

Abstract
Objectives: Forkhead/winged helix transcription factor p3 (Foxp3) increases in CD4+CD25+Treg cells during sepsis; however, related mechanisms are unclear. Our study aimed to explore the possible molecular mechanisms of high expression of Foxp3 in Treg cells during sepsis. Methods: Sepsis was induced by cecal ligation and puncture (CLP) method. CD4+CD25+Treg cells were isolated from peripheral blood and identified by flow cytometry (FCM). Treg cells were cultured with or without adenosine, adenosine agonist, adenosine antagonist, SMAD family member 3 (Smad3) agonist (transforming growth factor (TGF)-β1), or C-Jun N-Terminal Kinase (JNK) inhibitor. Expression levels of Foxp3 and activator protein 1 (AP-1) were determined. The binding of c-Fos or c-Jun to the Foxp3 promoter was then evaluated by the chromatin immunoprecipitation (ChIP) assay and quantified by quantitative real-time PCR (qRT-PCR). The mRNA and protein levels of Foxp3 were determined after transfection with siRNA against c-Fos, Fra-2, c-Jun or JunD. Results: Pharmacological inhibition of both adenosine and JNK reduced Foxp3 protein levels. JNK/AP-1 activation was involved in increased levels of Foxp3 protein in CD4+CD25+Treg cells. AP-1 regulated activity of Foxp3 promoter in Treg cells, and the induction of c-Fos or c-Jun activity leads to elevated transcription of Foxp3 gene. Knockdown of c-Fos, Fra-2, c-Jun, or JunD levels also reduced Foxp3 expression. Conclusion: We confirm that adenosine plays significant roles in the high expression of Foxp3. Adenosine promotes Foxp3 expression in Treg cells during sepsis via JNK/AP-1 pathway.
Keywords: Sepsis, adenosine, Foxp3, JNK/AP-1 pathway, regulatory T cells
Introduction
Sepsis is defined as a systemic inflammatory response syndrome (SIRS) caused by bacterial, fungal or viral origin infection [1]. It is one of the most common reasons for death in critically illpatients despite significant progress have been made in antibiotic and other supportive therapies [2]. Sepsis still represents a main challenge in treatment. It has been well demonstrated that sepsis progresses when the initial appropriate inflammatory response becomes extreme, leading to extensive and intense hyper inflammatory response [3,4]. However, this shift often results in an immunosuppressive state, which has long been regarded as a key factor in late mortality in patients with sepsis [5].
In recent years, adaptive immune system has been paid a great attention on the development of sepsis [6,7]. Among all the T lymphocyte subpopulations, regulatory T cells (Tregs) CD4+CD25+ have been shown to be critical in maintaining of immunologic homeostasis and tolerance [8,9]. These cells exhibit a pronounced antiinflammatory activity via directly inhibiting other immune cells and producinghigh levels of soluble CD25 and other inflammatory factors [10-12]. But their development and function are regulated by forkhead/winged helix transcription factor p3 (Foxp3) that is specifically expressed on CD4+CD25+Treg cells. Foxp3 has been considered as the control gene for the differentiation and function of Treg cells, as well as regulation of intracellular molecules related to effector T cell [11,13]. There is accumulating evidence indicating that markedly increased levels of Tregs in patients and animals during the early onset of sepsis [14-16]. Besides, expression levels of Foxp3+ are also increased in CD4+CD25+Tregs cells in patients with sepsis [17]. However, rare little information is available on the contribution of mechanism of high expression of Foxp3+ in CD4+CD25+Treg cells in sepsis patients.
In this study, a murine cecal ligation and puncture (CLP) model was used to explore the possible mechanism of high expression of Foxp3 in CD4+CD25+Treg cells. Better and deeper understanding of knowledge and management processes in sepsis may contribute to tailored therapeutic approaches for sepsis.
Materials and methods
Experimental animals
Twenty adult female Sprague-Dawley (SD) rats (8-12 weeks old, weighing 200-250 g) were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. (Chinese Academy of Sciences, Shanghai, China). The rats were kept in a conventional, temperature-controlled (22 ± 0.5°C)facility with a 14:10-hour light-dark cycle and fed with food and water ad libitum. This study was performed according to the Guide for the Care and Use of Medical Laboratory Animals (Ministry of Health, PR China, 1998) and the guidelines of the Laboratory Animal Ethical Commission of our university.
