

Abstract
Bronchiolitis obliterans syndrome (BOS), characterized by progressive airflow obstruction, is the main barrier to long-term graft survival after lung transplantation. Despite extensive studies, the mechanisms underlying BOS remain poorly understood, and targeted interventions have not yet been fully developed. In the present study, we employed a mouse model of tracheal transplantation and demonstrated that blockade of HMGB1 alone or combined with heparanase (HPSE) attenuates the development of BOS. It was noted that HMGB1 was first passively released from necrotic/damaged cells as a result of early unavoidable allograft injuries, leading to macrophage infiltration along with HMGB1 active secretion. Mechanistic studies revealed that extracellular HMGB1 acted through its receptor, RAGE, to activate NF-κB, which then bound to the HPSE promoter to transcribe its expression. The enhanced HPSE next released HS-bonded latent TGF-β from myofibroblast ECM by cleaving HS chains to promote the initiation and progression of BOS. Together, our data suggest that HMGB1 and HPSE could be viable targets for prevention and intervention of fibrotic diseases such BOS after lung transplantation.
Keywords: Lung transplantation, BOS, HMGB1, RAGE/NF-κB pathway, HPSE, TGF-β
Introduction
Lung transplantation is a life-saving procedure for patients with end-stage respiratory diseases. However, chronic lung allograft dysfunction (CLAD) is the major factor limiting long-term graft survival [1]. Bronchiolitis obliterans syndrome (BOS) is a classic subtype of CLAD characterized by the fibro-proliferative tissue remodeling and increased airway extracellular matrix (ECM) deposition with resultant lung dysfunction [2]. The pathogenesis of BOS is complex and includes both alloimmune and nonalloimmune pathways [3]. Although allograft accommodation has, in some cases, been associated with non-specific immunosuppression, BOS remains one of the major factors limiting long-term graft survival as the deterioration of graft performance occurs within 5 years in approximately 50% of patients who receive transplant surgery [4]. Therefore, gaining a better understanding of the mechanisms underlying BOS would be critical to achieve long-term lung allograft survival in clinical settings.
Nontraditional proinflammatory mediators and their receptors have been the focus of recent research into the mechanism of CLAD [5], and of which, high mobility group box-1 (HMGB1), a typical damage-associated protein, is particularly of interest. Previous studies including ours demonstrated that HMGB1 is implicated in organ injury and inflammatory response in the setting of organ transplantation [6]. HMGB1 is a chromatin-associated protein, but it can be passively released from necrotic/damaged cells [7,8], or secreted by the activated immune cells such as macrophages, dendritic cells (DCs) and natural killer (NK) cells [9,10]. In the extracellular milieu, HMGB1 promotes the activation of proinflammatory pathways by interacting with the pattern recognition receptors (i.e., TLR2/TLR4) and receptor for advanced glycation end products (RAGE) [11,12]. More recently, we demonstrated feasible evidence indicating that HMGB1 promotes fibrotic signaling in unilateral ureteral obstruction (UUO)-induced renal fibrosis [13]. Given that RAGE is constitutively expressed at high levels in the lung [14], our data support that HMGB1 is also implicated in the pathogenesis of BOS.
It has been well recognized that TGF-β plays an essential role in the initiation and progression of BOS [15,16]. In general, TGF-β is stored with a prodomain in the ECM as a latent complex [17], and release of active TGF-β is regulated by heparanase (HPSE), an endo-β-D-glucuronidase, which cleaves the heparan sulfate (HS) saccharide chains in the ECM [18]. Importantly, our pilot studies revealed that HMGB1 potently stimulated fibroblasts expressing high levels of HPSE, which prompted us to hypothesize that extracellular HMGB1 implicates in the initiation and progress of BOS by regulating HPSE activity and TGF-β signaling. To test this hypothesis, a murine heterotopic tracheal transplantation model was employed to mimic lung transplantation in clinical settings. Our studies revealed that HMGB1 acts through RAGE-dependent NF-κB pathway to promote HPSE transcription, which then triggers the release of TGF-β from ECM associated with the initiation and progression of BOS.
Materials and methods
Animals
Both C57BL/6J (B6, H-2b) and BALB/c (H-2d) mice (male, 8- to 12-wk old) were purchased from the Animal Experimental Center of Wuhan University and housed in the SPF facility of Tongji Medical College. All procedures were conducted in accordance with the NIH guidelines for the care and use of laboratory animals and were approved by the Tongji Hospital Institutional Animal Care and Use Committee (IACUC).
Reagents and antibodies
The primary antibodies that were used includ-ed the followings: rabbit anti-TGF-β, anti-p65, anti-HMGB1, anti-latent-HPSE, anti-active-HPSE, anti-GAPDH and anti-isotype IgG antibodies (Santa Cruz Technologies, Dallas, Texas, USA); rabbit anti-collagen I antibody (Millipore, Etobicoke, Ontario, Canada); and mouse anti-F4/80 and anti-α-smooth muscle actin (α-SMA) (Abcam, Cambridge, MA, USA). The secondary antibodies used for immunostaining included Alexa Fluor 488 donkey anti-rabbit IgG and Alexa Fluor 594 donkey anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA). The NF-κB inhibitor, JSH-23, was obtained from Selleck (Houston, TX, USA). All other reagents were purchased from Sigma (St. Louis, MO, USA), unless otherwise stated.
Heterotopic trachea transplantation
Heterotopic trachea transplantation (HTT) was performed according to an adaptation of the Hertz method [19]. Briefly, donor mice (BALB/c, H-2d) were euthanized with 100% CO2, and the trachea was freed by blunt dissection for transplantation. Recipient mice (B6, H-2b) were anaesthetized with pentobarbital sodium (Merck Chemicals, Shanghai, China, 100 mg/kg, i.p.), and the trachea was placed into a pouch created over the posterior upper back area of the mice.
HMGB1 neutralizing antibody treatment
NZB/W mouse-derived monoclonal anti-HMGB1 neutralizing antibody (isotype IgG) was kindly provided by Dr. Deng-Ping Yin, Department of General Surgery, University of Chicago. Recipient mice were administered 10 mg/kg anti-HMGB1 antibodies intraperitoneally (i.p.) one day before the transplant surgery and then twice a week for the first month post-transplantation. B6 littermates injected with an equal amount of mouse IgG were used as controls (n = 15 mice/group).
