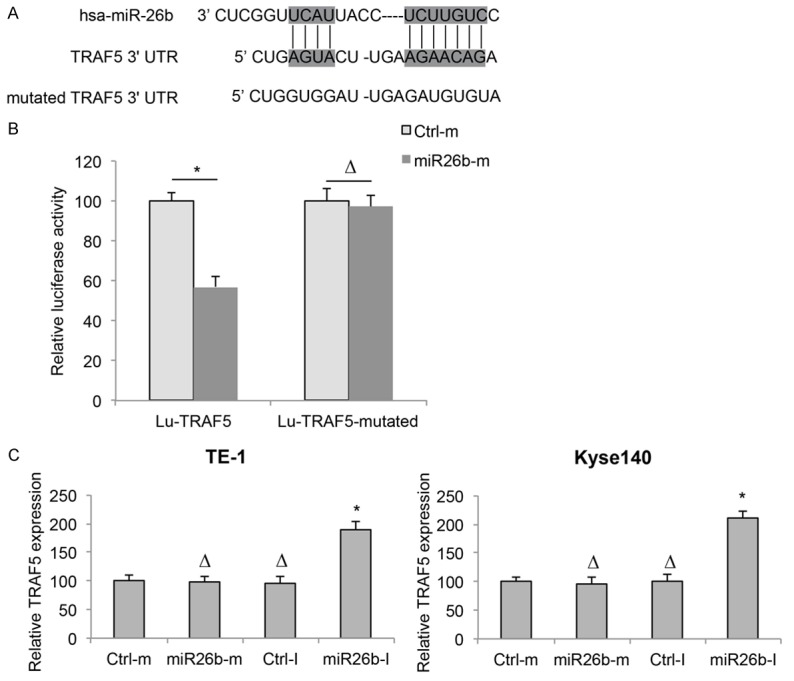
TRAF5 is downstream target of miR-26b in ESCC. A. Diagrams were shown for the sequence in the 3’ untranslated region (3’ UTR) of human TRAF5 gene, which was predicted to be bound by hsa-miR-26b. A mutated 3’ UTR sequence was created to nullify the binding of hsa-miR-26b. B. TRAF5 3’ UTR, and its mutated sequence were cloned into pmiR-REPORT to create two luciferase reporter, Lu-TRAF5 and Lu-TRAR5-mutated. In HEK293T cells, Lu-TRAF5 or Lu-TRAR5-mutated was co-transfected with Ctrl-m or miR26b-m for 48 h. Relative luciferase activities were measured using a dual-luciferase reporter assay (*P < 0.05, ΔP > 0.05). C. TE-1 and Kyse140 cells were transfected with Ctrl-m, miR26b-m, Ctrl-I or miR26b-I. Expressions of TRAF5 were measured by qRT-PCR (*P < 0.05, ΔP > 0.05).
We then investigated whether TRAF5 was directly regulated by miR-26b in ESCC cell lines, by examining TRAF5 expressions in miRNA-transfected TE-1 and Kyse140 cells. The analysis of qRT-PCR demonstrated that, TRAF5 was upregulated by miR26b-I, whereas unaffected by miR26b-m in both TE-1 and Kyse140 cells (Figure 6C, *P < 0.05, ΔP > 0.05). These results suggest that miR-26b downregulation inversely modulated the expression of TRAF5 in ESCC.
TRAF5 downregulation reversed the effect of miR-26b downregulation on cancer migration in ESCC
As we discovered that TRAF5 was negatively regulated by miR-26b, we wondered whether TRAF5 had direct impact on the modulation of miR-26b in ESCC. To examine this hypothesis, TE-1 and Kyse140 cells were transfected with a TRAF5-specific siRNA (TRAF5-siRNA) to knock down the endogenous gene expression of TRAF5. Other ESCC cells were transfected with a control scrambled-siRNA (S-siRNA). The knockdown efficacy was confirmed by qRT-PCR (Figure 7A, *P < 0.05).
Figure 7.