Induction of sepsis model
Polymicrobial sepsis model was induced using a CLP method according to a previous study [18], but with a minor modifications. All the rats were anesthetized by thiopental (25 mg/kg).The abdomen was shaven, and a midline laparotomy was performed. Thereafter, the cecum was ligated just below the ileocecal valve andpunctured twice through the cecum distal to the point of ligation by using a 12 G needle. The cecum was put into the abdomen and the incision was closed. After 24 h of CLP, the rats weresacrificed by cervical dislocation under deep anesthesia and blood was collected. The samples were anticoagulated with ethylenediaminetetraacetic acid.
Flow cytometry (FCM) analysis
After the samples were harvested, lymphocytes were separated by centrifugation on a lymphocyte separation medium. The lymphocytes were calculated, and the cell concentration was adjusted to 1×106/mL. The α CD4 (RM4-5) fluorochrome-labeled antibody (BD Biosciences, San Diego, CA), antibody directed against CD25 (clone 7D4) (eBioscience, San Diego, CA), and intranuclear phycoerythrin-conjugated α Foxp3 antibodies (3G3) (eBioscience, San Diego, CA) were separately added to the diluted lymphocytes according to the manufacturer’s instructions. FCM was performed by a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA), and CellQuest software (Becton Dickinson, Bedford, MA) was employed for data analysis.
Drugs
Treg cells was cultured with or without the following inhibitor or agonist for 24 h: SMAD family member 3 (Smad3) agonist, transforming growth factor (TGF)-β1 (1 ng/ml) (R&D Systems); C-Jun N-Terminal Kinase (JNK) inhibitor (SP600125, 10 µM) (LC laboratories); Adenosine (10 µM) (R&D Systems); Selective A2A receptor adenosine agonist (CGS21680, 100 nM) (LC laboratories); Selective A2A receptor adenosine antagonist, 8-(3-chlorostyryl) caffeine (CSC, 500 nM) (Sigma-Aldrich).
Small interference RNA (siRNA)
Cells were plated on 60-mm dish. After 24 hours of incubation, siRNA against c-Fos, Fra-2, c-Jun, JunD or control (nontargeting) siRNA were transfected into cells at a final density of 50 nM using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s guidelines. The si-c-Fos, si-Fra-2, si-c-Jun and si-JunD were all purchased from GenaPharma Shanghai, China. At 48 h after the transfection, the cells were harvested for further protein expression.
Western blotting
Protein was extracted from the cells and determined using a BCA assay kit (Perbio, Cramlington, UK). The samples (20 μg per lane) were separated on a 10-12% sodium dodecyl sulfate (SDS)-polyacrylamide gel and blotted onto polyvinylidene difluoride (PVDF) membranes (Bedford, MA, USA), blocked in 5% defatted milk powder in phosphate buffer saline (PBS) and incubated overnight at 4°C with the following antibodies: anti-Foxp3 antibody, anti-pJNK antibody, anti-JNK antibody, anti-p-Smad3 antibody, anti-Smad3 antibody, anti-p-c-Fos antibody, anti-c-Fos antibody, anti-FosB antibody, anti-p-Fra-1 antibody, anti-Fra-1 antibody, anti-Fra-2 antibody, anti-p-c-Jun antibody, anti-c-Jun antibody, anti-p-JunB antibody, anti-JunB antibody, anti-JunD antibody, and GAPDH. All the antibodies were purchased from Cell Signaling Technologies (Berverly, MA). Thereafter, the membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature. Enhanced chemiluminescence and densitometric analysis were finally performed.
Chromatin immunoprecipitation (ChIP) assay
A chromatin immunoprecipitation (ChIP) assay for c-Fos and c-Jun binding to the Foxp3 site was undertaken using Treg cells and a Simple ChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology) following the manufacturer’s protocol. ChIP were performed with cross-linked chromatin starved overnight, and 10 μl of c-Fos monoclonal antibody (mAb), c-Jun mAb or 2 μl of normal rabbit IgG was performed using protein A-Sepharose beads. Precipitated DNA was quantified by quantitative real-time PCR (qRT-PCR) with Foxp3-specific primers using an ABI 7700Prism (Applied Biosystems) with the SYBR Green PCR core reagent kit. The amount of immunoprecipitate, expressed as the signal relative to the total amount of input chromatin, was determined.
qRT-PCR
After 48 h of transfection with si-c-Fos, si-Fra-2, si-c-Jun or si-JunD, mRNA levels of Foxp3 were analyzed using qRT-PCR. Total RNA was extracted using a Trizol reagent (Invitrogen Corp., Carlsbad, CA) according to the manufacturer’s instructions. First strand complementary DNA (cDNA) was synthesized using the Superscript III reverse transcriptase (Invitrogen). The mRNAs levels were assessed using SYBR green-based qRT-PCR. GAPDH gene was used as a reference control. The primer sequences were as follows: 5’-primer-TTCCCTGGCATTTCCCATCC-3’ and reverse, 5’-primer-GCAGGCAGAGACACCATTCT-3’.
