

Abstract
It has been reported that CREPT acts as a highly expressed oncogene in a variety of tumors, affecting cyclin D1 signal pathways. However, the distribution and clinical significance of CREPT in NSCLC remains poorly understood. Our study focused on the role of CREPT on the regulation ofnon-small cell lung cancer (NSCLC). We found that CREPT mRNA and protein expression was significantly increased in NSCLC compared with adjacent lung tissues and was increased in various NSCLC cell lines compared with the normal human bronchial epithelial (HBE) cell line. siRNA-induced knockingdown of CREPT significantly inhibited the proliferation and migration of NSCLC cell lines by arresting cell cycle in S phase. Moreover, CREPT knocking down affected the expression of cell cycle proteins including c-mycand CDC25A. Finally, we found there were obvious correlations between CREPT with c-myc expression in histological type, differentiation, and pTNM stages of NSCLC (P<0.05, rs>0.3). Immunohistofluorescence studies demonstrated a co-localization phenomenon when CREPT and c-myc were expressed. Thus, we propose that CREPT may promote NSCLC cell growth and migration through the c-myc and CDC25A signaling molecules.
Keywords: CREPT, c-myc, CDC25A, proliferation, migration, co-localization
Introduction
Lung cancer remains the leading cause of cancer-related deaths in the worldwide [1]. Non-small cell lung cancer (NSCLC) represents the approximately 85% cases of diagnosed lung cancer and is associated with a relatively poor (15%) overall 5-year survival rate [2]. Therefore, novel strategies are needed to treat NSCLC. Understanding the molecular profiles of NSCLC, as well as elucidating the roles of oncogenes and tumor suppressors in the development of this malignancy, is expected to identify aberrant signaling pathways and molecular targets for therapy [3].
CREPT (cell-cycle related and expression-elevated protein in tumor) (Gene_ID 58490, Genbank NM_021215), a novel gene also called RPRD1B (regulation of nuclear pre-mRNA domain containing protein 1B) and C20ORF77, was recently identified to promote tumorigenesis through up-regulation of the expression of genes related to cell cycle. Prior research has demonstrated that CREPT is highly expressed in a variety of tumors and correlated with tumor stage, histology type, and a poor survival rate [4-6]. CREPT enhances the expression of cyclin D1 by promoting the formation of a chromatin loop by interacting with RNA polymerase II (RNAPII) [7].
Given all these elements, we speculated that CREPT may promote NSCLC cellular survival and proliferation and be a common tumor specific marker on the cell surface; however, the activated mechanism of CREPT and whether or not it further promotes NSCLC progression are all unclear. Thus, this research will focused on its detailed functions in NSCLC and provided a basis for further study on the development mechanism of NSCLC with CREPT, in order to proposea new clinical therapeutic target.
In this study, we confirmed that CREPT is highly expressed in NSCLC tissues and cell lines. Subsequently, we employed the newly developed lentivirus-delivered small interfering RNA (siRNA) technique to observe the effect of inhibiting CREPT on human NSCLC cells proliferation and migration.
Materials and methods
Patients and tissue samples
Paraffin-embedded tissue specimens from 72 patients with confirmed NSCLC, collected from 2006 to 2010, were analyzed; all tissue samples came from an archived thoracic oncology tissue repository housed at the Department of Thoracic Surgery of Tangdu Hospital affiliated to the Fourth Military Medical University (Xi’an, China). In addition, 24 fresh NSCLC tissue specimens and the paired adjacent normal lung tissues were obtained from patients undergoing radical surgery at the same center. These fresh specimens were placed in a 0.1% diethylpyrocarbonate (DEPC) water-treated freezing tube and stored at -80°C. All samples were reviewed by pathologists. Histological classification of tumors was performed according to the World Health Organization criteria. All tumors were staged according to the pathological tumor/node/metastasis (p-TNM) classification (7th edition) of the International Union against Cancer [8]. No patient had received chemotherapy, radiotherapy, biotherapy, or any other operation before undergoing lung cancer surgery.
The study protocol was approved by the Regional Ethics Committee for Clinical Research of the Fourth Military Medical University. All patients provided written informed consent for the use of their medical records and tissue specimens for research purposes.
Cell culture
The following human NSCLC cell lines were employed: squamous cell carcinoma (SCC): Calu-1, H520; adenocarcinoma (ADC): A549, H838, SK-LU-1, SPC-A-1 and normal human bronchial epithelial (HBE) cells. All cell lines were purchased from ATCC and maintained in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and cultured under a humidified atmosphere of 5% CO2 at 37°C.
Quantitative real-time RT-PCR
Total RNA from fresh tissue and cell lines was extracted using Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions. The cDNA synthesis was performed using a reverse transcription kit (Thermo Fisher Scientific, USA) using the Random Hexamer Primer, and Revert AidM-MuLV Reverse Transcriptase [9]. RT-PCR experiments were conducted using SYBR Green Premix Ex Taq II kit (Takara, Japan)according to the manufacturer’s instructions. The relative amount of mRNA expression in the target genes was calculated by the comparative ΔCt method using β-actin as a control [10]. The average ΔCT was calculated for both target gene and β-actin; ΔCT was determined as the mean of the triplicate CT values for target gene minus the mean of the triplicate CT values for β-actin [11]. Initial denaturation was conducted at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 61°C for 30 s, and final extension at 72°C for 50 s.
CREPT-primer: forward 5’-TCCGCAAAGCCAAATCAAATA-3’ and reverse 5’-CTGTATGAACTCGCCGCCA-3’. c-myc-primer: forward 5’-CAAGAGGCGAACACACAACGTCT-3’ and reverse 5’-AACTGTTCTCGTCGTTTCCGCAA-3’. CDC25A-primer: forward 5’-AAGCGTGTCATTGTTGTGTTTC-3’ and reverse 5’-GCTCAGGGTAGTGGAGTTTGG-3’. β-actin-primer: forward 5’-TGGCACCCAGCACAATGAA-3’ and reverse 5’-CTAAGTCATAGTCCGCCTAGAAGCA-3’.
Protein isolation and Western blot
Cells and fresh tissue samples (100 mg) were cut into small pieces, homogenized, and lysed, then placed on ice for 1 h in a radio-immunoprecipitation assay (RIPA) buffer (Cell Signaling) containing complete protease inhibitor cocktail (Roche). The samples were centrifuged at 14,000 ×g for 20 minthen the supernatant was harvested. The protein concentrations were determined using aBCA assay kit (Pierce). A 50 μg aliquot ofeach protein sample was mixed with loading buffer (CW0027A, CW Bio) and denatured at 65°C for 30 min. The protein was separated by SDS-polyacrylamide gel electrophoresis (PAGE) using 12% separation gel and 5% spacer gel, which were then transferred onto polyvinylidene fluoride (PVDF) membranes (GE healthcare) by electroblotting (Bio-RAD) at 2.5 mA/cm2 for 35 min. Membranes were blocked with 9% skimmed milk powder in Tris-buffered saline (TBST: 25 mM Tris/HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20) for 3 h at room temperature. Next, the membranes were incubated overnight at 4°C with the primary antibodies (anti-CREPT rabbit polyclonal antibody, 1:2000, GeneTex; anti-c-myc rabbit polyclonal antibody, 1:1500, Cell Signaling; anti-CDC25A rabbit polyclonal antibody, 1:5000, proteintech; and anti-β-actin rabbit monoclonal antibody, 1:1000, CW Bio). The membranes were washed with TBST buffer 6times for 5 min, and then subsequently incubated with the secondary antibody (goat anti-rabbit antibodies, 1:5000, CW Bio) at 37°C for 1 h, followed by color development using the Millipore chromogenic kit for Western blot analysis (Billerica), followed by observation under a chemiluminescence analyzer (Bio-Rad).