In vivo OGT2115 treatment
Similar as above, recipient mice were injected with OGT2115 (Tocris Bioscience, Bristol, UK, 20 mg/kg, i.p.) one day before allograft implantation followed by twice a week for the first month post-transplantation alone or combined with HMGB1 neutralizing antibodies. B6 littermates injected with an equal volume of PBS or control IgG were used as controls (n = 15 for each group).
Cell culture
Human lung fibroblasts (MRC-5, ATCC, Manassas, VA, USA) were cultured in minimal essential medium (MEM) (Genom, Hangzhou, China) with penicillin-streptomycin (Genom) and 10% fetal calf serum (FBS, Hyclone, Logan, UT, USA) at 37°C in 5% CO2 with change of medium every other day. For the common treatment group, cells were incubated in serum-free media for 12 hours before treating with an HPSE inhibitor (OGT2115, 0.5 µM) for another half hour, after which recombinant HMGB1 (PeproTech, Rocky Hill, New Jersey, USA, 100 ng/ml) was added into the media. An equal volume of PBS was used as the control for OGT2115 treatment. To detect latent TGF-β in myofibroblast-derived ECM, MRC-5 cells were incubated in serum-free media for 12 hours before treatment with recombinant HMGB1 (100 ng/ml) for 24 hours, after which the cells were washed with PBS and then treated with OGT2115 (0.5 µM) for 24 hours, or addition of an equal volume of PBS to serve as controls.
Histological and immunohistochemical analysis
Grafts harvested from the recipient mice were fixed with 4% formalin at room temperature for 24 hours. The formalin-fixed tissues were then embedded in paraffin and cut into 5-μm sections for hematoxylin and eosin (H&E) or immunohistochemical staining using the established techniques [20,21].
Morphological studies
Morphological analysis was performed by two independent investigators with Image-Pro Plus 6.0 software. For each graft, 6 randomly selected sections were analyzed by the same observer, and a mean value was generated to represent the average measurements for each outcome assessed. Luminal occlusion was defined as the obliteration of the inner cartilage space by abnormal proliferative fibrous tissues, and the percent occluded area was calculated as follows: (area within cartilage-area within residual lumen)/area within cartilage × 100%. Airway epithelial cell-secreted mucus located within the cartilage ring was not classified as obliteration [22].
Immunofluorescence analysis
MRC-5 cells were grown, treated and fixed in 24-well plates as indicated above. After fixation, the cells were rinsed with PBS and permeabilized in blocking buffer (5% goat serum + 0.5% BSA + 1% Triton X-100). The samples were next incubated with the primary antibody for 1 hour at room temperature and washed with PBS before applying the secondary antibody for another 60 minutes [23]. DAPI (Invitrogen, Carlsbad, CA, USA) was then added for nuclear staining, and Prolong Gold (Invitrogen) was used to preserve the signal. For paraffin-embedded sections, the samples were de-paraffinized and re-hydrated as indicated and boiled in borate saline buffer (pH 8.0) for antigen retrieval before the process of applying the primary antibody [24]. Confocal images were taken on a laser scanning confocal microscope (Carl Zeiss LSM 710, Germany) and processed with the ZEN 2009 software (Carl Zeiss, Germany).
Wound healing assay
GFP-labeled MRC-5 cells were seeded into six-well plates (5 × 105 cells per well) and incubated overnight. After incubation, the cells were washed with PBS, and wounds were created using a sterilized pipette tip. Wound closure was monitored at 0 and 24 hours using a camera-equipped inverted microscope [25]. The quantitation of wound closure was determined using the Image-Pro Plus 6.0 software.
Proliferation assay
MRC-5 cell proliferation assay was performed according to protocols described elsewhere [26]. Briefly, the cell pellets were resuspended in 5 μM CFSE (5, 6-carboxyfluorescein diacetatesuccinimidyl ester) at a concentration of 0.2-2 × 107 cells/ml and incubated at 37°C for 8 minutes. After washes with PBS, the cells were treated with recombinant HMGB1 and/or OGT2115 as described above. Cell proliferation was determined by flow cytometry analysis of the CFSE positive cells, and data were presented as the means ± SEM of values from triplicate samples.
Western blot analysis
Western blotting was performed as previously described [27]. Briefly, cell or tissue lysates (10 µg) were fractionated by SDS-PAGE and probed with the corresponding primary antibodies. Immunoreactive bands were visualized using an enhanced chemiluminescence system as reported [28].
ELISA
TGF-β levels were measured in cell culture supernatants using a commercial ELISA kit (eBioscience, SanDiego, CA, USA) as previously described [29].
Real-time PCR
Total RNA was extracted from cell samples and reverse transcribed into cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. mRNA levels were quantified using RT-PCR with HPSE specific primers (forward: 5’-GCA CAA ACA CTG ACA ATC CAA G-3’; reverse: 5’-AAA AGG ATA GGG TAA CCG CAA-3’). GAPDH (forward: 5’-GTG GTC TCC TCT GAC TTC AAC A-3’; reverse: 5’-CCA CCA CCC TGT TGC TGT AGA-3’) was used for normalization. Relative expression levels for the target gene were calculated using the 2-ΔΔCt approach as previously described [30].
Chromatin immunoprecipitation (ChIP) assay
MRC-5 cells were treated with PBS or recombinant HMGB1 (100 ng/ml) for 1 hour. The cells were harvested and subjected to ChIP assay using a ChIP assay kit (Haimen, JS, China) as previously described [27]. Anti-p65 or rabbit anti-isotype IgG antibody were used to precipitate the protein-DNA complexes. The resulting products were used as templates to amplify the HPSE promoter region flanking the NF-κB binding site with following primers: forward, 5’-AAT GTT GAG CAA CAT CAC AAT AC-3’; and reverse, 5’-GTG TCA GAA TCG GGA TGT GAA GTG TC-3’.