Statistical analysis
All experiments were repeated in triplicate with similar results. The collected data were represented as the mean ± standard deviation (SD). Statistical analyses were performed using statistic package for social science (SPSS, version 18.0, Chicago, IL) software. One-way analysis of variance (ANOVA) was used to calculate the P-values. A statistical significance was defined when P < 0.05.
Results
Foxp3 was promoted by adenosine
To explore the effect of adenosine on the expression of Foxp3, we examined the expression of Foxp3 after administration of adenosine, selective A2A receptor adenosine agonist (CGS21680) or selective A2A receptor adenosine antagonist (CSC) to the CD4+CD25+Treg cells. As shown in Figure 1, the expression of Foxp3 was significantly increased by adenosine compared to the control group (P < 0.05). In addition, the levels of Foxp3 were higher in the CGS21680 group, but lower in the CSC group, indicating that adenosine promoted the expression of Foxp3. However, there were no significant differences between the control group and the CGS21680+CSC group, suggesting that the effect of adenosine on the expression of Foxp3 was offset by simultaneous application of agonist and antagonist.
Figure 1.

Foxp3 expression in the CD4+CD25+Treg cellsafter administration of adenosine, adenosine agonist (CGS21680) or adenosine antagonist (CSC).Treg, regulatory T cells; Foxp3, forkhead/winged helix transcription factor p3.
Foxp3 was associated with JNK activation
To investigate the possible mechanism of adenosine on the expression of Foxp3, the relationships between the expression of Foxp3 and the activation of JNK or Smad3 were investigated.We found that both the levels of Foxp3 and p-JNK were markedly elevated by adenosine or agonist of adenosine (CGS21680), but were decreased by JNK inhibitor (SP600125) compared with the control group (P < 0.05). Moreover, simultaneous application of SP600125 with adenosine or agonist of adenosine had no effect on the levels of p-JNK compared with only application of SP600125, but could significantly increase the levels of Foxp3. But the levels of Foxp3 were still lower than that only application of adenosine or agonist of adenosine (Figure 2A). Although the levels of Foxp3 and p-Smad3 were markedly increased by Smad3 agonist (TGF-β1) (P < 0.05), the levels of p-Smad3 were not affected by adenosine or its agonist (Figure 2B). These results demonstrated that the expression of Foxp3 was associated with the activation of JNK.
Figure 2.
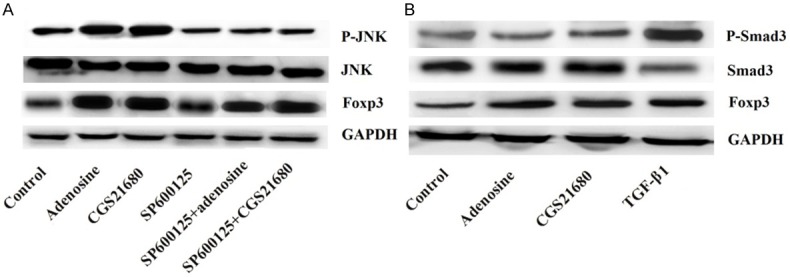
Expressions of Foxp3 are associated with JNK but not Smad3 activation. A. Expressions of Foxp3 are associated with JNK activation; B. Expressions of Foxp3 are not associated with Smad3 activation. Foxp3, forkhead/winged helix transcription factor p3; JNK, C-Jun N-Terminal Kinase; Smad3, SMAD family member 3.
Foxp3 was involved in activation of the JNK/activator protein 1 (AP-1) pathway
To characterize the involved signaling pathway of the effect of adenosine on the expression of Foxp3, we studied the effect of adenosine on activation of AP-1. Western blotting analysis revealed that no significant differences were found in the levels of FosB, Fra-1, p-JunB, and JunB among the different groups. However, SP600125 was capable of significantly increasing c-Fos, p-Fra-1, and c-Jun expression but suppressing p-c-Fos, Fra-2, p-c-Jun, and Jun-D expression (P < 0.05) (Figure 3). In addition, these abilities were reversed by adenosine or its agonist. Overall, these results suggested that the JNK pathway and the transcription factor AP-1 contributed to the effect of adenosine on the expression of Foxp3.