Lentivirus vectors for CREPT RNA interfering
To observe the effect of inhibiting CREPT on human NSCLC cells, we employed the newly developed lentivirus-delivered RNA interfering technique. hu6-MCS-CMV-EGFP-Lentivirus (Genechem, China) was used to express small interfering RNAs (siRNAs) targeting the CREPT sequence (Genbank no. NM_021215). Anon-targeting sequence was used as a lentivirus negativecontrol (NC). Targeted oligonucleotide sequences were: siRNA-1: ATGTCTGTTACTAGCAGAA; siRNA-2: AAGATGTTTCTCTATTGGA; siRNA-3: CTCTTAATACCTGTTACTA and TTCTCCGAACGTGTCACGT for the lentivirus negativecontrol. Preliminary experiment results showed that siRNA-1 significantly inhibited CPEPT expression and was therefore, selected as anoptimal siRNA for following experiments.NSCLC cell lines, Calu-1 (SCC) and H838 (ADC), were infected with CREPT-siRNA lentivirus and with NC lentivirus. Cells were plated in 6-well plates (5×104 cells/well), grown to 60% confluence, and treated with tittered viral supernatant ata multiplicity of infection (MOI) of 20 for 12 h without toxiceffect observed. Then,the media was changed to RPMI 1640 medium supplemented with 10% FBS. The interference efficiency of the template was detected by RT-PCR and Western blot analysis.The Calu-1 and H838 cells transfected with the CREPT-siRNA lentivirus or NC lentivirus were designated as Calu-1-siRNA or Calu-1-NC and H838-siRNA or H838-NC, respectively. Non-transfected cells were also included as a positive control and designated Calu-1-Control or H838-Control.
MTT assay
After 5 days of siRNAs infection, cell viability and proliferation were evaluated by a modifiedMTT assay. Calu-1 and H838 cells in exponential growth were trypsinized and counted, then plated in 96-well plates at a final concentration of 4×103 cells/200 μL/well in RPMI 1640 medium supplemented with 10% FBS. After seeding, cell viability was assessed at days 1, 2, 3, 4, 5 and 6. An aliquot of 20 μL of MTT (5 mg/ml) was added to each well. After an additional 4 h of incubation at 37°C, 150 μL of DMSO was added to each well, and the optical density (OD) of each well was measured at 570 nm using an ELISA reader (Thermo, USA) [12]. Growth curves were portrayed based on the OD value.
Colony formation assay
A colony formation was performed to assess the anchorage-independent growth ability of cells, as a characteristic of in vitro tumorigenicity [10]. After 5 days of lentiviral infection, the Calu-1 and H838 cells transfected and untreated were trypsinized, counted and their 500 cells were seeded in 60 mm plates in RPMI 1640 medium supplemented with 10% FBS at 37°C. All cells were grown for 3 weeks, with medium changed during thesecond week. Next, the plates were fixed with 4% formaldehyde and stained with Giemsa and washed twice with phosphate buffered saline (PBS). The images were obtained and visible colonies (>100 μm) with more than 50 cells counted by manual means.
Analysis of cell cycle and apoptosis
To ascertain the impact of CREPT silencing on Calu-1 and H838 cells, cell cycle phase and apoptosis were analyzed using flow cytometry. After 5 days of lentiviral infection, the Calu-1 and H838 cells transfected and untreated were harvested, washed twice and re-suspended in ice-cold PBS. To analyze the cell cycle, 1×106 cells were fixed with 75% ice-cold ethanol for 30 min, and stained with propidium iodide (PI, 50 μg/ml) in the presence of RNase (100 μg/ml). For analysis of apoptosis, the cells were stained with 100 μL binding buffer containing 5 μL Annexin V-FITC, or V-PE at 37°C in the dark, for 15 min. The resulting cells were analyzed using flow cytometer (Beckman Coulter, USA).
Scratch wound healing assay
Cell migration was measured using a scratch assay determined, as described previously [13]. After 5 days of lentiviral infection, bothtransfected and untreated Calu-1 and H838 cells were harvested and their 5×105 cells were plated in 6-well plates. Cells were incubated overnight. After incubation, the platesyielded cells of 80% confluence, then the monolayer was scraped in a straight line to create a “scratch” using a 200 μL pipette tip. After removing debris and adding fresh media containing no FBS, cells were photographed at 0 h, 24 h, and 48 h. The migration distance was measured and assessed using image J software at 3 different sites from each wound area of scratch, at each time point. The healing degree was calculated by cell relative migration area for each treatment.
Transwell invasion and migration assay
To further examined the cell invasion and migration, the transwell invasion and migration was performed using 8 μm pore size transwell chambers (Corning, USA) in vitro following the manufacturer’s instructions. In brief, the matrigel (5 mg/ml, Corning, USA) was diluted into 1 mg/ml in ice-cold RPMI 1640 medium supplemented with 10% FBS. An aliquot of 200 μL diluted matrigel was added to the upper transwell chambers and incubated at 37°C for 4 h for gelling. A total of 1×105 cells in 400 μL media supplemented with no FBS were plated in the upper chamber and 600 μL RPMI 1640 medium supplemented with 10% FBS was covered on the bottom chambers as chemoattractant. After incubation at 37°C for 48 h, the non-invasive cells in the top surface were carefully removed with a cotton swab. The invasive cells that had traversed to the bottom surface were fixed in dehydrated alcohol for 30 min and stained with 4 mg/ml crystal violet for 10 min. To quantify the traversed cells, cell counting was obtained by photographing 5 random fields under microscope at 400× magnification [14].
The migration assay was performed in a similar strategy with chamber membrane without coating with matrigel.
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde for 30 min and blocked in 0.5% Trion X-100 for 15 min. The cells were washed 3 times (5 min for each) with PBS after each step. The tumor samples were fixed with 10% formaldehyde and embedded in paraffin. Sections were sliced at 4 μm thickness, deparaffinized with a series of xylene washes, and rehydrated through a graded series of alcohol rinses. Microwave antigen retrieval was performed at 750 W for 5 min and 450 W for 15 min in 0.1 M citrate buffer (pH 6.0) to enhance the immunoreactivity. To block the endogenous peroxidase activity, all the cells and tumor samples were incubated in 3% hydrogen peroxidase at room temperature for 30 min and washed with PBS three times for 5 min. All the cells and tumor samples were incubated with 10% normal goat serum for 30 min at room temperature to block nonspecific antibody reaction, followed by incubation in a humidified chamber overnight at 4°C with anti-CREPT rabbit polyclonal antibody (1:300, GeneTex). After an additional series of washes, the samples were stained with goat anti-rabbit (Cy3, Zhuangzhi bio) at 37°C for 4 min, then each sample was washed with PBS three times, for 5 min. Then, the samples were incubated with anti-c-myc mouse polyclonal antibody (1:160, Abcam) overnight at 4°C. After an additional series of washes, the samples were stained with goat anti-mouse (Alexa Fluor 488, Zhuangzhi bio) at 37°C for 50 min. After the final washing, the samples were mounted in 50% glycerol (in PBS) and visualized under a fluorescence microscope (Leica DM4000B, Germany).
