

Abstract
Elevated levels of advanced glycation endproducts (AGEs) is an important risk factor for atherosclerosis. Dysfunction of endothelial progenitor cells (EPCs), which is essential for re-endothelialization and neovascularization, is a hallmark of atherosclerosis. However, it remains unclear whether and how AGEs acts on EPCs to promote pathogenesis of atherosclerosis. In this study, EPCs were exposed to different concentrations of AGEs. The expression of NADPH and Rac1 was measured to investigate the involvement of NADPH oxidase pathway. ROS was examined to indicate the level of oxidative stress in EPCs. Total JNK and p-JNK were determined by Western blotting. Cell apoptosis was evaluated by both TUNEL staining and flow cytometry. Cell proliferation was measured by 3H thymidine uptake. The results showed that treatment of EPCs with AGEs increased the levels of ROS in EPCs. Mechanistically, AGEs increased the activity of NADPH oxidase and the expression of Rac1, a major component of NADPH. Importantly, treatment of EPCs with AGEs activated the JNK signaling pathway, which was closely associated with cell apoptosis and inhibition of proliferation. Our results suggest that the RAGE activation by AGEs in EPCs upregulates intracellular ROS levels, which contributes to increased activity of NADPH oxidase and expression of Rac1, thus promoting cellular apoptosis and inhibiting proliferation. Mechanistically, AGEs binding to the receptor RAGE in EPCs is associated with hyperactivity of JNK signaling pathway, which is downstream of ROS. Our findings suggest that dysregulation of the AGEs/RAGE axis in EPCs may promote atherosclerosis and identify the NADPH/ROS/JNK signaling axis as a potential target for therapeutic intervention.
Keywords: Advanced glycation endproducts, endothelial progenitor cells, NADPH oxidase, JNK pathway, apoptosis
Introduction
Glycation is defined as a process that single sugar molecules bond to protein(s) or lipid(s) to generate advanced glycation endproducts (AGEs). Increased formation of AGEs and binding to their cell surface receptor, receptor for AGEs (RAGE), cause irreversible tissue damages and activate the expression of a variety of cytokines. RAGE is expressed in a wide array of cell types, such as vascular cells, inflammatory cells, glomerular epithelial cells (podocytes), neurons of the central and peripheral nervous systems. RAGE is usually expressed at low levels in tissues in adult. Whereas, expression of RAGE is elevated in pathologies such as cardiovascular disease, diabetes and inflammation and may play an important role in the progression of atherosclerosis [1-3].
Reactive oxygen species (ROS) are constitutively generated and converted to nontoxic metabolites under normal physiological conditions [4]. Elevated levels of ROS exceeding the capacity of cellular antioxidant defense systems result in oxidative stress and contribute to the pathology of several chronic diseases, including cardiovascular dysfunctions, atherosclerosis, diabetes mellitus and reperfusion injury [5-7]. Previous studies indicate that ligand-RAGE interaction triggers ROS generation and activates downstream signaling pathways and gene expression. The primary mechanism by which RAGE generates oxidative stress may via activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in artery endothelial cells, artery smooth muscle cells and corneal epithelial cells [8-10], resulting in cell apoptosis [11]. Mechanistically, AGEs activates Mitogen-activated protein kinase (MAPK) pathways, a family of serine-threonine protein kinases. Notably, activation of C-jun N terminal kinase (JNK), major subfamilies of MAPK pathways, has been shown to result in cell apoptosis and dysfunction [12]. However, it is still not fully understood how ROS generation by AGEs/RAGE interaction might promote atherosclerosis.
Endothelial progenitor cells (EPCs) are bone-marrow-derived stem cells that differentiate into endothelial cells. EPCs contribute to tissue repair and neovasculature homeostasis via both angiogenesis and arteriogenesis, thus representing critical subsets of cells for atherosclerosis progression and treatment [13,14]. It is well appreciated that these progenitor cells are able to repair the injured vessel endothelium and enhance neovascularization of ischemic tissues. However, reduced numbers of EPCs are associated with endothelial dysfunction and elevated risk of adverse cardiovascular events. Although previous data showed that AGEs induced apoptosis of EPCs [15,16], the molecular mechanism underlying impairment of EPC function in atherosclerosis remains largely unknown.
To address this issue, EPCs were treated with different concentrations of AGEs to determine the levels of oxidative stress. To investigate potential underlying mechanism, we measured the activity of RAGE, NADPH oxidase and JNK pathways in these EPCs. In addition, cell proliferation and apoptosis were also examined. Our findings suggest that AGEs/RAGE interaction may activate RAGE-NADPH oxidase to elevate ROS levels, which then promote apoptosis and inhibit cell proliferation of EPCs via JNK pathways to accelerate the process of atherosclerosis.
Methods
Ethics statement
Animal experiments were approved by the Institutional Animal Care and Use Committee of the Third Military Medical University (Chongqing, China). All the procedures complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
Animals
Twenty male Sprague-Dawley rats (150-160 g) were obtained from the Laboratory Animal Center of the Third Military Medical University (Chongqing, China). Before experimentation, all rats were kept in an animal facility under controlled temperature conditions (21°C) with 12:12 hours light:dark cycle. Rats were housed five per cage and received standard laboratory diet and water. After one week of acclimatization, rats were anesthetized with ketamine (50 mg/kg) before being killed by decapitation. Following decapitation, bone marrow was carefully removed and processed according to the desired experiments.