RNA interference
Small-interfering RNA (siRNA) duplexes for silencing human RAGE were designed using the RNAi Designer and synthesized by Ribobio (Guangzhou, Guangdong, China). The sequence for RAGE siRNA was 5’-CAC UGC AGU CGG AGC UAA UGG-3’. A scramble siRNA (5’-GUA CCG CAC GUC AUU CGC AUC-3’) was served as a control. siRNA transfection was performed with Lipofectamine RNAi Max (Invitrogen, Carlsbad, USA) according to the established techniques [25].
Statistical analysis
All experiments were performed in triplicate, and statistical analyses were conducted using the SPSS version 16.0 software. Data were analyzed using one-way analysis of variance (ANOVA) with Tukey’s HSD post-hoc test for multiple comparisons. A p-value of less than 0.05 was considered statistically significant.
Results
HMGB1 stimulates fibroblast transdifferentiation by inducing HPSE expression
We first sought to examine the role of HMGB1 in lung fibroblast transdifferentiation, in which we stimulated MRC-5 cells (human normal lung fibroblasts) with recombinant HMGB1. HMGB1 potently stimulated the transdifferentiation of fibroblasts into myofibroblasts as manifested by the induction of high levels of α-SMA and collagen I expression (Figure 1A). More interestingly, we also found that HMGB1 induced a significant increase for both active and latent HPSE (Figure 1B). To confirm these results, we then conducted real-time PCR analysis and demonstrated that HMGB1 induced high levels of HPSE mRNA (Figure 1C). To address whether HPSE is required for HMGB1-induced fibroblast transdifferentiation, OGT2115, an HPSE inhibitor, was added into the culture. Remarkably, OGT2115 significantly repressed HMGB1-induced α-SMA and collagen I expression (Figure 1D), indicating that HMGB1-induced fibroblast transdifferentiation likely depends on HPSE activity.
Figure 1.
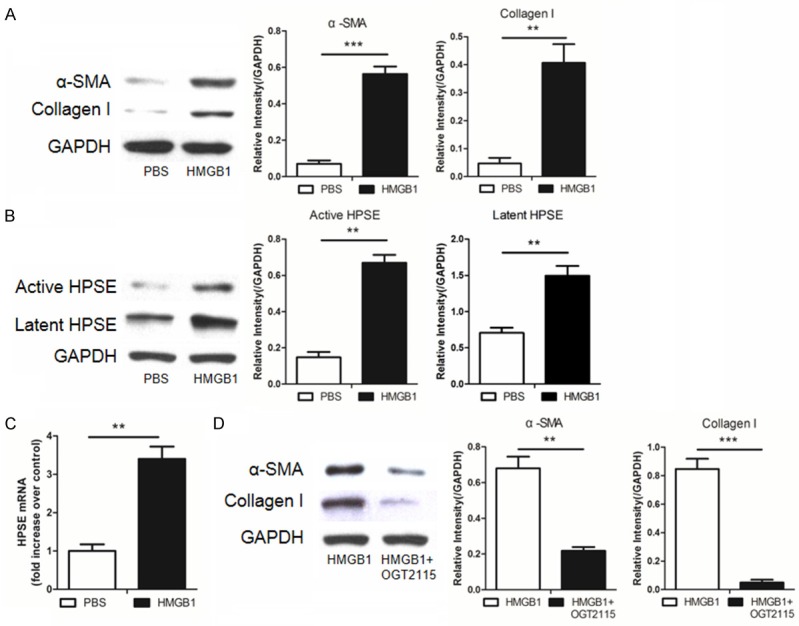
HMGB1-induced fibroblast transdifferentiation depends on HPSE activity. MRC-5 cells were cultured with recombinant HMGB1 (100 ng/ml) or OGT2115 (0.5 µM) for 24 hours and then subjected to following analysis. A. Western blot results for analysis of HMGB1-induced α-SMA and collagen I expression. B. HMGB1 stimulated MRC-5 cells expression of significantly higher levels of active and latent HPSE. C. Real-time PCR for analysis of the HPSE mRNA levels after HMGB1 stimulation. D. Addition of OGT2115, an HPSE inhibitor, attenuated HMGB1 induced α-SMA and collagen I expression. *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
To confirm the above observations, we conducted immunostaining of stimulated MRC-5 cells. Indeed, HMGB1 induced high levels of α-SMA expression, while α-SMA was almost undetectable upon the addition of OGT2115 (Figure 2A). We then assessed the impact of HMGB1 on MRC-5 cell proliferation. CFSE based flow cytometry analysis revealed that HMGB1 significantly stimulated MRC-5 cell proliferation, which was potently repressed by OGT2115 (Figure 2B). Consistent with these results, HMGB1 significantly promoted the migration of MRC-5 cells, while OGT2115 almost diminished HMGB1-induced migration as determined by cell scratch assays (Figure 2C). Of interestingly note, addition of OGT2115 resulted in the accumulation of latent HPSE along with a significant reduction for active HPSE (Figure 2D). Given that OGT2115 did not show perceptible impact on HPSE transcription as determined by real-time PCR analysis (Figure 2E), these data indicate that OGT2115 inhibits HPSE activity by attenuating its maturation. Taken together, our results indicate that HMGB1 promotes fibroblast transdifferentiation, migration and proliferation in an HPSE-dependent manner, which prompted us to hypothesize that extracellular HMGB1 implicates in the initiation and progress of BOS by regulating HPSE activity.
Figure 2.