Immunohistochemistry staining
Paraffin-embedded tissues were cut into 4 μm sections for deparaffinized and rehydrated through a graded series of ethanol solution. Microwave antigen retrieval was performed at 750 W for 5 min and 450 W for 12 min in 0.1 M citrate buffer (pH 6.0) to enhance immunoreactivity. To block the endogenous peroxidase activity, the slides were incubated in 3% hydrogen peroxidase at room temperature for 20 min and washed with PBS (phosphate-buffered saline) three times for 5 min. The slides were incubated with 10% normal goat serum for 30 min at room temperature to block nonspecific antibody reaction, followed by incubation in a humidified chamber overnight at 4°C with the primary antibodies (anti-CREPT rabbit polyclonal antibody, 1:300, GeneTex; anti-c-myc rabbit polyclonal antibody, 1:160, Cell Signaling; and anti-CDC25A rabbit polyclonal antibody, 1:400, proteintech). After washing with PBS three times for 5 min, the slides were incubated for 50 min at 37°C with an EnVision+ - labeled polymer. Peroxidase activity was visualized with the DAB Elite kit (Dako, Denmark), and the slides were counterstained with hematoxylin. To confirm the specificity of the immunostaining, blank controls were obtained by replacing the primary antibody with PBS.
Evaluation of immunohistochemical staining
Five random fields from each section were viewed under a light microscope (Leica DM4000B, Germany) at 400× magnification. Specimens were scored according to the staining intensity and the percentage of positive cells. The results were scored based on the following criteria: a) the percentage of positive cells (≤5%: 0; 6-25%: 1; 26-50%: 2; 51-75%: 3; and >75%: 4); b) the staining intensity (no color: 0; yellow: 1; brown: 2; and tan: 3); and c) the two grades were multiplied together and specimens were assigned to one of 4 levels: 0: negative (-); 1-4: weakly positive (+); 5-8: moderately positive (++); 9-12: strongly positive (+++) [9,15]. To investigate the correlation of protein expression, - and + was defined as low expression, and, ++ and +++ were defined as high expression.
All slides were assessed by 3 independent investigators who were blinded to the clinical features and outcomes. The final immunohistochemical staining score reported is the average of the scores from the three investigators.
Statistical analysis
Each experiment was performed in triplicate. Bands from Western blot were quantized using Quantity One software (Bio-Rad, USA). Relative protein levels were calculated relative to the amount of β-actin respectively. All statistical analyses were performed with SPSS 15.0 software (SPSS, Inc., Chicago, IL). All values in the text and figures are expressed as the mean ± SD of these observations. Student’s t-test was used for raw data analysis. A P-value <0.05 was considered statistically significant. Spearman’s rank correlation coefficients have been calculated for the assessment of overall concordance, and rs was expressed the relevant coefficient, and when P<0.05, rs>0.3 was considered of positive relevance.
Results
Patient characteristics
Paraffin-embedded tissues from 72 patients with confirmed NSCLC were diagnosed with SCC (n=39, 54.17%) and ADC (n=33, 45.83%).Histopathologic diagnoses included: well differentiation (n=10, 13.89%), moderate differentiation (n=35, 48.61%) and poor differentiation (n=27, 37.5%) tumors. Postoperative staging evaluation demonstrated: stage I (n=13, 18.06%), stage II (n=29, 40.28%), stage III (n=24, 33.33%) and stage IV (n=6, 8.33%).
CREPT is over expressed in NSCLC tissue samples and cell lines
We examined the expression of CREPT in 24 paired fresh tumor tissue and the adjacent normal lung tissues from patients with NSCLC by using RT-PCR and Western blot to analyze the CREPT mRNA and protein level, respectively. The results showed that the CREPT mRNA and protein level in tumor tissue were significantly higher than those in the adjacent normal lung tissues (P<0.05, Figure 1A and 1B). To further confirm these observations, we examined the expression of CREPT in cell lines. The CREPT mRNA and protein level were clearly elevated in the six NSCLC cell lines, SCC: Calu-1, H520 and ADC: A549, H838, SK-LU-1, SPC-A-1, which were higher than those in the normal human bronchial epithelial (HBE) cells (P<0.01, Figure 1C and 1D). These results indicated an association of the CREPT over expression with NSCLC.
Figure 1.
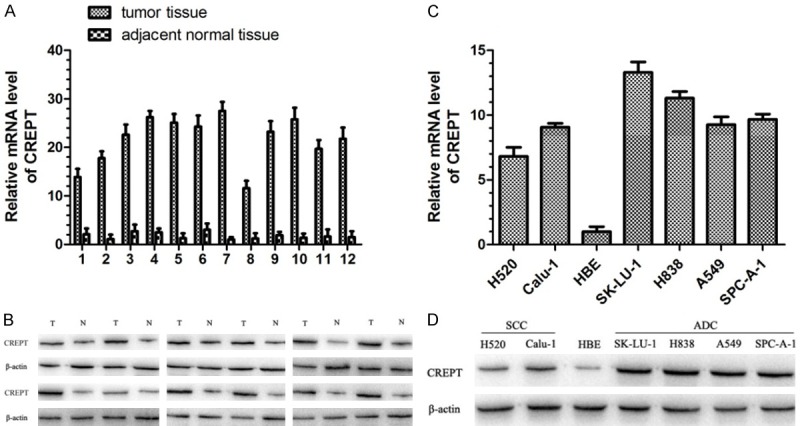
RT-PCR and Western blot analysis results showed that the expression of CREPT mRNA and protein was significantly higher in fresh tumor tissue and six NSCLC cell lines than those in paired adjacent normal lung tissues and the normal human bronchial epithelial (HBE) cells. A. Expression of CREPT mRNA was higher in tumor tissue than that in paired adjacent normal lung tissues. Data are expressed as mean ± SD. B. Expression of CREPT protein was higher in tumor tissue (T) than that in paired adjacent normal lung tissues (N), and anti-β-actin antibody was used as an internal control. C. Expression of CREPT mRNA was higher in six NSCLC cell lines than that in the normal human bronchial epithelial (HBE) cells. Data are expressed as mean ± SD. D. Expression of CREPT protein was higher in six NSCLC cell lines than that in the normal human bronchial epithelial (HBE) cells, and anti-β-actin antibody was used as an internal control.
Silencing of CREPT by lentivirus-delivered RNA interfering
To further explore the mechanism of CREPT in NSCLC, NSCLC cell lines Calu-1 (SCC) and H838 (ADC) were infected with CREPT-siRNA lentivirus and NC lentivirus. The infection efficiency of green fluorescent protein (GFP) in Calu-1 and H838 cells was about 80-85% after 3 days of infection at a multiplicity of infection (MOI) of 20 (Figure 2A). Cells were harvested after 5 days of infection; RT-PCR and Western blot analysis were used to determine the knock-down efficiency, respectively. Compared with theNC-siRNA group and blank control (nosiRNA) group, the mRNA and protein expressions of CREPT were significantly inhibited in cells transfected with CREPT-siRNA (P<0.05, Figure 2B and 2C). The successful establishment of a CREPT gene silencing lentivirus provided a useful tool for investigating the function of CREPT in NSCLC cell lines.