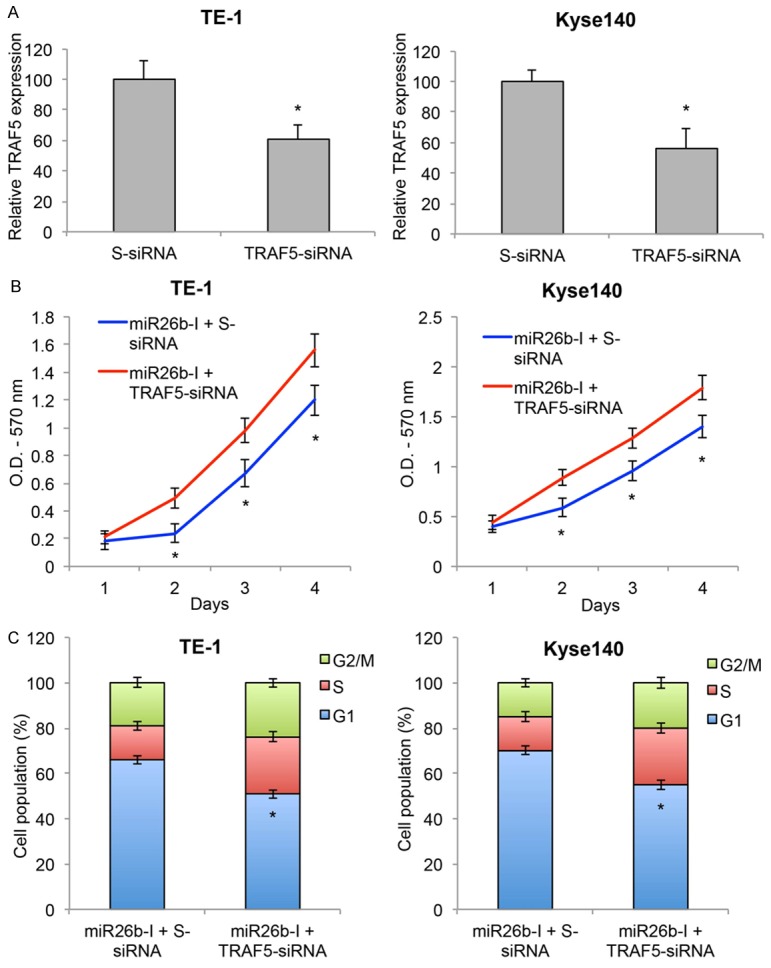
TRAF5 inversely regulated miR-26b-modulated cancer proliferation and cell cycle transition in ESCC. A. ESCC cell lines TE-1 and Kyse140 were transfected with TRAF5 specific siRNA (TRAF5-siRNA) or a control scrambled-siRNA (S-siRNA). The transfection efficiencies were verified by qRT-PCR (*P < 0.05). B. TE-1 and Kyse140 were pre-transfected with TRAF5-siRNA or S-siRNA for 24 h, then transfected with miR26b-I for another 48 h. The in vitro growth of TE-1 and Kyse140 cells were examined for 4 consecutive days using a proliferation assay (*P < 0.05). C. Post-transfection cell cycle progression was examined by a cell-cycle assay. Cell populations at G1, S and G2/M stages were calculated (*P < 0.05).
Then, TE-1 and Kyse140 cells were pre-transfected with S-siRNA or TRAF5-siRNA for 24 h, followed by 2nd transfection of miR26b-I. 48 h later, a cell proliferation assay showed that TRAF5-siRNA pre-transfection significantly reversed the inhibition of miR26b-I on ESCC in vitro cell growth (Figure 7B, *P < 0.05). In addition, a cell-cycle assay also demonstrated that TRAF5-siRNA pre-transfection significantly reversed the G1-stage arrest of ESCC cells induced by miR26b-I (Figure 7C, *P < 0.05). Therefore, these results suggest that TRAF5 downregulation could reverse the inhibitory effect of miR-26b downregulation on cancer proliferation and cell-cycle transition in ESCC cells.