Cell isolation, culture and treatment
Mononuclear cells were isolated from the bone marrow by density-gradient centrifugation and washed three times with phosphate buffered saline (PBS). Cells were seeded into 24-well plates at a density of 1 × 106 cells per well and cultured in Dulbecco’s modification of Eagle’s medium (DMEM) (Hyclone, USA) supplemented with 20% fetal calf serum (Hyclone, USA) and vascular endothelial growth factor (VEGF) (50 ng/mL). After four days, non-adherent cells were removed and adherent cells were cultured for additional seven days as previously described [17]. Cells were incubated with various concentrations (0, 100, 200 and 400 µg/ml) of AGEs (Jingmei BioTech, China) for 24 hours. siRNA transfection was carried out according to the manufacturer’s protocol for the RAGE-siRNA kit (Ambion, USA). The following siRNA sequence was used: sense: 5’-CCCAGAAGCUAGAAUGGAAtt-3’; antisense: 5’-UUCCAUUCUAGCUUCUGGGtt-3’ [16].
Cell staining and selection
Cultured cells were first incubated with DiI-labeled acetylated low-density lipoprotein (LDL) (Molecular Probe, Life Technologies, USA) at 37°C for 12 hours and then fixed with 2% paraformaldehyde for 10 minutes. The cells were then incubated with 10 μg/ml FITC-UEA-I (GeneTex, Irvine, USA) at 37°C for 1 hour. After staining, samples were viewed with laser confocal microscopy. Cells demonstrating double-positive fluorescence were identified as differentiating EPCs. Two or three independent investigators evaluated the number of EPCs per well by counting 15 randomly selected high-power fields [16].
Measurements of intracellular ROS
To determine intracellular ROS formation, the fluorescent intensity of H2DCF-DA (Sigma, USA) was measured by flow cytometry analysis following the manufacturer’s protocol [16]. Isolated EPCs were collected by centrifugation and resuspended in DMEM. H2DCF-DA and EPCs were mixed together in the dark at room temperature for 15 minutes. The mean fluorescence intensity of EPCs for each sample was quantified by flow cytometry analysis. EPCs mixed with PBS were used as controls.
NADPH oxidase activity assay
NADPH oxidase activity was determined by spectrophotometry analysis according to the manufacturer’s protocol (Jingmei BioTech, China). The activity of NADPH was assessed by optical density (OD). When the activity of enzymes increases, the optical density is also increased. The OD value was measured at 450 nm. Diphenyliodonium iodide (DPI) (Jingmei BioTech, China) was also used as an NADPH inhibitor.
Western blot
Total proteins from EPCs were extracted on ice with cell lysis buffer (Cell Signaling Technology, USA). Twenty μg of proteins were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were blocked with 2% fat-free milk in PBS at room temperature for 1 hours and probed with primary antibodies overnight at 4°C (Rabbit anti-rat Rac1 and p-JNK, Boisynthesis BioTech, USA) in PBS containing 0.1% Tween 20 (PBST) and 1% fat-free milk. Membranes were then washed four times in PBST and incubated with goat anti-rabbit IgG-horseradish peroxidase (Beyotime Institute of Biotechnology, China). Signals were developed using ECL reagents (Amersham Pharmacia Biotech, USA).
Detection of apoptosis
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was used to detect apoptosis. EPCs from each sample were stained according to the manufacturer’s instructions (Roche, Switzerland). Apoptosis was also investigated with Annexin V and Propidium Iodide Apoptosis Detection Kit (Jingmei BioTech, China) coupled with flow cytometry analysis according to the manufacturer’s instructions.
Caspase-3 activity assay
Caspase-3 activity was determined using a caspase-3 activity kit (Beyotime, China) according to the manufacturer’s protocol, which is based on the ability of caspase-3 to change acetyl-Asp-Glu-Val-Asp p-nitroanilide into the yellow formazan product, p-nitroaniline. Caspase-3 activity was quantified by the spectrophotometer at an absorbance of 405 nm.
Cyt-C activity assay
Cyt-C was determined by spectrophotometry analysis according to the manufacturer’s protocol (Boyao, China). The activity of Cyt-C was assessed by optical density. When the activity of Cyt-C increases, the optical density is also increased. The OD value was measured at 450 nm.
3H thymidine uptake
EPCs were cultured in DMEM with 3H thymidine (1 uCi/ml) for 6 hours. Incubations were terminated by removing the culture medium and washed with cold PBS three times followed by the addition of ice-cold 10% TCA for 20 minutes. Next, the cells were washed three times with ice-cold water. Then, they were lysed with 1 ml 0.5 M NaOH for 30 minutes at 37°C. After solubilization, 0.5 ml/well aliquot samples were removed, transferred to scintillation vials with 10 ml of scintillation cocktail already added to each vial and radioactivity was quantified by the liquid scintillation counter.
Statistical analysis
For statistical analysis, all values are expressed as mean ± SD. Differences among groups were assessed by an independent-sample t-test for single comparisons and multiple comparisons between groups were assessed by ANOVA. Probability values < 0.05 were considered statistically significant. Data analysis was carried out with the Statistical Package for Social Sciences (SPSS version 18.0).