Figure 2.
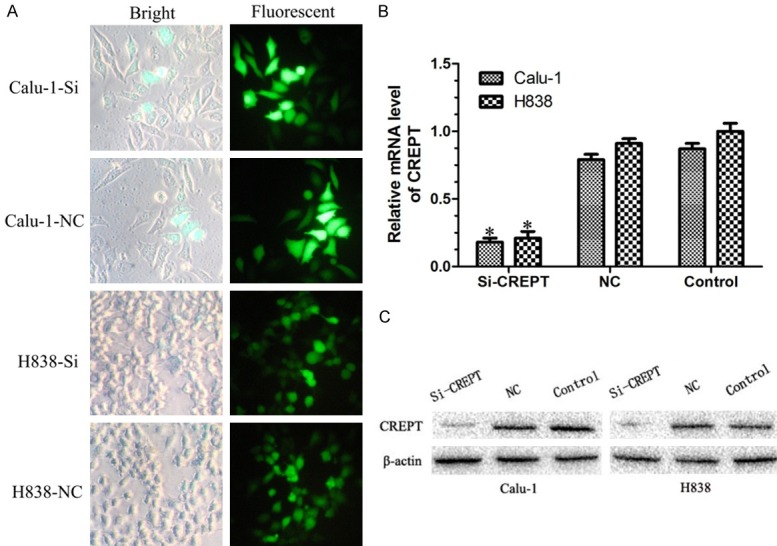
The expressions of CREPT were significantly inhibited in cells transfected with CREPT-siRNA. A. Micrograph of Calu-1 and H838 cells infected with CREPT-siRNA for three days in bright and fluorescent fields (400× magnification). More than 80% of cells expressed GFP. B. RT-PCR results showing the CREPT mRNA levels were inhibited in cells transfected with CREPT-siRNA as compared with NC-siRNA and blank control. C. Western blot analysis showing the expression levels of CREPT protein in cells transfected with CREPT-siRNA was remarkable lower than that of NC-siRNA and blank control cells. * indicates P<0.05.
Down regulation of CREPT expression inhibited the growth in NSCLC cell lines
To investigate the possible role of crept in the growth of Calu-1 and H838 cells, MTT assays and colony formation assays were performed. MTT assays results showed the cell viability was significantly lower in cells infected with CREPT-siRNA compared to cells infected with NC-siRNA and blank control (no siRNA) cells following a 6-day growth (P<0.01, Figure 3A and 3B). The difference was more pronounced with time-dependent manner. Colony formation assays also showed the colony-forming ability was dramatically decreased in cells infected with CREPT-siRNA compared to cells infected with NC-siRNA and blank control (P<0.05, Figure 3C and 3D). Therefore, the low viability and the low colony number for cells treated with CREPT-siRNA demonstrate that down-regulation of CREPT expression, which inhibited the growth of human NSCLC cells in vitro.
Figure 3.

Down regulation of CREPT expression inhibited the proliferation of Calu-1 and H838 cells. A, B. MTT assays suggest that CREPT-siRNA depressed the growth curves of Calu-1 and H838 cells as compared with NC-siRNA and blank contro with time-dependent manner. C, D. Colony formation assays showed that the CREPT-siRNA inhibited the number of cells and clones as compared with NC-siRNA and blank control. Data are the number of colonies formed expressed as mean ± SD. E-H. The percentage of cells in different phases of the cell cycle was determined. G1-phase population was significantly increased, whereas the S-phase population was reduced in the CREPT-siRNA groups compared to the NC-siRNA and blank control groups (P<0.05). I-K. Knockdown of CREPT expression increased cell apoptosis in the CREPT-siRNA groups compared to the NC-siRNA and blank control groups (P<0.01).* indicates P<0.05, ** indicates P<0.01.
Knocking down of CREPT arrests the cell cycle in G1 phase and induces cell apoptosis
To determine whether CREPT is necessary for cell cycle and apoptosis progression of Calu-1 and H838 cells, we assessed the cell cycle and apoptosis phases in cells by flow cytometry. As shown in Figure 3, the percentages of Calu-1 and H838 cells in the G1 phase in the CREPT-siRNA-transfected groups were much higher than those in the cells infected with NC-siRNA and blank control (Calu-1: CREPT-siRNA 69.06 ± 2.96% vs. NC 51.95 ± 6.85% and Control 48.96 ± 4.89%; H838: CREPT-siRNA 68.47 ± 3.60% vs. NC 52.23 ± 3.16% and Control 52.99 ± 14.98%, P<0.05, Figure 3E-H). In addition, compared with the NC-siRNA-transfected and blank control groups, the percentages of Calu-1 and H838 cells in the S phase in the CREPT-siRNA-transfected group were expected to decrease (Calu-1: CREPT-siRNA 16.24 ± 2.12% vs. NC 25.29 ± 4.29% and Control 34.36 ± 8.40%; H838: CREPT-siRNA 17.71 ± 1.08% vs. NC 25.32 ± 2.32% and Control 27.42 ± 6.15%, P<0.05, Figure 3E-H). Cell apoptosis was significantly increased in the CREPT-siRNA groups compared to the NC-siRNA and blank control groups (Calu-1: CREPT-siRNA 10.20 ± 2.80% vs. NC 2.57 ±0.44% and Control 2.78 ± 0.68%, P<0.01; H838: CREPT-siRNA 10.58 ± 3.29% vs. NC 2.77 ± 0.54% and Control 2.88 ± 0.73%, P<0.01, Figure 3I-K). Taken together, these data indicated that inhibitionof CREPT exhibited a specific inhibitory effect on NSCLC proliferation, as induction of a G1 phase arrest, inhibition of S phase entry, and by inducing apoptosis.
Knockdown of CREPT decreases the expression of cell cycle proteins
In order to elucidate the underlying molecular mechanism of how CREPT affects the cell cycle, we detected the expression of cell cycle-related molecules in CREPT-siRNA-transfected cells, such as c-myc and CDC25A by RT-PCR and Western blot analysis. The RT-PCR results showed that the mRNA levels of c-myc and CDC25A were significantly decreased in CREPT-siRNA-transfected cells compared with those in the NC-siRNA and blank control cells (P<0.05, Figure 4A and 4B). Furthermore, we examined the protein levels of c-myc and CDC25A by Western blot analysis. As shown in Figure 4C, the inhibition of CREPT down regulated c-myc and CDC25A protein expression in CREPT-siRNA-transfected cells compared with that in the NC-siRNA and blank control cells. Taken these finding stogether, we suggested that CREPT is important for cell cycle regulation and may be partly mediated by affecting cell-cycle protein expression, including c-myc and CDC25A.
Figure 4.