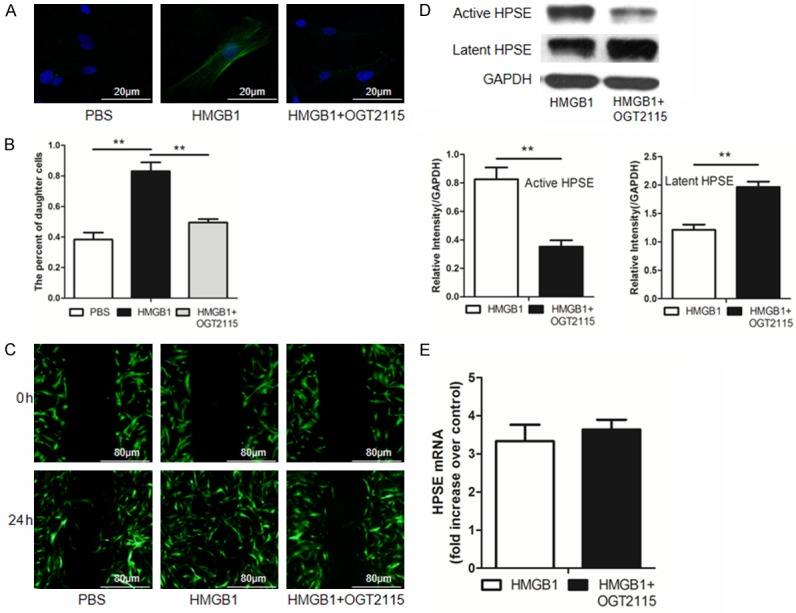
HMGB1 promotes fibroblast transdifferentiation, migration and proliferation in an HPSE dependent manner. Similar as above, MRC-5 cells were treated with recombinant HMGB1 (100 ng/ml) or HMGB1 along with OGT2115 (0.5 µM). A. Results for immunosatining of α-SMA, a marker for fibroblast-myofibroblast differentiation. Nucleus stained in blue, while α-SMA stained in green. B. Flow cytometry results for analysis of MRC-5 proliferation. C. Results for cell scratch assays for analysis of cell migration. D. Addition of OGT2115 resulted in the accumulation of latent HPSE but reduction of active HPSE in HMGB1 stimulated MRC-5 cells. E. Addition of OGT2115 did not affect HPSE mRNA levels. **, p < 0.01.
Detection of extracellular HMGB1 and HPSE expression in allografts during BOS development
It was suggested that ischemia/reperfusion is one of major pathophysiological component of allograft malfunction after transplantation. We first checked passive HMGB1 release in allografts after insult of tracheal harvest. To this end, we stained HMGB1 in tracheal allografts following 4 hours of cold ischemic insult. HMGB1 was solely localized in the nuclei of freshly collected trachea, while certain cells manifested cytoplasmic HMGB1 in ischemia insulted trachea (Figure 3A), indicating ischemia induced cell damage along with HMGB1 passive release. To further address the role of HMGB1 in post-transplant airway lesions, a mouse model of tracheal transplantation was employed for the study. Remar-kably, larger amount of HMGB1 passive release was even noted after 24 hours of tracheal transplantation (Figure 3B). Next, we examined active HMGB1 secretion by allograft infiltrated immune cells such as macrophages, in which we co-stained trachea sections with HMGB1 and F4/80, a marker for macrophages. Indeed, the F4/80 positive macrophages were characterized by the significant HMGB1 cytoplasmic translocation (Figure 3C), indicating HMGB1 active secretion.
Figure 3.
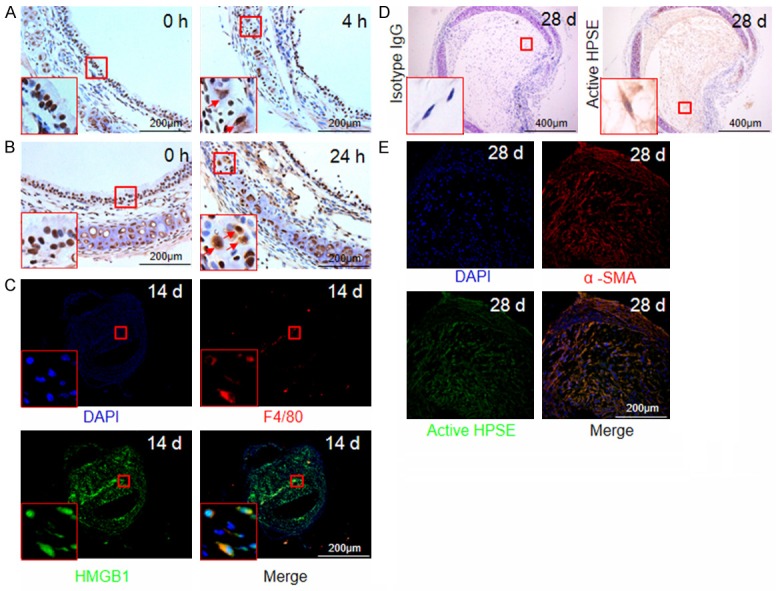
Tracheal allografts manifest HMGB1 passive release and active secretion along with altered HPSE expression. A. Cold ischemic insult (4 hours) resulted in HMGB1 passive release as indicated by arrows within the inset picture. B. Detection of HMGB1 passive release from tracheal allografts (indicated by arrows within the inset picture) after 24 hours of transplantation. C. Co-immunostaining for analysis of HMGB1 active secretion by allograft infiltrated macrophages after day 14 of transplantation. HMGB1 stained in green, macrophages stained by F4/80 in red, and nucleus stained in blue by DAPI. D. Tracheal allografts with BOS onset were manifested by active HPSE expression. E. Results for co-immunostaining of α-SMA and HPSE in tracheal grafts at day 28 of transplantation. Active HPSE was stained green, α-SMA was stained in red, and nucleus was stained in blue.
Given the role of HPSE played in ECM remodeling [18,31], we next investigated the impact of HPSE on BOS, which is manifested by a marked increase in ECM deposition. HPSE was noted to be widely distributed in the lumen of allografts after day 28 of transplantation (Figure 3D), and more interestingly, HPSE was highly expressed by the α-SMA positive myofibroblasts (Figure 3E). Collectively, our data support that HMGB1 and HPSE are likely involved in the initiation and progression of BOS.
Blockade of HMGB1 or inhibition of HPSE activity attenuate BOS
We then assessed the impact of HMGB1 or HPSE on BOS. The recipient mice were administered with an HMGB1-neutralizing antibody (HMGB1 Ab group) or OGT2115 (OGT2115 group). In line with our expectation, the luminal occlusion area of allografts were both significantly reduced in the HMGB1 Ab group (23.33%±6.51%) and OGT2115 group (46.33%±9.07%) as compared with the control group (92.4%±8.23%). Moreover, the protective effect was more significant once recipient mice treated with combined HMGB1 neutralizing antibody and OGT2115 (7%±5.29%) (Figure 4). Collectively, these data support that neutralizing HMGB1 or inhibiting HPSE activity attenuate BOS development.
Figure 4.