Figure 3.
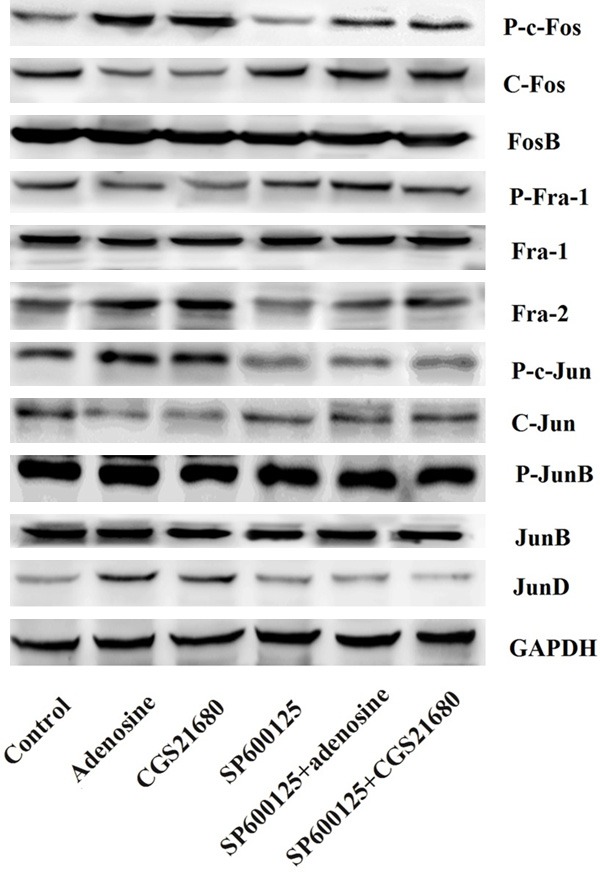
Expressions of Foxp3 is involved in activation of the JNK/AP-1 pathway. Foxp3, forkhead/winged helix transcription factor p3; C-Jun N-Terminal Kinase; AP-1, activator protein 1 JNK.
Both c-Fos and c-Jun can bind to the Foxp3 promoters to promote its transcription
The binding of c-Fos and c-Jun to the Foxp3 promoter was identified by the ChIP assay, as well as the effect of si-c-Fos or si-c-Jun on expression of Foxp3. Sonicated chromatin was precipitated with antibodies against c-Fos or c-Jun, with non-immune rabbit IgG as a control. The region of the Foxp3 promoter pulled down by immunoprecipitation was evaluated by qRT-PCR using Foxp3-specific primers. As shown in Figure 4A and 4B, signal relative to input for both c-Fos and c-Jun was significantly higher than those in the IgG group (P < 0.05). Besides, binding of c-Fos or c-Jun to the AP-1 region containing Foxp3 promoter was respectively decreased in cells treated with si-c-Fos or si-c-Jun (P < 0.05) (Figure 4C). These results suggested that the binding of c-Fos or c-Jun to the Foxp3 promoter was necessary and could promote the expression of Foxp3 in CD4+CD25+Treg cells.
Figure 4.
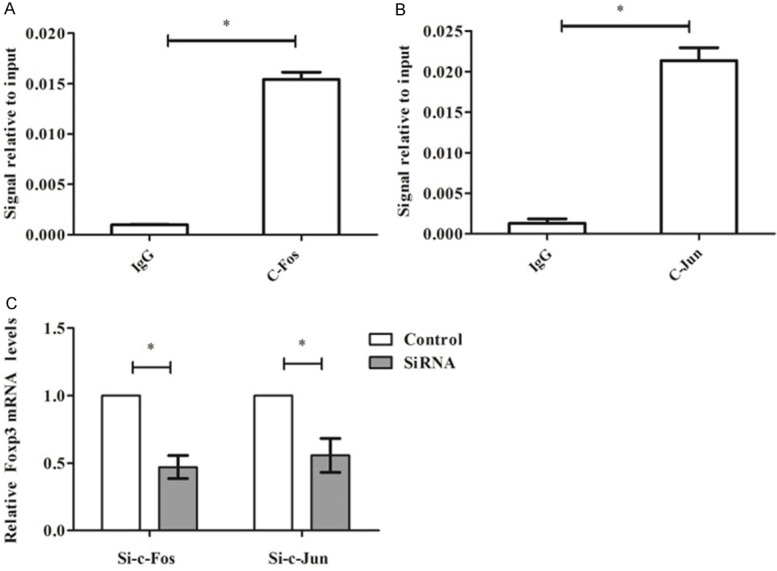
C-Fos or c-Jun can bind to the Foxp3 promoters to promote its transcription. A. Signal relative to input for c-Fos; B. Signal relative to input forc-Jun; C. Relative Foxp3 mRNA level after transfection with si-c-Fos or si-c-Jun. Foxp3, forkhead/winged helix transcription factor p3; si, small interference. *P < 0.05 compared with IgG or control group.