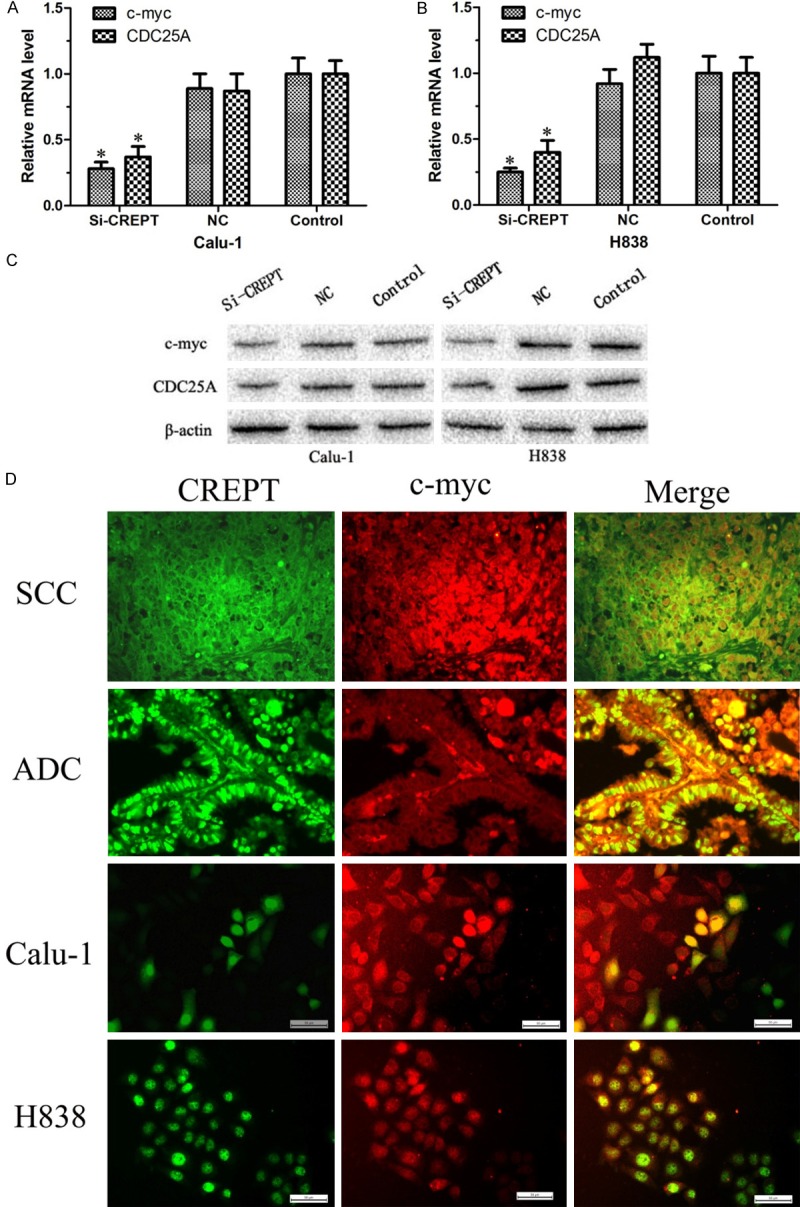
Cell cycle-related molecules were assayed by RT-PCR and Western blot analysis and the co-localization of CREPT protein expression and c-myc protein expression was observed. A, B. RT-PCR results found that knockdown of CREPT resulted in down regulation of c-myc and CDC25A mRNA levels. C. Western blot analysis showed that c-myc and CDC25A protein expression was down regulated in knocking down CREPT cells. D. Immunofluorescence staining results showing the co-localization of CREPT protein expression and c-myc protein expression was observed both in cells and tissues, ADC and SCC. ADC: adenocarcinoma; SCC: squamous cell carcinoma (400× magnification). * indicates P<0.05.
CREPT-siRNA significantly impaired the ability of migration and invasion in NSCLC cells
Given the ability of CREPT to promote NSCLC cell growth and proliferation, we were interested in exploring its potential effects on migration and invasion in NSCLC cell. To test these possible effects, scratch wound healing assay, we performed transwell invasion and a migration assay. In scratch wound healing assays, the results displayed that Calu-1 and H838 cells using CREPT-siRNA was decreased in migration by an average of 55% and 49%, respectively, when compared with cells infected with NC-siRNA and blank control (P<0.05, Figure 5A-D). In order to further prove this effect, we used a transwell migration assay. As measured by the numbers of cells migrating through the chamber membrane, CREPT-siRNA-transfected groups (74 ± 16, 69 ± 13) demonstrated a noticeable decrease in mobility compared to those of the NC-siRNA and blank control groups (NC: 283 ± 35, 271 ± 29, Control: 295 ± 26, 307 ± 32, P<0.01, Figure 5E and 5F) in Calu-1 and H838 cells, respectively. The invasion capability associated with CREPT expression was examined with transwell chambers coated with matrigel. As expected, the numbers of cells migrating through the membrane were greatly reduced when CREPT was inhibited (Calu-1: CREPT-siRNA 40 ± 11 vs. NC 137 ± 15 and Control 125 ± 21, P<0.01; H838: CREPT-siRNA 36 ± 7 vs. NC 102 ± 18 and Control 117 ± 23, P<0.01, Figure 5G and 5H). Therefore, the inhibition ofCREPT significantly hindered NSCLC cells migration and invasion.
Figure 5.
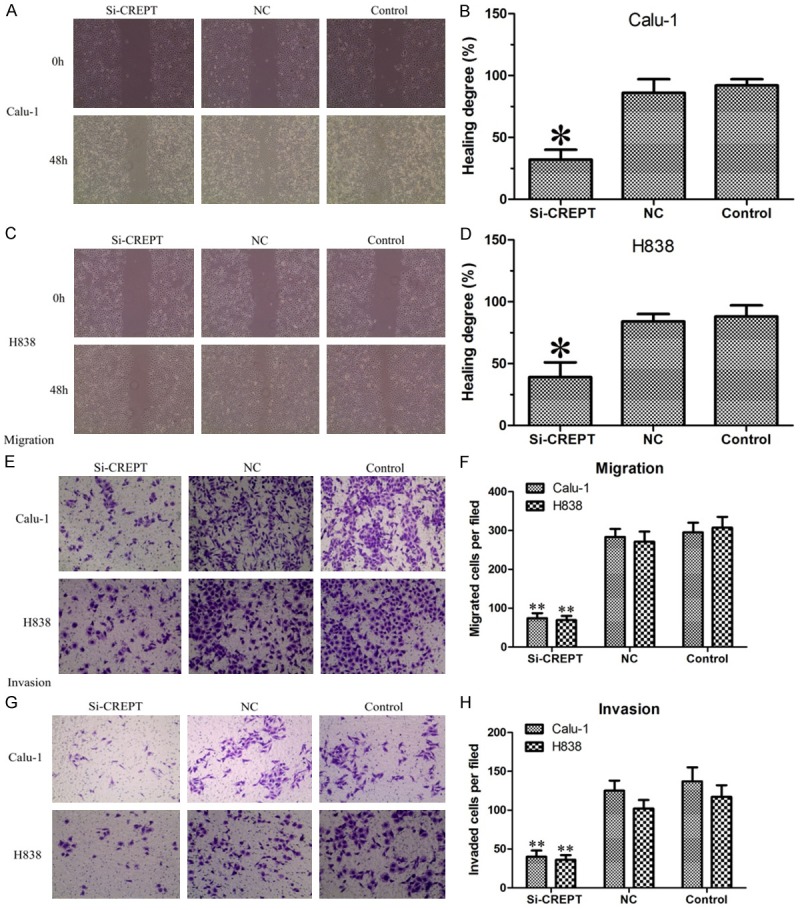
Scratch wound healing assay, transwell migration and invasion assay proved that CREPT-siRNA significantly impaired the ability of migration and invasion in Calu-1 and H838 cells. A-D. Scratch wound healing assay results showed that NC-siRNA and blank control cells migrated and the open area was almost healed after 48 hours. However, the healing of the open area was markedly attenuated when CREPT was knocked down. E, F. Transwell migration assay showed that the numbers of cells migrating through the membrane were greatly reduced when CREPT was knocked down. G, H. Transwell invasion assay showed that when CREPT was knocked down the numbers of cells migrating through the membrane were greatly reduced. * indicates P<0.05, ** indicates P<0.01.