TRAF5 downregulation reversed the effect of miR-26b downregulation on cancer migration in ESCC
After double-transfection in TE-1 and Kyse140 cells, we also examined ESCC migration capability through an in vitro invasion assay. Once cells reached ~80% confluence, we used a cell scraper to create a linear wound area, and then allow ESCC cells to grow for 24 h. By comparing double-transfected TE-1 cells at the onset (0 h) and the end (24 h) of invasion assay, we noticed that significantly more cells invaded into the wound area with TRAF5-siRNA pre-transfection (Figure 8A). Quantitative measurement confirmed this observation by showing that invasion capability of TE-1 cells was significantly recovered by TRAF5-siRNA pre-transfection (Figure 8B, *P < 0.05). Similar results were obtained from double-transfected Kyse140 cells. Transmit-light images showed that more Kyse140 cells invaded into wound area by TRAF5-siRNA pre-transfection (Figure 8C). Quantification indicated that invasion capability of Kyse140 cells was significantly improved by TRAF5-siRNA pre-transfection (Figure 8D, *P < 0.05). These results suggest TRAF5 downregulation could reverse the inhibitory effect of miR-26b downregulation on cancer migration in ESCC cells.
Figure 8.
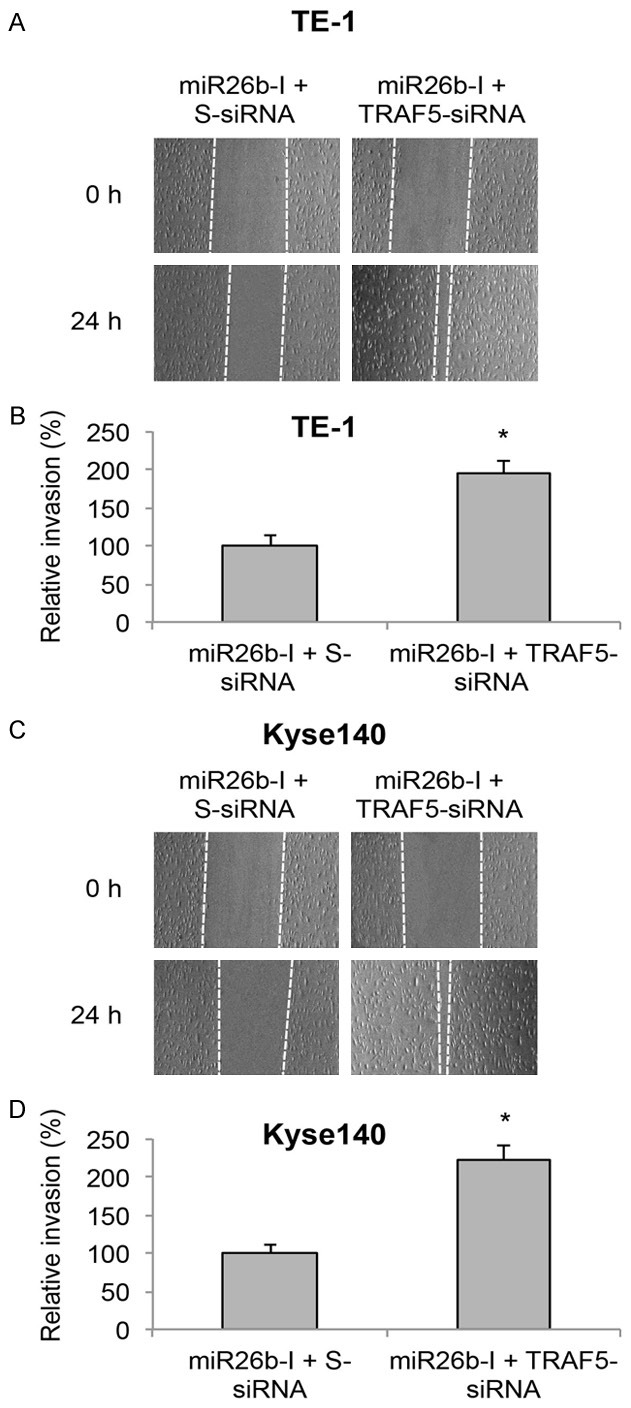
TRAF5 inversely regulated miR-26b-modulated cancer migration in ESCC. A. TE-1 cell lines were pre-transfected with TRAF5-siRNA or S-siRNA for 24 h, then transfected with miR26b-I for another 48 h. An in vitro invasion assay was performed by creating a linear wound area on cell monolayer and monitored for 24 h. B. Invasion capability in miRNA-transfected TE-1 cells was measured by comparing the travelling distances of TE-1 cells invaded into the wound areas (ΔP > 0.05). C, D. Kyse140 cells were treated the same way as TE-1 cells and the invasion capability was measured (ΔP > 0.05).