Results
AGEs induced-upregulation of intracellular ROS in EPCs is dependent on the receptor RAGE
Bone marrow derived mononuclear cells from male Sprague-Dawley rats were collected, cultured in DMEM for seven days and stained by FITC-UEA-I and DiI-acLDL. FITC-UEA-I and DiI-acLDL double staining was seen in 83.4 ± 5.8% of attached cells, demonstrating that the cultured cells had endothelial cell characteristics. Thus, after in vitro culture, the isolated MNCs differentiated into EPCs [16].
To examine whether AGEs affects RAGE expression, EPCs were treated with AGEs at different doses, and RAGE protein was detected by Western blotting. RAGE protein levels were significantly induced by 272% in response to AGEs concentrations of 400 µg/ml. Silenced RAGE by siRNA can obstruct this effect (Figure 1A). To determine the effects of AGEs on ROS production in EPCs, the fluorescence intensity of H2DCF-DA was quantified using both fluorescence microscopy and flow cytometry analysis. Normal EPCs showed a low base level of ROS. Treatment with AGEs significantly increased the generation of ROS in EPCs, whereas silencing RAGE prevented AGEs induced ROS formation (Figure 1B). Consistent with the fluorescence microscopy results, flow cytometry analysis also revealed a stepwise increase in ROS expression with increasing AGEs concentration (n = 5, p < 0.05, Figure 1C).
Figure 1.
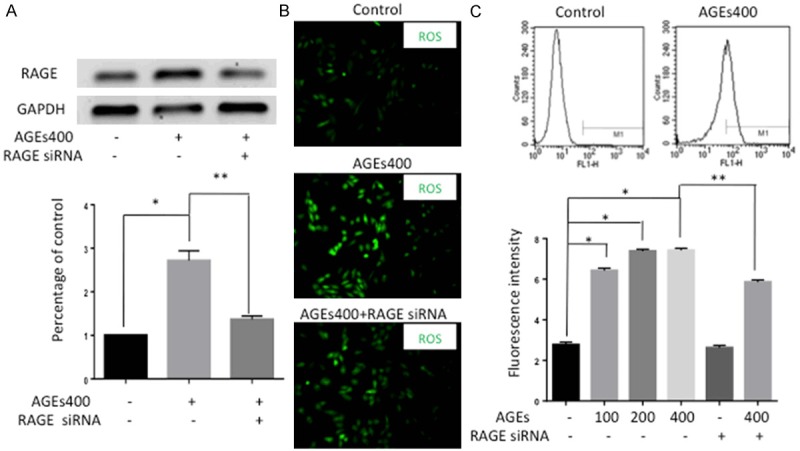
Activation of AGEs/RAGE signaling axis upregulates ROS level in EPCs. A. Western blot analysis showed that AGEs increased RAGE protein expression and that silencing RAGE by siRNA can obstruct this effect. Significances of differences are as indicated: n = 5; *P < 0.05 vs control; **P < 0.05 vs AGEs400. B. The fluorescence intensity of ROS was detected by fluorescence microscopy. Normal EPCs showed low ROS production without AGEs. Treatment of EPCs with AGEs increased the generation of ROS and blocking RAGE with siRNA weakened this effect (×400). C. The fluorescence intensity of ROS was also detected by flow-cytometry analysis. The fluorescence intensity increased with increasing AGEs concentrations and the effect was attenuated by RAGE siRNA. Significances of differences are as indicated: n = 5; *P < 0.05 vs control; **P < 0.05 vs AGEs400.
AGEs/RAGE signaling axis increases the activity of NADPH oxidase and the expression of Rac1 in EPCs
Previous study has demonstrated that AGEs may take effects via the RAGE-NADPH oxidase pathway. To confirm this, we assessed the activity of NADPH oxidase by ELISA. As shown in Figure 2A, AGEs treatment significantly increased the OD value of NADPH oxidase (n = 5, P < 0.05), and this increase in OD was apparently abolished by downregulation of RAGE. Because the optical density increases as the enzyme activity level increases, these results suggest that AGEs increase NADPH oxidase in EPCs. Consistent with the ELISA results, Western blot analysis revealed an increasing in Rac1 protein level with AGEs co-cultured (Figure 2B).
Figure 2.
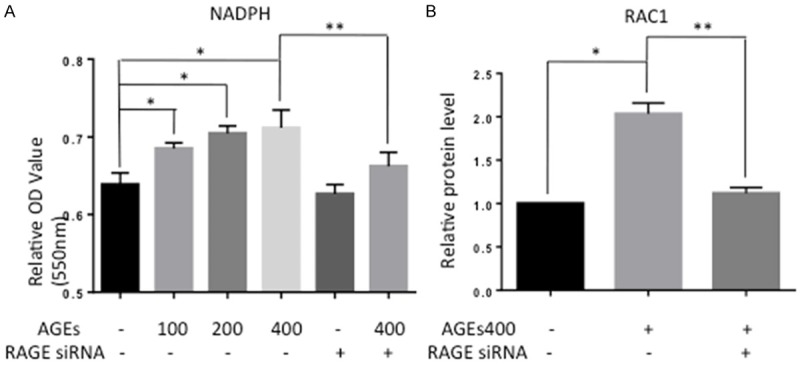
Treatment of EPCs with AGEs increased the activity of NADPH oxidase and the protein level of Rac1. A. AGEs increased the optical density, indicating that AGEs increased the activity of NADPH oxidase. Blocking RAGE prevented this effect. Significances of differences are as indicated: n = 5; *P < 0.05 vs control; **P < 0.05 vs AGEs400. B. Rac1 protein levels were assessed by Western blot. AGEs up-regulated Rac1 expression. Blocking RAGE prevented this effect. Significances of differences are as indicated: n = 5; *P < 0.05 vs control; **P < 0.05 vs AGEs400.