The co-localization of CREPT with c-myc protein expression
CREPT and c-myc are mainly located in the cell nucleus and to some extentin cytoplasm. Furthermore, we performed an immunostaining assay to investigate whether CREPT and c-myc is co-localized. Preliminary experimental results showed that the co-localization phenomenon of CREPT and c-myc protein in NSCLC existed both in ADC and SCC (Figure 4D). In addition, the same method was used in NSCLC cell lines; the co-localization phenomenon of CREPT and c-myc protein expression was exhibited both in cell Calu-1 and H838 (Figure 4D). These results indicate that CREPT may be interacts with c-myc in the nucleus, however, it need be identified by further research.
The correlation of CREPT with c-myc, CDC25A expression in NSCLC
In order to investigate the role of CREPT in NSCLC with c-myc and the influence of CDC25A on the downstream signaling pathway, the correlation of CREPT with c-myc, CDC25A expression rate and intensity according to immunohistochemical staining of these proteins were analyzed (Figure 6, Table 1). There was anobvious correlation of CREPT with c-myc, CDC25A expression in NSCLC, ADC, and SCC (P<0.05, rs>0.434). Similar correlations were displayed in moderate differentiation (P<0.05, rs>0.378) and pTNM stages ofNSCLC (P<0.05, rs>0.352).
Figure 6.
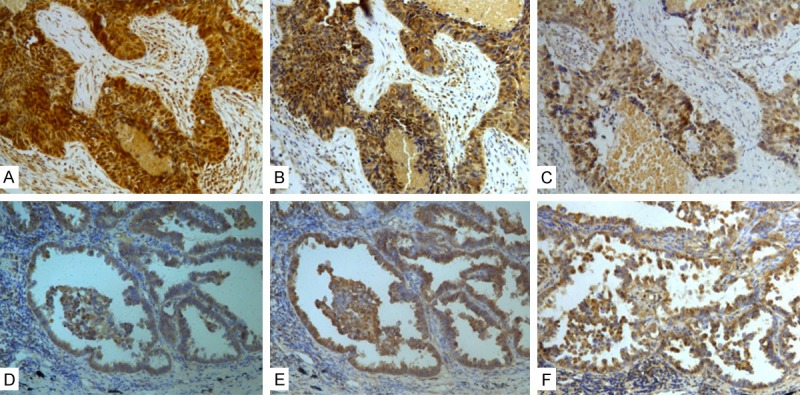
Expression of CREPT, c-myc and CDC25A by immunohistochemistry in NSCLC samples. A. Positive CREPT staining in SCC. B. Positive c-myc staining in SCC. C. Positive CDC25A staining in SCC. D. Positive CREPT staining in ADC. E. Positive c-myc staining in ADC. F. Positive CDC25A staining in ADC. (200× magnification).
Table 1.
The correlation of CREPT with c-myc and CDC25A expression in NSCLC
| Group | CREPT | c-myc | CDC25A | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Low | High | Statistics | Low | High | Statistics | |||
| NSCLC (n=72) | Low | 15 | 8 | rs=0.439 | 20 | 7 | rs=0.462 | |
| High | 10 | 39 | P=0.000 | 12 | 33 | P=0.111 | ||
| Histological type | ADC (n=33) | Low | 8 | 3 | rs=0.434 | 11 | 2 | rs=0.583 |
| High | 6 | 16 | P=0.024 | 5 | 15 | P=0.001 | ||
| SCC (n=39) | Low | 7 | 5 | rs=0.446 | 9 | 5 | rs=0.354 | |
| High | 4 | 23 | P=0.009 | 7 | 18 | P=0.043 | ||
| Differentiation | Well+Moderate (n=45) | Low | 11 | 5 | rs=0.399 | 17 | 6 | rs=0.378 |
| High | 8 | 21 | P=0.012 | 8 | 14 | P=0,017 | ||
| Poor (n=27) | Low | 4 | 3 | rs=0.497 | 3 | 1 | rs=0.467 | |
| High | 2 | 18 | P=0.024 | 4 | 19 | P=0.042 | ||
| pTNM stages | I-II (n=42) | Low | 10 | 6 | rs=0.352 | 14 | 4 | rs=0.440 |
| High | 7 | 19 | P=0.029 | 8 | 16 | P=0.006 | ||
| III-IV (n=30) | Low | 5 | 2 | rs=0,558 | 6 | 3 | rs=0.463 | |
| High | 3 | 20 | P=0.007 | 4 | 17 | P=0.003 | ||
Discussion
CREPT (cell-cycle related and expression-elevated protein in tumor, also named RPR1B) is a novel gene that belongs a new family of pro-teins within the RPR domain and was recently identified to promote tumorigenesis by up-regulating the expression of genes related to the cell cycle. CREPT has been reported to be an elevated expressed oncogene in a variety of tumors including colon, lung, liver, breast, prostate, stomach, uterine endometrium, and cervix cancers [4]. CREPT over expression level has been closely correlated with the degree of differentiation, tumor stage, histology type and depth of invasion. [5,6]. In retroperitoneal leiomyosarcomas, (78.9%) patients displayed positive expressions of CREPT, Ki-67, and PCNA. The expression of CREPT correlated with mitotic count and tumor size. The patients lacking CREPT expression exhibited significantly longer overall postoperative survival (median, 60.0 months) than the patients displaying CREPT expression (median, 33.0 months), and CREPT expression correlated with distant recurrence within 5 years after surgery [5]. Therefore, we hypothesized that CREPT might be a key factor in neoplasia and its development [6].
Yet, the expression and activated mechanism of CREPT in NSCLC and whether further promote NSCLC progression are still all unclear. So, in this report, we first detected the mRNA and protein expression of CREPT in NSCLC tissue samples and cell lines. The results showed that the CREPT mRNA and protein level in tumor tissue were significantly higher than in the adjacent normal lung tissues. To further confirm these observations, we examined the expression of CREPT in cells. The CREPT mRNA and protein level were clearly elevated in the 6 NSCLC cell lines compared to that in the normal human bronchial epithelial (HBE) cells. Therefore, whether the CREPT can regulate the NSCLC progression and the possible mechanism require investigation.