Discussions
A recent study demonstrated that miR-26a was downregulated in EAC and miR-26a directly regulated cancer proliferation and cell cycle in EAC cell line EA OE33 cells [9]. In the present study, we examined the gene expression profiles of miR-26 family, including miR-26a and miR-26b in another type of human esophageal cancer, ESCC. We used qRT-PCR to analyze both ESCC cell lines and ESCC tumors samples. To our surprise, we found that miR-26a and miR-26b were differentially expressed in ESCC, as miR-26b was aberrantly overexpressed in ESCC cell lines and ESCC tumors whereas miR-26a was equally expressed between normal esophageal epithelial cell and ESCC cell lines as well as ESCC tumor and non-tumor tissues. These results revealed that miR-26a and miR-26b, though closely associated to each other in many of the biological systems, are distinctly expressed in EAC and ESCC sub-types of human esophageal cancer. Though the complex signaling pathways contributing to the differential expression of miR-26 family during esophageal tumorigenesis is largely unknown, this specific expression pattern may be used as a potential biomarker to differentiate ESCC from EAC in the diagnosis for patients with esophageal cancer.
In addition, our study discovered that, miR-26b, but not miR-26a was predominantly upregulated in ESCC. Interestingly, the differential expressing pattern of miR-26a vs. miR-26b was also seen in other biological system. During the process of myogenesis, miR-26a was found to be differentially expressed from myoblasts to myotubes whereas miR-26b was unchanged during this transition [18]. Further examination demonstrated that overexpressing miR-26a post-transcriptionally suppressed Ezh2 gene and induced skeletal muscle cell differentiation [18]. Therefore, our results may suggest that miR-26b and miR-26a have different functional roles in the regulation of cancer development in ESCC.
The following functional experiments confirmed our hypothesis on different functional roles of miR-26a and miR-26b in ESCC. Using synthetic miRNA oligonucleotides, we transfected ESCC cell lines TE-1 and Kyse140 with both miR-26a and miR-26b mimics and inhibitors, and analyzed their effects on cancer proliferation, cell-cycle progression and cancer migration. We found that only the downregulation of miR-26b had tumor-suppressive effect, whereas miR-26b upregulation, miR-26a downregulation or miR-26a upregulation had no meaningful regulatory effect on ESCC cancer development. The phenomenon of only one of the miR-26 family having predominant functional effect was also seen in other tumors. For example, upregulating miR-26a was able to suppress EZH2 gene to induce cancer apoptosis in nasopharyngeal carcinoma, whereas overexpressing miR-26b had no such effect [19]. Thus, the findings of our study showing miR-26b was upregulated in ESCC and miR-26b downregulation had tumor suppressive effect in ESCC, suggest that miR-26b is mainly acting as a tumor oncogene in ESCC.
Also in the present study, we demonstrated that, by luciferase reporter assay and qRT-PCR, TRAF5 was the direct downstream target gene of miR-26b in ESCC. Most importantly, we showed that siRNA-mediated TRAF5 knockdown was able to reverse the inhibitory effect of miR-26b downregulation on ESCC proliferation, cell-cycle transition and cancer migration. Therefore, those results suggest that the predominant signaling pathway associated with miR-26b regulation on ESCC development was through TRAF5. It would be interesting to learn, possibly through future experiments, whether other members of the TRAF family, such as TRAF1, -2 or -6 may also participate in the regulation of miR-26b in ESCC.
In summary, we discovered a novel epigenetic mechanism of tumor oncogene miR-26b in human ESCC, mainly through the inhibition on TRAF5 signaling pathway. Those findings may facilitate identifying novel biomarker for early diagnosis or developing new clinical treatments to improve the prognosis in patients suffered from esophageal cancer.
Disclosure of conflict of interest
None.