Upregulation of NADPH oxidase activity is essential for AGEs and the receptor RAGE to promote ROS generation in EPCs
NADPH oxidase transfers electrons from NADPH to molecular oxygen and produces ROS. NADPH oxidase may play a critical role in AGEs-induced ROS in EPCs. To confirm this, we block NADPH oxidase by DPI, a NADPH pharmacological inhibitor. As shown in Figure 3, DPI reduced AGEs induced activation of ROS (n = 5, p < 0.05), similar to block RAGE by siRNA. These results indicate that treatment of EPCs with AGEs results in ROS generation that occurs at least partially through the RAGE-NADPH pathway.
Figure 3.
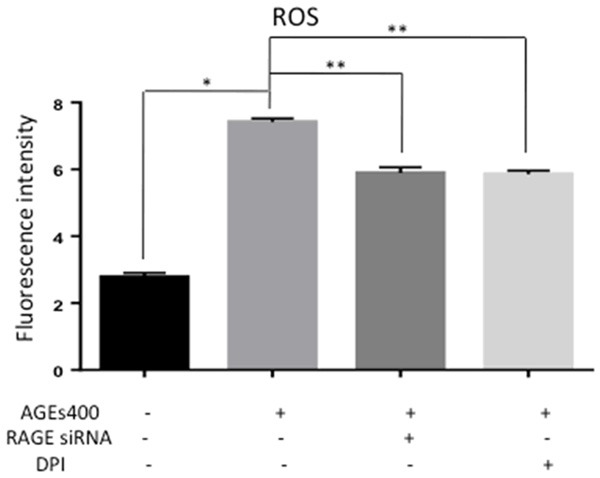
NADPH inhibition decrease ROS generation of EPCs. AGEs increased the optical density while co-cultured with DPI prevented this effect similar to block RAGE by siRNA. Significances of differences are as indicated: n = 5; *P < 0.05 vs control; **P < 0.05 vs AGEs400.
AGEs/RAGE axis induces apoptosis of EPCs by promoting NADPH oxidase activity
After incubation with AGEs, EPC apoptosis was assessed by TUNEL staining, flow cytometry analysis, Cyt-C activity assay and caspase-3 activity assay. In TUNEL staining, AGEs increased EPCs apoptosis, and this effect was apparently abolished by downregulation of RAGE or inhibition of NADPH (Figure 4A). Consistent with the TUNEL staining results, data from flow-cytometry (Figure 4B), Cyt-C activity assay (Figure 4C) and caspase-3 activity assay (Figure 4D) also showed the same results (n = 5, P < 0.05). Results from the 3H thymidine uptake assay showed that AGEs inhibited the proliferative activity of EPCs. Downregulation of RAGE or inhibition of NADPH can block this effect (n = 5, P < 0.05) (Figure 4E).
Figure 4.
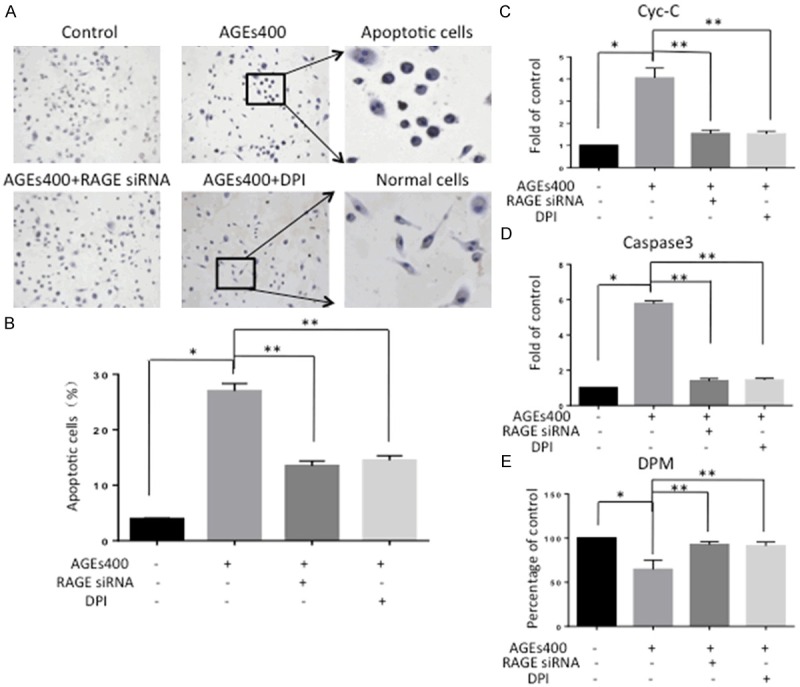
AGEs induces apoptosis and inhibits proliferation of EPCs. (A) In the TUNEL assay, nuclei in apoptosis cells appear brown. Co-culturing with AGEs increases apoptosis compared with the control group. Blocking RAGE or culturing with DPI prevented this effect. (B) Cells expressing annexin V were counted by flow-cytometry analysis. The number of EPCs undergoing apoptosis increased with AGEs, and the effect was attenuated by RAGE silencing and NADPH inhibition. Cyt-C activity assay (C) and caspase-3 activity assay (D) also showed the same results. Significances of differences are as indicated: n = 5; *P < 0.05 vs control; **P < 0.05 vs AGEs400. (E) Cell proliferation was evaluated by 3H thymidine incorporation assay. Co-cultured with AGEs inhibits EPCs proliferation. SiRNA and DPI decreased the inhibitory effects of AGEs. Significances of differences are as indicated: n = 5; *P < 0.05 vs control; **P < 0.05 vs AGEs400.