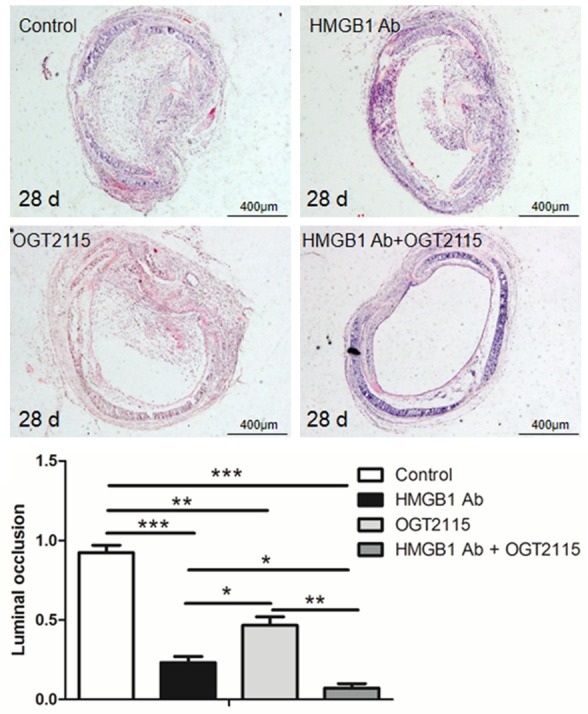
Blockade of HMGB1 along with HPSE attenuates BOS development. Tracheal allografts were harvested at day 28 of implantation, and the luminal occlusion ratio was assessed as described for each group (n = 15 for each group). *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
HMGB1 acts through RAGE to induce HPSE transcription
It has been well demonstrated that extracellular HMGB1 can act through receptors TLR2/4 or RAGE [32]. We thus next analyzed the expression of HMGB1 receptors after recombinant HMGB1 stimulation. Western blot analysis revealed that HMGB1 induced a significant increase for the expression of RAGE but not TLR2 and TLR4 (Figure 5A). We then used an siRNA to knockdown the expression of RAGE in MRC-5 cells (Figure 5B). Indeed, knockdown of RAGE expression attenuated HMGB1-induced active HPSE and latent HPSE (Figure 5C). These data suggest that HMGB1 acts predominantly through receptor RAGE to induce the expression of HPSE.
Figure 5.

HMGB1 acts through its receptor RAGE to induce HPSE expression. A. HMGB1 preferentially stimulated RAGE expression in MRC-5 cells. B. An siRNA potently repressed RAGE expression in MRC-5 cells. C. Knockdown of RAGE by siRNA attenuated HMGB1 induced HPSE expression in MRC-5 cells. **, p < 0.01, and ***, p < 0.001.
The HMGB1-RAGE axis stimulates NF-κB binding to the HPSE promoter
To address the signaling pathway by which HMGB1 induces HPSE expression, we first examined NF-κB activity. Western blot analysis showed that HMGB1 induced NF-κB activation by promoting p65 nuclear translocation (Figure 6A). More interestingly, ChIP assay revealed that p65 could directly bind to HPSE promoter (Figure 6B). In consistent with these results, knockdown of RAGE down-regulated HMGB1-induced p65 nuclear translocation (Figure 6C). To further confirm this observation, JSH-23, an NF-κB inhibitor (JSH-23), was added into MRC-5 cultures along with recombinant HMGB1 as above. Indeed, repression of NF-κB activity attenuated HMGB1-induced levels for active and latent HPSE (Figure 6D). Together, our data indicate that HMGB1 binds to its cognate receptor RAGE, and by which it activates NF-κB to enhance HPSE expression.
Figure 6.

The HMGB1-RAGE axis activates NF-κB to promote HPSE expression. MRC-5 cells were treated with 100 ng/ml HMGB1 alone, or combined with RAGE siRNA, or combined with NFκB inhibitor JSH-23 as described, and then subjected to following analyses. A. HMGB1 potently stimulated NF-κB subunit p65 nuclear translocation. B. ChIP assays revealed that NF-κB selectively binds to the HPSE promoter. C. Knockdown of RAGE by siRNA in MRC-5 cells repressed HMGB1 induced p65 nuclear translocation. D. Suppression of NF-κB activity by JSH-23 attenuated HMGB1 induced active and latent HPSE expression. *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
HPSE cleaves HS chains to release latent TGF-β from ECM
Finally, we sought to address the mechanism by which HPSE exacerbates fibrosis. During the course of fibrosis, myofibroblasts secret latent TGF-β, which is stored in the ECM through its binding to ECM components such as HS saccharide chains [33]. This is evidence that autocrine TGF-β formation as the stimulus for constitutive α-SMA expression in cultured lung myofibroblasts [34], we thus postulated that HPSE promotes fibrosis by enhancing ECM stored TGF-β release through the cleavage of HS saccharide chains. We first conducted immunostaining to assess the content of latent TGF-β in the myofibroblast-derived ECM in the presence of OGT2115 or PBS. In accordance with our hypothesis, OGT2115 increased the content of latent TGF-β in the ECM compared with controls (Figure 7A). Next, the culture supernatants were collected for analysis of active TGF-β by ELISA. In contrast to the immunostaining results, addition of OGT2115 significantly decreased the release of active TGF-β from ECM into the supernatants (Figure 7B). Taken together, our data demonstrate that HMGB1 activates NF-κB through its cognate receptor RAGE to enhance HPSE transcription, which then promotes TGF-β release from ECM to exacerbate fibrosis.
Figure 7.

HPSE promotes TGF-β release from myofibroblast-derived ECM. HMGB1 stimulated myofibroblasts were treated with OGT2115 (0.5 µM) or an equal volume of PBS for 24 hours, and then subjected to analysis of latent and active TGF-β levels. A. Suppression of HPSE activity resulted in the accumulation of latent TGF-β content in myofibroblast-derived ECM. Latent TGF-β stained in red, while nuclei stained in blue. B. Results for analysis of TGF-β release from ECM into the cell culture supernatant by ELISA. OGT2115 significantly reduced the content of TGF-β in the culture supernatant. **, p < 0.01.