To further explore the relationship between CREPT and NSCLC, NSCLC cell lines were infected with CREPT-siRNA lentivirus. In our study, we found that both in SCC and ADC NSCLC cells, lacking of CREPT significantly depressed the growth curves and inhibited the number of cells and clones. Flow cytometry suggested that CREPT-siRNA arrests the cell cycle in G1 phase and induces cell apoptosis. In conclusion, knocking down CREPT decreases the proliferation and growth of NSCLC Cells. Previous studies reported that RPRD1A and RPRD1B appear to bind preferentially to RNAP II with a phosphorylated CTD [16]. Binding of RPRD1A and RPRD1B to RNAP II results in a specific reduction of RNAP II with S5P and S7P at target gene promoters [17,18]. Dongdong Lu et al. found that CREPT regulates cyclin D1 expression by binding to its promoter (B) and terminator (F), enhancing its transcription both in vivo and in vitro, and interacting with RNA polymerase II (RNAPII) [4]. Cell-cycle-related proteins precisely regulate cell proliferation. Cyclin D/CDK4 or cyclin D/CDK6 complex forms in early G1 phase and functions during the whole G1 to S phase transition [19]. Cyclin D1 controls the G1/S transition of the cell cycle and is tightly regulated by activators or repressors, which together keep cells growing normally [20]. The cell cycle-related factors, such as cyclin D1, cyclin E, CDK6, CDK4, and CDK2 increased dramatically when CREPT was over expressed. In the nude mice model, CREPT overexpression significantly accelerated the tumor xenograft growth, accompanied by elevated cyclin D1 and Ki-67 [6]. Our results and their conclusion are consistent.
The cell cycle-related factors, CDC25A and c-myc play an important role in cell proliferation regulation. CDC25A is essential for transition from G1 to S phase [21]. The CDC25A protein phosphatase is an example of a key cell-cycle regulator that is over produced in many human cancers [22]. CDC25A drives the cell cycle forward by activating cyclin-dependent proteinkinases (Cdks). The importance of CDC25A regulation is underscored by the observation that its overproduction leads to accelerated entry of cells into both S phase and mitosisand failure to regulate CDC25A during a checkpoint response causes bypass of DNA damage and replication checkpoints, resulting in enhanced DNA damage [23,24]. The c-myc proto-oncogene belongs to a family of related genes implicated in the control of normal cell proliferation and the induction of neoplasia [25]. CDC25A expression was raised following activation of c-myc and the CDC25A gene contains three Myc/Max binding sites within the first two introns. These can direct Myc-dependent transcription to from a heterologous promoter [21].
Increasing evidence has shown that CREPT promotes the transcriptional activity of the Wnt/β-catenin pathway. CREPT interacts with both β-catenin and TCF4, and enhances the activity of the β-catenin-TCF4 complex to initiate transcription of Wnt target genes, including cyclin D1 and c-myc, resulting in up-regulated cell proliferation and invasion [26-28]. Moreover, CREPT was shown to occupy at TCF4 binding sites (TBS) of the promoters of Wnt-targeted genes under Wnt stimulation. CREPT localizes to the TBS in both CCND1 and c-myc promoters; Wnt stimulation enhances occupancy, and CREPT is important for the occupancy of β-catenin and TCF4 on the promoter of c-myc [20].
Taking all of these data in to account, we speculate that there may be a new CREPT/c-myc/CDC25A pathway different from CREPT/cyclin D1 pathway. To verify our conjecture, we tested the expression of c-myc and CDC25A by RT-PCR and Western blot analysis in CREPT-siRNA-transfected cells. The results found that inhibitionof CREPT decreases the expression of cell cycle factor c-myc and CDC25A. Furthermore, we noticed that there was a co-localization phenomenon of CREPT with c-myc protein expression both in NSCLC cells and tissues. In addition, we also found that there were obvious correlation of CREPT with c-myc, CDC25A expression in NSCLC, ADC and SCC. Commendable correlations were also displayed in moderate differentiation and pTNM stages in NSCLC. Thus, according to our experimental results, oursurmise of this possible pathway is highly convincing. However, the underlying molecular mechanism of this possible CREPT/c-myc/CDC25A pathway is still unclear and need requires further investigation.
As discussed above, lacking of CREPT significantly depressed the cell growth and inhibited cells proliferation. We further characterized the biological function of CREPT in migration and invasion of NSCLC cells. Our results showed that inhibition of CREPT decreased migration by an average of 55% and 49% in scratch wound healing assays and the numbers of cells migrating through the membrane (with and without matrigel-coating) were greatly reduced when CREPT was inhibited. Thus, CREPT-siRNA significantly impaired the ability of migration and invasion in NSCLC cells. However, the mechanism of how CREPT regulates NSCLC migration and invasion is remains unclear. A BCAT1 (branched chain aminotransferase 1 gene, also known as ECA39) gene, may play a certain role in this process. BCAT1 was identified from a c-myc-induced tumor and has been proven to be directly regulated by c-myc through its binding to the specific DNA sequence, CACGTG [29]. Suppression of BCAT1 in glioma cell lines blocked the excretion of glutamate and has led to reduced proliferation and invasiveness in vitro, as well as significant decreases in tumor growth in a glioblastoma xenograft model [30]. A proteomics approach identified the cytoskeletal proteins actin and cdc42 as down-regulated in c-myc reconstituted fibroblasts, which suggest that c-myc plays a role in enhancing fibroblast motility [31]. As mentioned above, we found that CREPT can regulate the expression of c-myc in NSCLC. Thus, we can infer that CREPT may regulate the NSCLC migration and invasion through c-myc and BCAT1. However, more evidence is required to determine the importance of CREPT in NSCLC cells on migration and invasion. The interaction of CREPT with metastasis-associated protein such as Cd44v6, CEA, E-cadherin, MMP-2, MMP-9 and β-Catenin is still unclear and needs to be tested.
CREPT is a highly conserved oncogene, which is highly expressed in tumors and accelerates tumor development. Taken together the results in this study, we found that CREPT expression was significantly increased in NSCLC tissues and various NSCLC cell lines. Then, for the first time we have identified that siRNA-induced knocking down of CREPT significantly inhibited the proliferation and migration of NSCLC cell lines. Moreover, CREPT knocking down depressed the expression of cell cycle proteins including c-myc and CDC25A. Furthermore, we noticed that there was a co-localization phenomenon of CREPT with c-myc protein expression in NSCLC. In addition, we also found that there were obvious correlation of CREPT with c-myc, CDC25A expression in NSCLC. Thus, we suggested that CREPT may promote NSCLC cell growth through the c-myc and CDC25A signaling pathway. We believe that CREPT should be another target for tumor diagnosis and therapy development. However, the mechanism of NSCLC progression by CREPT regulating needs to be examined further. The pertinence of the over expression CREPT levels and poor prognosis in NSCLC is still not fully defined. To compensate for these shortcomings, we intend to carry out further multicenter clinical studies, expand the sample size, and enrich the means of detection.