AGEs stimulation of the receptor RAGE in EPCs activates the JNK signaling pathway
JNK signaling is one of the important signaling pathways activated by ROS. JNK signaling is a major stress response pathway and induces cell apoptosis in response to various stressful conditions. AGEs induced-cell apoptosis may activate JNK signaling. To test this hypothesis, we assessed p-JNK by Western blot. AGEs treatment significantly increased the protein level of p-JNK in EPCs, while siRNA blocking RAGE apparently prevented JNK activation (Figure 5).
Figure 5.
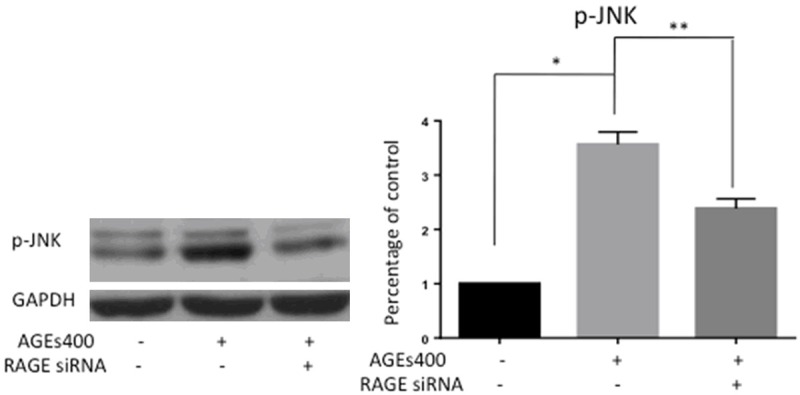
AGEs treatment activates the JNK signaling pathway in EPCs. EPCs were cultured for 24 hours with AGEs at a dose of 400 µg/mL. Protein levels of p-JNK were assessed by Western blot. The graphs show quantification of p-JNK as assessed. Significances of differences are as indicated: n = 5; *P < 0.05 vs control; **P < 0.05 vs AGEs400.
Discussion
In the current study, we established EPCs by in vitro culturing bone marrow derived mononuclear cells from male Sprague-Dawley rats. We found that treatment of EPCs with AGEs significantly increased the generation of ROS and the activity of NADPH oxidase and inhibited EPCs proliferation in a concentration-dependent manner. Intriguingly, treatment of EPCs with AGEs resulted in activation of the JNK signaling pathway and cell apoptosis.
Numerous epidemiological studies suggest that diabetes is associated with cardiovascular complications and accelerated atherosclerosis [18-20]. The proposed acceleration of atherosclerosis progression by diabetes has not been fully investigated. Furthermore, the underlying mechanism associating diabetes with atherosclerosis progression cannot simply be explained by the traditional cardiovascular risk factors. AGEs have been found to rapidly exacerbate symptoms in patients with diabetes and cardiovascular disease. Accumulation of AGEs is a characteristic feature of aging. It was recently reported that interaction of AGEs with their receptor could activate complex signaling pathways, causing increased generation of ROS [15]. Under normal physiological conditions, ROS are generated and converted to nontoxic metabolites. Low levels of ROS regulate various signal transduction pathways involved in cell survival, differentiation and growth. High levels of ROS are considered harmful metabolic products and have been implicated in the pathogenesis of cardiovascular diseases. When the rate of ROS formation exceeds the capacity of cellular antioxidative defenses, a state of oxidative stress occurs that can lead to cell death [21-23]. Thus, deregulation of AGEs may promote progression of diabetes and cardiovascular disease by increasing the generation of ROS through an unknown underlying mechanism.
Diabetes affects the number and function of circulating progenitor cells [24-26], and these were recently found to be regulated by AGEs [27,28]. Atherosclerosis is a progressive and dynamic disease arising from endothelial dysfunction. Progenitor cells circulate in the vessel, take part in re-endothelialization and neovascularization and restore vessel endothelial function. The capacity of EPCs to restore endothelial function may be due to their low ROS levels and reduced sensitivity to ROS-induced cell apoptosis. In our study, AGEs, in a concentration-dependent manner, caused the formation of ROS in EPCs. Thus, accumulation of AGEs might deteriorate the conditions of diabetes and cardiovascular disease by damaging EPCs via increased ROS generation.