Discussion
Despite extensive study, the exact molecular mechanisms underlying CLAD, especially BOS, have yet to be fully addressed. Recent studies including ours demonstrated reasonable evidence for a role of HMGB1 in allograft rejection [6], we thus hypothesized that HMGB1 may play a critical role in the pathogenesis of BOS after lung transplantation. In this report, we identified extracellular HMGB1 as an essential molecule to promote the initiation and progression of BOS after lung transplantation. Our studies also highlighted a previously unknown signaling network that links HMGB1, RAGE, HPSE and TGF-β release from ECM during the course of fibrotic processes such as in the setting of BOS.
In this study, a mouse tracheal transplantation model was employed to demonstrate the impact of HMGB1 on lung allograft chronic rejection. Previous studies have shown that HMGB1 can be either passively released from damaged/necrotic cells or actively secreted by activated immune cells such as macrophages, DCs or NK cells [35]. Indeed, a portion of HMGB1 was passively released from necrotic or damaged tracheal allograft cells, which suffered a number of inevitable insults at the early stage of posttransplantation. The other portion of HMGB1 detected was actively secreted by the infiltrated macrophages. To dissect the precise impact of HMGB1 on BOS development, recipient mice were administered with an HMGB1 neutralizing antibody and then assessed BOS severity after day 28 day of transplantation. Remarkably, blockade of HMGB1 resulted in a significant reduction in the luminal occlusion area of allografts as compared with that of controls.
It was interestingly noted that HMGB1 also stimulated myofibroblasts expression of high levels of HPSE, an endoglycosidase that cleaves HS chains associated with ECM remodeling [36]. There is suggestive evidence that HPSE may modulate the signaling of pro-fibrotic factors such as TGF-β [18]. We thus hypothesized that HMGB1 exacerbates BOS by enhancing HPSE expression. To address this notion, we first examined HPSE expression in tracheal allografts upon BOS onset, and found that HPSE was highly expressed in the airway lumen of allografts. Further analysis revealed that HPSE was mainly expressed by myofibroblasts, which play a central role in fibroproliferative airway remodeling during the course of BOS development after lung transplantation by producing excessive amounts of ECM [37]. To further address this question, we conducted tracheal transplantation along with administration of OGT2115, an HPSE inhibitor, and then assessed BOS severity as above. The results showed that inhibition of HPSE significantly ameliorated the severity of BOS.
To address the cross-talk between HMGB1 and HPSE during fibroblast-myofibroblast transition in the setting of BOS, we first examined the impact of HPSE activity on HMGB1-induced myofibroblast differentiation, migration and proliferation. Indeed, inhibition of HPSE activity not only attenuated HMGB1-induced myofibroblast differentiation, but also inhibited HMGB1-induced proliferation and migration. Given that HMGB1 significantly increased the expression of active HPSE and latent HPSE along with enhanced expression of the fibrotic markers collagen I and α-SMA, our data support that HPSE is downstream of HMGB1 signaling.
To date, TLR2, TLR4 and RAGE have been identified as the receptors for extracellular HMGB1 [38]. However, it seems that RAGE is most likely the primary receptor for HMGB1 in myofibroblast differentiation since HMGB1 failed to manifest a perceptible impact on TLR2 and TLR4 expression but enhanced RAGE levels by one-fold. Indeed, knockdown of RAGE significantly attenuated HMGB1-induced HPSE expression. Upon binding to its receptor RAGE, HMGB1 activates NF-κB as manifested by the nuclear translocation of its subunit p65. ChIP assays demonstrated evidence that NF-κB directly binds to the HPSE promoter to transcribe HPSE expression, which was further supported by the observation that suppression of NF-κB activity significantly attenuated HMGB1-induced active and latent HPSE expressions.
It has been well demonstrated that HS chains form a key component of ECM and that breakdown of HS chains is carried out by HPSE [39]. Previous studies have shown that TGF-β can be produced by myofibroblasts, secreted predominantly in its inactive latent form and deposited in the ECM through binding to HS chains [33]. Given that the TGF-β autocrine signaling loop initiates and maintains the differentiation of fibroblasts into myofibroblasts [40], we speculated that HMGB1-induced HPSE may play a role to cleave HS chains from latent TGF-β in the myofibroblast ECM. Indeed, HMGB1 treated myofibroblasts stored more latent TGF-β in the ECM and less active TGF-β in the cell supernatant after treatment with an HPSE inhibitor.
In summary, the development of BOS after lung transplantation is a complex process that involves a number of factors. In the current report, we addressed the impact of HMGB1 on the pathology of a mouse BOS model. We demonstrated evidence supporting that HMGB1 is implicated in the initiation and progression of BOS, as manifested by the HMGB1 passive release at the early stage of tracheal transplantation and active secretion during the course of BOS. We further characterized that HMGB1 activates NF-κB through its cognate receptor RAGE to transcribe HPSE expression, which then cleaves HS chains to release autocrine TGF-β from myofibroblast ECM. The active autocrine TGF-β finally initiates and maintains the differentiation of fibroblasts into myofibroblasts to exacerbate BOS (Figure 8). Given the complexity of BOS pathoetiology, this new insight may have great potential to develop novel therapies against fibrotic diseases, especially in the context of BOS after lung transplantation.
Figure 8.