Acknowledgements
We would like to thank the members of our research team (Thoracic surgery department laboratory) for their help in providing experimental, technical support and everyone who collaborated to ensure that the study proceeded smoothly.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kabbout M, Garcia MM, Fujimoto J, Liu DD, Woods D, Chow CW, Mendoza G, Momin AA, James BP, Solis L, Behrens C, Lee JJ, Wistuba II, Kadara H. ETS2 mediated tumor suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clin Cancer Res. 2013;19:3383–3395. doi: 10.1158/1078-0432.CCR-13-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu D, Wu Y, Wang Y, Ren F, Wang D, Su F, Zhang Y, Yang X, Jin G, Hao X, He D, Zhai Y, Irwin DM, Hu J, Sung JJ, Yu J, Jia B, Chang Z. CREPT accelerates tumorigenesis by regulating the transcription of cell-cycle-related genes. Cancer Cell. 2012;21:92–104. doi: 10.1016/j.ccr.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 5.She Y, Liang J, Chen L, Qiu Y, Liu N, Zhao X, Huang X, Wang Y, Ren F, Chang Z, Li P. CREPT expression correlates with poor prognosis in patients with retroperitoneal leiomyosarcoma. Int J Clin Exp Pathol. 2014;7:6596–6605. [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Qiu H, Hu W, Li S, Yu J. RPRD1B promotes tumor growth by accelerating the cell cycle in endometrial cancer. Oncol Rep. 2014;31:1389–1395. doi: 10.3892/or.2014.2990. [DOI] [PubMed] [Google Scholar]
- 7.Ren F, Wang R, Zhang Y, Liu C, Wang Y, Hu J, Zhang L, Chang Z. Characterization of a monoclonal antibody against CREPT, a novel protein highly expressed in tumors. Monoclon Antib Immunodiagn Immunother. 2014;33:401–408. doi: 10.1089/mab.2014.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldstraw P, Crowley J, Chansky K, Giroux DJ, Groome PA, Rami-Porta R, Postmus PE, Rusch V, Sobin L International Association for the Study of Lung Cancer International Staging Committee; Participating Institutions. The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J Thorac Oncol. 2007;2:706–714. doi: 10.1097/JTO.0b013e31812f3c1a. [DOI] [PubMed] [Google Scholar]
- 9.Yang G, Wang XJ, Huang LJ, Zhou YA, Tian F, Zhao JB, Chen P, Liu BY, Wen MM, Li XF, Zhang ZP. High ABCG4 Expression Is Associated with Poor Prognosis in Non-Small-Cell Lung Cancer Patients Treated with Cisplatin-Based Chemotherapy. PLoS One. 2015;10:e0135576. doi: 10.1371/journal.pone.0135576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhan W, Han T, Zhang C, Xie C, Gan M, Deng K, Fu M, Wang JB. TRIM59 Promotes the Proliferation and Migration of Non-Small Cell Lung Cancer Cells by Upregulating Cell Cycle Related Proteins. PLoS One. 2015;10:e0142596. doi: 10.1371/journal.pone.0142596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y, Su Z, Huang Y, Sun T, Chen S, Wu T, Chen G, Xie X, Li B, Du Z. The Zfx gene is expressed in human gliomas and is important in the proliferation and apoptosis of the human malignant glioma cell line U251. J Exp Clin Cancer Res. 2011;30:114. doi: 10.1186/1756-9966-30-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Wang J, Shen B, Peng C, Zheng M. The ABCC4 gene is a promising target for pancreatic cancer therapy. Gene. 2012;491:194–199. doi: 10.1016/j.gene.2011.09.029. [DOI] [PubMed] [Google Scholar]
- 13.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Yang HJ, Gao SS, Wang M, Shi Y, Cheng BF, Feng ZW. Identification of a novel role of RING finger protein 11 promoting the metastasis of murine melanoma cells. Am J Transl Res. 2015;7:1629–1635. [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, Zhou Y, Zhang Z, Tian F, Ma N, Liu T, Gu Z, Wang Y. Upregulated fascin1 in non-small cell lung cancer promotes the migration and invasiveness, but not proliferation. Cancer Lett. 2010;290:238–247. doi: 10.1016/j.canlet.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Ni Z, Xu C, Guo X, Hunter GO, Kuznetsova OV, Tempel W, Marcon E, Zhong G, Guo H, Kuo WH, Li J, Young P, Olsen JB, Wan C, Loppnau P, El Bakkouri M, Senisterra GA, He H, Huang H, Sidhu SS, Emili A, Murphy S, Mosley AL, Arrowsmith CH, Min J, Greenblatt JF. RPRD1A and RPRD1B are human RNA polymerase II C-terminal domain scaffolds for Ser5 dephosphorylation. Nat Struct Mol Biol. 2014;21:686–695. doi: 10.1038/nsmb.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ni Z, Olsen JB, Guo X, Zhong G, Ruan ED, Marcon E, Young P, Guo H, Li J, Moffat J, Emili A, Greenblatt JF. Control of the RNA polymerase II phosphorylation state in promoter regions by CTD interaction domain-containing proteins RPRD1A and RPRD1B. Transcription. 2011;2:237–242. doi: 10.4161/trns.2.5.17803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mei K, Jin Z, Ren F, Wang Y, Chang Z, Wang X. Structural basis for the recognition of RNA polymerase II C-terminal domain by CREPT and p15RS. Sci China Life Sci. 2014;57:97–106. doi: 10.1007/s11427-013-4589-7. [DOI] [PubMed] [Google Scholar]
- 19.Lee MH, Yang HY. Regulators of G1 cyclin-dependent kinases and cancers. Cancer Metastasis Rev. 2003;22:435–449. doi: 10.1023/a:1023785332315. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Liu C, Duan X, Ren F, Li S, Jin Z, Wang Y, Feng Y, Liu Z, Chang Z. CREPT/RPRD1B, a recently identified novel protein highly expressed in tumors, enhances the beta-catenin. TCF4 transcriptional activity in response to Wnt signaling. J Biol Chem. 2014;289:22589–22599. doi: 10.1074/jbc.M114.560979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature. 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- 22.Kristjansdottir K, Rudolph J. Cdc25 phosphatases and cancer. Chem Biol. 2004;11:1043–1051. doi: 10.1016/j.chembiol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Hoffmann I, Draetta G, Karsenti E. Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J. 1994;13:4302–4310. doi: 10.1002/j.1460-2075.1994.tb06750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, Piwnica-Worms H. GSK-3 beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 26.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 27.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 29.Zhou W, Feng X, Ren C, Jiang X, Liu W, Huang W, Liu Z, Li Z, Zeng L, Wang L, Zhu B, Shi J, Liu J, Zhang C, Liu Y, Yao K. Over-expression of BCAT1, a c-Myc target gene, induces cell proliferation, migration and invasion in nasopharyngeal carcinoma. Mol Cancer. 2013;12:53. doi: 10.1186/1476-4598-12-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tonjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, Pleier SV, Bai AH, Karra D, Piro RM, Felsberg J, Addington A, Lemke D, Weibrecht I, Hovestadt V, Rolli CG, Campos B, Turcan S, Sturm D, Witt H, Chan TA, Herold-Mende C, Kemkemer R, Konig R, Schmidt K, Hull WE, Pfister SM, Jugold M, Hutson SM, Plass C, Okun JG, Reifenberger G, Lichter P, Radlwimmer B. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med. 2013;19:901–908. doi: 10.1038/nm.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]