Accumulating evidence has demonstrated that AGEs play an important role in the oxidative stress of diverse cell types via the RAGE-NADPH oxidase pathway [29,30]. NADPH oxidase transfers electrons from NADPH to molecular oxygen and produces ROS. We attempted to investigate the influence of AGEs on NADPH oxidase in EPCs to understand the underlying mechanisms of intracellular ROS production by AGEs. Our results showed that treatment of cultured EPCs with AGEs resulted in a concentration-dependent increase in NADPH oxidase activity. Rac1, a member of the Rho family of GTPases, is critical for the activation of the NADPH oxidase complex [31]. Rac1 activation is related to oxidative stress. Our results also showed that AGEs increased Rac1 protein level in cultured EPCs. Meanwhile, we found that the activation of NADPH oxidase and increase of Rac1 expression by AGEs could be weakened by silencing RAGE with siRNA. This result suggested that NADPH oxidase is a downstream target of RAGE in the AGEs-induced ROS pathway in rat EPCs. Similar to the siRNA knockdown of RAGE, we found that DPI prevented the induction of NADPH oxidase activity by AGEs. However, both RAGE siRNA and DPI only partially inhibited the induction of ROS by AGEs. These results indicate that there are additional mechanisms, besides NADPH oxidase, by which AGEs and RAGE induce intracellular ROS production.
The JNK pathway responds strongly to a variety of stress signals including oxidative stress, and oxidative stress-induced activation of JNK signaling is an important step in the induction of cell apoptosis [32,33]. ROS are the common mediators of intracellular second messengers. When the formation of ROS exceeds the capacity of the intracellular antioxidative defense system, the JNK pathway is activated and its activation eventually leads to apoptosis [34,35]. Our results indicate that JNK phosphorylation and apoptosis are induced in EPCs cultured with AGEs, suggesting that activation of JNK signaling pathway may be responsible for the AGEs-induced apoptosis.
Our data showed that AGEs could promote apoptosis of EPCs and could be attenuated by blocking RAGE. EPCs can differentiate into mature ECs and restore endothelial function. The number of healthy EPCs correlates with endothelial function and inversely correlates with the concentration of AGEs. Our findings suggest that high levels of AGEs may promote apoptosis and decrease the number of EPCs, leading to endothelial dysfunction. And these may accelerate the process of atherosclerosis.
Conclusions
We have demonstrated that AGEs-RAGE interaction induced oxidative stress, at least in part through activation of NADPH oxidase/Rac1, resulted in the activation of JNK pathway and ultimately led to apoptosis in EPCs (Figure 6). We speculate that AGEs induced excessive apoptosis reduces the ability of EPCs to maintain and restore epithelial function, contributing to pathologies associated with atherosclerosis. Our findings may provide novel therapeutic strategies for the clinical treatment of atherosclerosis.
Figure 6.
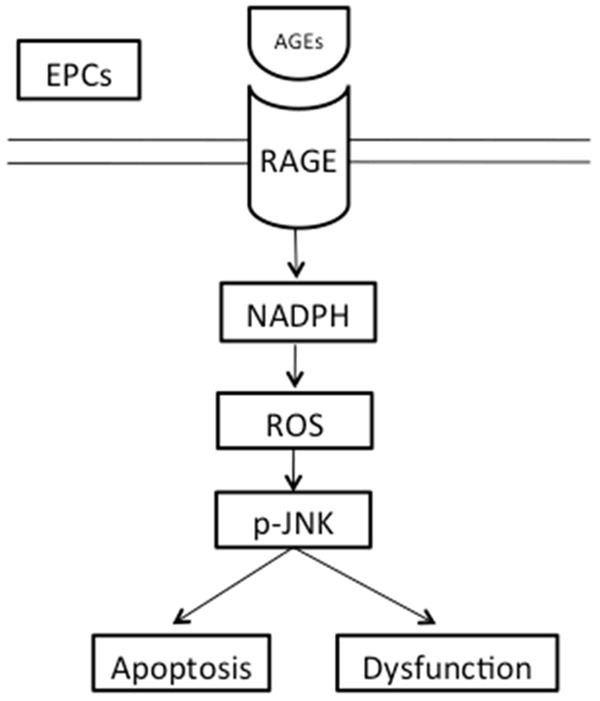
The sketch of RAGE-NADPH-JNK pathway. AGEs-RAGE interaction induced NADPH oxidase/Rac1 activation and increased oxidative stress, resulted in the activation of the JNK pathway and ultimately led to EPCs apoptosis and dysfunction.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Nos. 81100110, 81100134 and 81100149) and the Cardiovascular Disease Research Key Laboratory Foundation of Chongqing, China.
Disclosure of conflict of interest
None.
Authors’ contribution
JC and LH acquired and analyzed the data, and wrote the manuscript. JJ and SY analyzed data. JC, LH, JJ, SY, HT, BC, and MS acquired and researched the data. All authors read and approved the final manuscript.