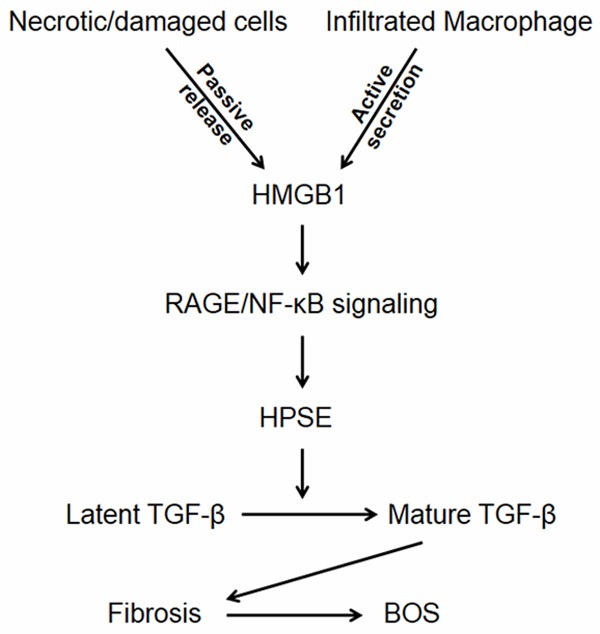
A schematic diagram shows the impact of HMGB1 on BOS development. During the course of BOS, HMGB1 is first passively released as a result of early unavoidable allograft injuries, which then initiates inflammatory response, leading to macrophage infiltration. The infiltrated macrophages further secrete copious amount of HMGB1. Extracellular HMGB1 acts through its receptor, RAGE, to activate NF-κB, which then binds to the HPSE promoter to transcribe its expression. The enhanced HPSE next cleaves latent TGF-β from myofibroblast ECM to promote the initiation and progression of BOS.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81130014, 81471046 and 81530024), and the Funding for Generation of Experimental Animals from the Hubei State Technology Department (2015BCE100), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_14R20), and the Innovative Funding for Translational Research from Tongji Hospital.
Disclosure of conflict of interest
None.
References
- 1.Weigt SS, DerHovanessian A, Wallace WD, Lynch JP 3rd, Belperio JA. Bronchiolitis obliterans syndrome: the Achilles’ heel of lung transplantation. Semin Respir Crit Care Med. 2013;34:336–351. doi: 10.1055/s-0033-1348467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilar PR, Michelson AP, Isakow W. Obliterative Bronchiolitis. Transplantation. 2016;100:272–283. doi: 10.1097/TP.0000000000000892. [DOI] [PubMed] [Google Scholar]
- 3.Estenne M, Maurer JR, Boehler A, Egan JJ, Frost A, Hertz M, Mallory GB, Snell GI, Yousem S. Bronchiolitis obliterans syndrome 2001: an update of the diagnostic criteria. J Heart Lung Transplant. 2002;21:297–310. doi: 10.1016/s1053-2498(02)00398-4. [DOI] [PubMed] [Google Scholar]
- 4.Christie JD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Dobbels F, Kirk R, Rahmel AO, Stehlik J, Hertz MI International Society of Heart and Lung Transplantation. The Registry of the International Society for Heart and Lung Transplantation: 29th adult lung and heart-lung transplant report-2012. J Heart Lung Transplant. 2012;31:1073–1086. doi: 10.1016/j.healun.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Li JH, Zhao B, Zhu XH, Wang L, Zou HJ, Chen S, Guo H, Ruan YL, Zheng F, Xiang Y, Ming CS, Gong FL, Chen G. Blockade of Extracellular HMGB1 Suppresses Xenoreactive B Cell Responses and Delays Acute Vascular Xenogeneic Rejection. Am J Transplant. 2015;15:2062–2074. doi: 10.1111/ajt.13275. [DOI] [PubMed] [Google Scholar]
- 6.Huang Y, Yin H, Han J, Huang B, Xu J, Zheng F, Tan Z, Fang M, Rui L, Chen D, Wang S, Zheng X, Wang CY, Gong F. Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am J Transplant. 2007;7:799–808. doi: 10.1111/j.1600-6143.2007.01734.x. [DOI] [PubMed] [Google Scholar]
- 7.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Muller S, Iannacone M, Traversari C, Bianchi ME, Manfredi AA. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5:825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bell CW, Jiang W, Reich CF 3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:C1318–1325. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- 9.Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med. 2004;255:332–343. doi: 10.1111/j.1365-2796.2003.01296.x. [DOI] [PubMed] [Google Scholar]
- 10.Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174:7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 11.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 12.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 13.Zhang M, Guo Y, Fu H, Hu S, Pan J, Wang Y, Cheng J, Song J, Yu Q, Zhang S, Xu JF, Pei G, Xiang X, Yang P, Wang CY. Chop deficiency prevents UUO-induced renal fibrosis by attenuating fibrotic signals originated from Hmgb1/TLR4/NFkappaB/IL-1beta signaling. Cell Death Dis. 2015;6:e1847. doi: 10.1038/cddis.2015.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buckley ST, Ehrhardt C. The receptor for advanced glycation end products (RAGE) and the lung. J Biomed Biotechnol. 2010;2010:917108. doi: 10.1155/2010/917108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Z, Ramachandran S, Gunasekaran M, Zhou F, Trulock E, Kreisel D, Hachem R, Mohanakumar T. MicroRNA-144 dysregulates the transforming growth factor-beta signaling cascade and contributes to the development of bronchiolitis obliterans syndrome after human lung transplantation. J Heart Lung Transplant. 2015;34:1154–1162. doi: 10.1016/j.healun.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frost AE. Bronchiolitis obliterans: the Achilles heel of lung transplantation. Verh K Acad Geneeskd Belg. 2002;64:303–319. discussion 319-322. [PubMed] [Google Scholar]
- 17.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-beta structure and activation. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masola V, Zaza G, Secchi MF, Gambaro G, Lupo A, Onisto M. Heparanase is a key player in renal fibrosis by regulating TGF-beta expression and activity. Biochim Biophys Acta. 2014;1843:2122–2128. doi: 10.1016/j.bbamcr.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Hertz MI, Jessurun J, King MB, Savik SK, Murray JJ. Reproduction of the obliterative bronchiolitis lesion after heterotopic transplantation of mouse airways. Am J Pathol. 1993;142:1945–1951. [PMC free article] [PubMed] [Google Scholar]