References
- 1.Napier KJ, Scheerer M, Misra S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol. 2014;6:112–120. doi: 10.4251/wjgo.v6.i5.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.Herszenyi L, Tulassay Z. Epidemiology of gastrointestinal and liver tumors. Eur Rev Med Pharmacol Sci. 2010;14:249–258. [PubMed] [Google Scholar]
- 4.Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400–412. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- 5.Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Genet Dev. 2005;15:563–568. doi: 10.1016/j.gde.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Huang J, Zhang SY, Gao YM, Liu YF, Liu YB, Zhao ZG, Yang K. MicroRNAs as oncogenes or tumour suppressors in oesophageal cancer: potential biomarkers and therapeutic targets. Cell Prolif. 2014;47:277–286. doi: 10.1111/cpr.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakai NS, Samia-Aly E, Barbera M, Fitzgerald RC. A review of the current understanding and clinical utility of miRNAs in esophageal cancer. Semin Cancer Biol. 2013;23:512–521. doi: 10.1016/j.semcancer.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Mayne GC, Hussey DJ, Watson DI. MicroRNAs and esophageal cancer--implications for pathogenesis and therapy. Curr Pharm Des. 2013;19:1211–1226. doi: 10.2174/138161213804805702. [DOI] [PubMed] [Google Scholar]
- 9.Zhang YF, Zhang AR, Zhang BC, Rao ZG, Gao JF, Lv MH, Wu YY, Wang SM, Wang RQ, Fang DC. MiR-26a regulates cell cycle and anoikis of human esophageal adenocarcinoma cells through Rb1-E2F1 signaling pathway. Mol Biol Rep. 2013;40:1711–1720. doi: 10.1007/s11033-012-2222-7. [DOI] [PubMed] [Google Scholar]
- 10.Jackson-Bernitsas DG, Ichikawa H, Takada Y, Myers JN, Lin XL, Darnay BG, Chaturvedi MM, Aggarwal BB. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene. 2007;26:1385–1397. doi: 10.1038/sj.onc.1209945. [DOI] [PubMed] [Google Scholar]
- 11.Ha H, Han D, Choi Y. TRAF-mediated TNFR-family signaling. Curr Protoc Immunol. 2009 doi: 10.1002/0471142735.im1109ds87. Chapter 11: Unit11 19D. [DOI] [PubMed] [Google Scholar]
- 12.Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Annu Rev Biophys. 2013;42:443–468. doi: 10.1146/annurev-biophys-083012-130338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tao T, Cheng C, Ji Y, Xu G, Zhang J, Zhang L, Shen A. Numbl inhibits glioma cell migration and invasion by suppressing TRAF5-mediated NF-kappaB activation. Mol Biol Cell. 2012;23:2635–2644. doi: 10.1091/mbc.E11-09-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu P, Gao Y, Huan J, Ge X, Tang Y, Shen W, Tian Y, Shen W, Zou S, Zhou J, Zhang S. Upregulation of PAX2 promotes the metastasis of esophageal cancer through interleukin-5. Cell Physiol Biochem. 2015;35:740–754. doi: 10.1159/000369734. [DOI] [PubMed] [Google Scholar]
- 15.Andl CD, Mizushima T, Nakagawa H, Oyama K, Harada H, Chruma K, Herlyn M, Rustgi AK. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem. 2003;278:1824–1830. doi: 10.1074/jbc.M209148200. [DOI] [PubMed] [Google Scholar]
- 16.Fleige S, Walf V, Huch S, Prgomet C, Sehm J, Pfaffl MW. Comparison of relative mRNA quantification models and the impact of RNA integrity in quantitative real-time RT-PCR. Biotechnol Lett. 2006;28:1601–1613. doi: 10.1007/s10529-006-9127-2. [DOI] [PubMed] [Google Scholar]
- 17.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 18.Wong CF, Tellam RL. MicroRNA-26a targets the histone methyltransferase Enhancer of Zeste homolog 2 during myogenesis. J Biol Chem. 2008;283:9836–9843. doi: 10.1074/jbc.M709614200. [DOI] [PubMed] [Google Scholar]
- 19.Alajez NM, Shi W, Hui AB, Bruce J, Lenarduzzi M, Ito E, Yue S, O’Sullivan B, Liu FF. Enhancer of Zeste homolog 2 (EZH2) is overexpressed in recurrent nasopharyngeal carcinoma and is regulated by miR-26a, miR-101, and miR-98. Cell Death Dis. 2010;1:e85. doi: 10.1038/cddis.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]