Abbreviations
- AGEs
advanced glycation endproducts
- DMEM
dulbecco’s modification of eagle’s medium
- DPI
Diphenyliodonium iodide
- EPCs
endothelial progenitor cells
- LDL
low-density lipoprotein
- NADPH
nicotinamide adenine dinucleotide phosphate
- OD
optical density
- PBS
phosphate buffered saline
- ROS
reactive oxygen species
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
References
- 1.Daffu G, Del Pozo CH, O’Shea KM, Ananthakrishnan R, Ramasamy R, Schmidt AM. Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int J Mol Sci. 2013;14:19891–19910. doi: 10.3390/ijms141019891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen MM Jr. Perspectives on RAGE signaling and its role in cardiovascular disease. Am J Med Genet A. 2013;161:2750–2755. doi: 10.1002/ajmg.a.36181. [DOI] [PubMed] [Google Scholar]
- 3.Ward MS, Fortheringham AK, Cooper ME, Forbes JM. Targeting advanced glycation endproducts and mitochondrial dysfunction in cardiovascular disease. Curr Opin Pharmacol. 2013;13:654–661. doi: 10.1016/j.coph.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Mangge H, Becker K, Fuchs D, Gostner JM. Antioxidants, inflammation and cardiovascular disease. World J Cardiol. 2014;6:462–477. doi: 10.4330/wjc.v6.i6.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magenta A, Greco S, Gaetano C, Martelli F. Oxidative stress and microRNAs in vascular diseases. Int J Mol Sci. 2013;14:17319–17346. doi: 10.3390/ijms140917319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sena CM, Pereira AM, Seiça R. Endothelial dysfunction-A major mediator of diabetic vascular disease. Biochim Biophys Acta. 2013;1832:2216–2231. doi: 10.1016/j.bbadis.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Gage MC, Yuldasheva NY, Viswambharan H, Sukumar P, Cubbon RM, Galloway S, Imrie H, Skromna A, Smith J, Jackson CL, Kearney MT, Wheatcroft SB. Endothelium-specific insulin resistance leads to accelerated atherosclerosis in areas with disturbed flow patterns: a role for reactive oxygen species. Atherosclerosis. 2013;230:131–139. doi: 10.1016/j.atherosclerosis.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 8.Zhong Y, Cheng CF, Luo YZ, Tian CW, Yang H, Liu BR, Chen MS, Chen YF, Liu SM. C-reactive protein stimulates RAGE expression in human coronary artery endothelial cells in vitro via ROS generation and ERK/NF-κB activation. Acta Pharmacol Sin. 2015;36:440–447. doi: 10.1038/aps.2014.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di BB, Li HW, Li WP, Shen XH, Sun ZJ, Wu X. Pioglitazone inhibits high glucose-induced expression of receptor for advanced glycation end products in coronary artery smooth muscle cells. Mol Med Rep. 2015;11:2601–2607. doi: 10.3892/mmr.2014.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi L, Yu X, Yang H, Wu X. Advanced glycation end products induce human corneal epithelial cells apoptosis through generation of reactive oxygen species and activation of JNK and p38 MAPK pathways. PLoS One. 2013;8:e66781. doi: 10.1371/journal.pone.0066781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niiya Y, Abumiya T, Yamagishi S, Takino J, Takeuchi M. Advance glycation end products increase permeability of brain microvascular endothelial cells through reactiveoxygen species-induced vascular endothelial growth factor expression. J Stroke Cerebrovasc Dis. 2012;21:293–298. doi: 10.1016/j.jstrokecerebrovasdis.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Yang Q, Chen C, Wu S, Zhang Y, Mao X, Wang W. Advance glycation end products downregulates peroxisome proliferator-activated receptor γ expression incultured rabbit chondrocyte through MAPK pathway. Eur J Pharmacol. 2010;649:108–114. doi: 10.1016/j.ejphar.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 13.Ii M, Takeshita K, Ibusuki K, Luedemann C, Wecker A, Eaton E, Thorne T, Asahara T, Liao JK, Losordo DW. Notch signaling regulates endothelial progenitor cell activity during recovery from arterial injury in hypercholesterolemic mice. Circulation. 2010;121:1104–1112. doi: 10.1161/CIRCULATIONAHA.105.553917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams WJ, Zhang Y, Cloutier J, Kuchimanchi P, Newton G, Sehrawat S, Aird WC, Mayadas TN, Luscinskas FW, García-Cardeña G. Functional vascular endothelium derived from human induced pluripotent stem cells. Stem Cell Reports. 2013;1:105–113. doi: 10.1016/j.stemcr.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Huang L, Song M, Yu S, Gao P, Jing J. C-reactive protein upregulates receptor for advanced glycation end products expression and alters antioxidant defenses in rat endothelial progenitor cells. J Cardiovasc Pharmacol. 2009;53:359–367. doi: 10.1097/FJC.0b013e31819b5438. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Song M, Yu S, Gao P, Yu Y, Wang H, Huang L. Advanced glycation endproducts alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation endproducts mediate overpression of cell oxidant stress. Mol Cell Biochem. 2010;335:137–146. doi: 10.1007/s11010-009-0250-y. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Jin J, Song M, Dong H, Zhao G, Huang L. C-reactive protein down-regulates endothelial nitric oxide synthase expression and promotes apoptosis in endothelial progenitor cells through receptor for advanced glycation end-products. Gene. 2012;496:128–135. doi: 10.1016/j.gene.2011.12.039. [DOI] [PubMed] [Google Scholar]