- 20.Yang P, Zhang Y, Pang J, Zhang S, Yu Q, He L, Wagner KU, Zhou Z, Wang CY. Loss of Jak2 impairs endothelial function by attenuating Raf-1/MEK1/Sp-1 signaling along with altered eNOS activities. Am J Pathol. 2013;183:617–625. doi: 10.1016/j.ajpath.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Fang J, He L, Wang SQ, Ma MJ, Liu HY, Zhu XH, Zhu P, Wei X, Wang CY. A simplified two-stitch sleeve technique for arterial anastomosis of cervical heterotopic cardiac transplantation in mice. Am J Transl Res. 2013;5:521–529. [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Zhou X, Zhan Z, Meng Q, Han Y, Shi Q, Tang J, Li J, Fan H, Liu Z. IL-25 regulates the polarization of macrophages and attenuates obliterative bronchiolitis in murine trachea transplantation models. Int Immunopharmacol. 2015;25:383–392. doi: 10.1016/j.intimp.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 23.Zhang S, Zhong J, Yang P, Gong F, Wang CY. HMGB1, an innate alarmin, in the pathogenesis of type 1 diabetes. Int J Clin Exp Pathol. 2009;3:24–38. [PMC free article] [PubMed] [Google Scholar]
- 24.Hu S, Zhang Y, Zhang M, Guo Y, Yang P, Zhang S, Simsekyilmaz S, Xu JF, Li J, Xiang X, Yu Q, Wang CY. Aloperine protects mice against ischemia reperfusion (IR)-induced renal injury by regulating PI3K/AKT/mTOR signaling and AP-1 activity. Mol Med. 2015 doi: 10.2119/molmed.2015.00056. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao X, Zhong J, Zhang S, Zhang Y, Yu Q, Yang P, Wang MH, Fulton DJ, Shi H, Dong Z, Wang D, Wang CY. Loss of methyl-CpG-binding domain protein 2 enhances endothelial angiogenesis and protects mice against hind-limb ischemic injury. Circulation. 2011;123:2964–2974. doi: 10.1161/CIRCULATIONAHA.110.966408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kucuksezer UC, Palomares O, Ruckert B, Jartti T, Puhakka T, Nandy A, Gemicioglu B, Fahrner HB, Jung A, Deniz G, Akdis CA, Akdis M. Triggering of specific Toll-like receptors and proinflammatory cytokines breaks allergen-specific T-cell tolerance in human tonsils and peripheral blood. J Allergy Clin Immunol. 2013;131:875–885. doi: 10.1016/j.jaci.2012.10.051. [DOI] [PubMed] [Google Scholar]
- 27.Zhong J, Yu Q, Yang P, Rao X, He L, Fang J, Tu Y, Zhang Z, Lai Q, Zhang S, Kuczma M, Kraj P, Xu JF, Gong F, Zhou J, Wen L, Eizirik DL, Du J, Wang W, Wang CY. MBD2 regulates TH17 differentiation and experimental autoimmune encephalomyelitis by controlling the homeostasis of T-bet/Hlx axis. J Autoimmun. 2014;53:95–104. doi: 10.1016/j.jaut.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M, He X, Cheng Z, Ao Q, Cao Y, Yang P, Su Y, Zhao J, Zhang S, Yu Q, Ning Q, Xiang X, Xiong W, Wang CY, Xu Y. Chop Deficiency Protects Mice against Bleomycin-induced Pulmonary Fibrosis by Attenuating M2 Macrophage Production. Mol Ther. 2016;24:915–25. doi: 10.1038/mt.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong J, Yang P, Muta K, Dong R, Marrero M, Gong F, Wang CY. Loss of Jak2 selectively suppresses DC-mediated innate immune response and protects mice from lethal dose of LPS-induced septic shock. PLoS One. 2010;5:e9593. doi: 10.1371/journal.pone.0009593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S, Lv JW, Yang P, Yu Q, Pang J, Wang Z, Guo H, Liu S, Hu J, Li J, Leng J, Huang Y, Ye Z, Wang CY. Loss of dicer exacerbates cyclophosphamide-induced bladder overactivity by enhancing purinergic signaling. Am J Pathol. 2012;181:937–946. doi: 10.1016/j.ajpath.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Chen L, Chen B, Meliton A, Liu SQ, Shi Y, Liu T, Deb DK, Solway J, Li YC. Chronic Activation of the Renin-Angiotensin System Induces Lung Fibrosis. Sci Rep. 2015;5:15561. doi: 10.1038/srep15561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han J, Zhong J, Wei W, Wang Y, Huang Y, Yang P, Purohit S, Dong Z, Wang MH, She JX, Gong F, Stern DM, Wang CY. Extracellular high-mobility group box 1 acts as an innate immune mediator to enhance autoimmune progression and diabetes onset in NOD mice. Diabetes. 2008;57:2118–2127. doi: 10.2337/db07-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Secchi MF, Masola V, Zaza G, Lupo A, Gambaro G, Onisto M. Recent data concerning heparanase: focus on fibrosis, inflammation and cancer. Biomol Concepts. 2015;6:415–421. doi: 10.1515/bmc-2015-0021. [DOI] [PubMed] [Google Scholar]
- 34.Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z, He L, Hu S, Wang Y, Lai Q, Yang P, Yu Q, Zhang S, Xiong F, Simsekyilmaz S, Ning Q, Li J, Zhang D, Zhang H, Xiang X, Zhou Z, Sun H, Wang CY. AAL exacerbates pro-inflammatory response in macrophages by regulating Mincle/Syk/Card9 signaling along with the Nlrp3 inflammasome assembly. Am J Transl Res. 2015;7:1812–1825. [PMC free article] [PubMed] [Google Scholar]
- 36.Hermano E, Meirovitz A, Meir K, Nussbaum G, Appelbaum L, Peretz T, Elkin M. Macrophage polarization in pancreatic carcinoma: role of heparanase enzyme. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato M, Hirayama S, Lara-Guerra H, Anraku M, Waddell TK, Liu M, Keshavjee S. MMP-dependent migration of extrapulmonary myofibroblast progenitors contributing to posttransplant airway fibrosis in the lung. Am J Transplant. 2009;9:1027–1036. doi: 10.1111/j.1600-6143.2009.02605.x. [DOI] [PubMed] [Google Scholar]
- 38.Duan L, Wang CY, Chen J, Gong Q, Zhu P, Zheng F, Tan Z, Gong F, Fang M. High-mobility group box 1 promotes early acute allograft rejection by enhancing IL-6-dependent Th17 alloreactive response. Lab Invest. 2011;91:43–53. doi: 10.1038/labinvest.2010.141. [DOI] [PubMed] [Google Scholar]
- 39.Wu L, Viola CM, Brzozowski AM, Davies GJ. Structural characterization of human heparanase reveals insights into substrate recognition. Nat Struct Mol Biol. 2015;22:1016–1022. doi: 10.1038/nsmb.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, Orimo A. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107:20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]