- 18.Blomster JI, Chow CK, Zoungas S, Woodward M, Patel A, Poulter NR, Marre M, Harrap S, Chalmers J, Hillis GS. The influence of physical activity on vascular complications and mortality in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2013;15:1008–1012. doi: 10.1111/dom.12122. [DOI] [PubMed] [Google Scholar]
- 19.Cohen Tervaert JW. Cardiovascular disease due to accelerated atherosclerosis in systemic vasculitides. Best Pract Res Clin Rheumatol. 2013;27:33–44. doi: 10.1016/j.berh.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 20.van Dooren FE, Nefs G, Schram MT, Verhey FR, Denollet J, Pouwer F. Depression and risk of mortality in people with diabetes mellitus: a systematic review and meta-analysis. PLoS One. 2013;8:e57058. doi: 10.1371/journal.pone.0057058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pitocco D, Tesauro M, Alessandro R, Ghirlanda G, Cardillo C. Oxidative stress in diabetes: implications for vascular and other complications. Int J Mol Sci. 2013;14:21525–21550. doi: 10.3390/ijms141121525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higashi Y, Sukhanov S, Anwar A, Shai SY, Delafontaine P. Aging, atherosclerosis, and IGF-1. J Gerontol A Biol Sci Med Sci. 2012;67:626–639. doi: 10.1093/gerona/gls102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salminen A, Ojala J, Kaarniranta K, Kauppinen A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell Mol Life Sci. 2012;69:2999–3013. doi: 10.1007/s00018-012-0962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Povsic TJ, Sloane R, Green JB, Zhou J, Pieper CF, Pearson MP, Peterson ED, Cohen HJ, Morey MC. Depletion of circulating progenitor cells precedes overt diabetes: A substudy from the VA enhanced fitness trial. J Diabetes Complications. 2013;27:633–636. doi: 10.1016/j.jdiacomp.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desouza CV. Does drug therapy reverse endothelial progenitor cell dysfunction in diabetes? J Diabetes Complications. 2013;27:519–525. doi: 10.1016/j.jdiacomp.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 26.Barthelmes D, Irhimeh MR, Gillies MC, Karimipour M, Zhou M, Zhu L, Shen WY. Diabetes impairs mobilization of mouse bone marrow-derived Lin(-)/VEGF-R2(+) progenitor cells. Blood Cells Mol Dis. 2013;51:163–173. doi: 10.1016/j.bcmd.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Stolzing A, Sellers D, Llewelyn O, Scutt A. Diabetes induced changes in rat mesenchymal stem cells. Cells Tissues Organs. 2010;191:453–465. doi: 10.1159/000281826. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Zhang X, Guan X, Cui X, Wang Y, Chu H, Cheng M. Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc Diabetol. 2012;11:46. doi: 10.1186/1475-2840-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guimarães EL, Empsen C, Geerts A, van Grunsven LA. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol. 2010;52:389–397. doi: 10.1016/j.jhep.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Niiya Y, Abumiya T, Yamagishi S, Takino J, Takeuchi M. Advanced glycation end products increase permeability of brain microvascular endothelial cells through reactive oxygen species-induced vascular endothelial growth factor expression. J Stroke Cerebrovasc Dis. 2012;21:293–298. doi: 10.1016/j.jstrokecerebrovasdis.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Elnakish MT, Hassanain HH, Janssen PM, Angelos MG, Khan M. Emerging role of oxidative stress in metabolic syndrome and cardiovascular diseases: important role of Rac/NADPH oxidase. J Pathol. 2013;231:290–300. doi: 10.1002/path.4255. [DOI] [PubMed] [Google Scholar]
- 32.Hu WS, Lin YM, Ho TJ, Chen RJ, Li YH, Tsai FJ, Tsai CH, Day CH, Chen TS, Huang CY. Genistein suppresses the isoproterenol-treated H9c2 cardiomyoblast cell apoptosis associated with P-38, Erk1/2, JNK, and NFκB signaling protein activation. Am J Chin Med. 2013;41:1125–1136. doi: 10.1142/S0192415X13500766. [DOI] [PubMed] [Google Scholar]
- 33.Sun PH, Ye L, Mason MD, Jiang WG. Receptor-like protein tyrosine phosphatase κ negatively regulates the apoptosis of prostate cancer cells via the JNK pathway. Int J Oncol. 2013;43:1560–1568. doi: 10.3892/ijo.2013.2082. [DOI] [PubMed] [Google Scholar]
- 34.Mukherjee S, Ghosh S, Choudhury S, Adhikary A, Manna K, Dey S, Sa G, Das T, Chattopadhyay S. Pomegranate reverses methotrexate-induced oxidative stress and apoptosis in hepatocytes by modulating Nrf2-NF-κB pathways. J Nutr Biochem. 2013;24:2040–2050. doi: 10.1016/j.jnutbio.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 35.Lu TH, Tseng TJ, Su CC, Tang FC, Yen CC, Liu YY, Yang CY, Wu CC, Chen KL, Hung DZ, Chen YW. Arsenic induces reactive oxygen species-caused neuronal cell apoptosis through JNK/ERK-mediated mitochondria-dependent and GRP 78/CHOP-regulated pathways. Toxicol Lett. 2013;224:130–140. doi: 10.1016/j.toxlet.2013.10.013. [DOI] [PubMed] [Google Scholar]