Knockdown of c-Fos, Fra-2,c-Jun, or JunD decreased expression of Foxp3
We tested whether knockdown of c-Fos, Fra-2, c-Jun, or JunD could decrease expression of Foxp3. Pre-incubation of CD4+CD25+Treg cells with specific siRNA (si-c-Fos, si-Fra-2, si-c-jun, or si-JunD) significantly decreased protein levels of c-Fos, Fra-2, c-jun, or JunD Foxp3 compared with the control group (P < 0.05) (Figure 5A). Besides, knockdown of c-Fos, Fra-2, c-Jun, or JunD considerably reduced the protein levels of Foxp3. The levels were the lowest when co-transfection with the four kinds of siRNA (Figure 5B).
Figure 5.
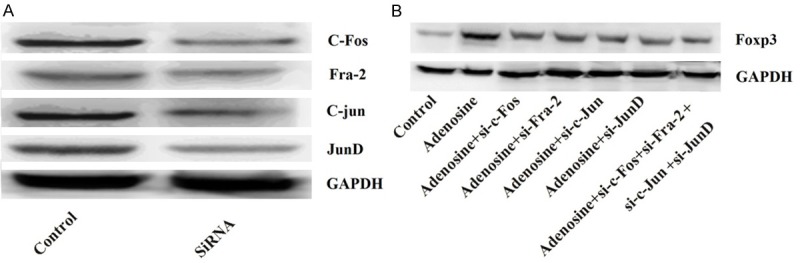
Knockdown of c-Fos, Fra-2, c-Jun, or JunD decreases expression of Foxp3. A. Expression levels of c-Fos, Fra-2, c-Jun, or JunD after transfection with siRNA; B. Expression levels of Foxp3 after knockdown of c-Fos, Fra-2, c-Jun, or JunD. Foxp3, forkhead/winged helix transcription factor p3; siRNA, small interference RNA.
Discussion
Researchers have proven the high expression of Foxp3 in Treg cells during the sepsis; however, the possible mechanism is still unclear. In the present study, we confirm that JNK/AP-1 activation is involved in the elevated levels of Foxp3 protein in CD4+CD25+Treg cells. We demonstrate that AP-1 regulates Foxp3 promoter activity in CD4+CD25+Treg cells and that the induction of c-Fos or c-Jun activity leads to increased transcription of the endogenous Foxp3 gene. Pharmacological inhibition of both adenosine and the JNK pathway reduced Foxp3 protein levels, and siRNA knockdown of c-Fos, Fra-2, c-Jun, or JunD levels also reduced Foxp3 expression.
It has been well demonstrated that severe sepsis and septic shock disrupt immune homeostasis by induction of an initial intense systemic inflammatory response, and then followed by a negative feedback of anti-inflammatory process [3,19]. Moreover, studies have confirmed that Tregs are associated with the pathogenesis of sepsis, which might be significantly contributed to the increased morbidity and mortality in patients with sepsis. In spite of CD4+CD25+ Treg cells are only a small part of the T lymphocyte lineage in the immune system, these cells have shown potent regulatory properties on cellular activation which make them criticalactors in the suppression of immune response during sepsis [20]. Clinical and experimental studies have suggested that sepsis exerts a relative and high elevation in Treg number and augmentation of its inhibitory function [21,22]. Considering this reason, it is necessary to be explicit about a better and deeper understanding of Tregs. The physiological action of these Treg cells will be benefit to develop new and targeted therapeutic strategies for regulating the immune response in severe sepsis and septic shock. Yet, studies on how to modulate Tregs for the improvement of sepsis and survival rate has been paid only little attention, and limited to experimental researches.
Treg cells can be distinguished from other activated T cells by expression of the transcriptional regulator Foxp3 that belongs to the family of X chromosomes. Foxp3 has been acted as a master switch gene for the differentiation, development, and phenotype of Tregs [23]because its mRNA and encoded protein are exclusively expressed on Tregs. But Foxp3 gene stable [24] and high expression level [25] are essential to these processes, whereas, mutations of Foxp3 gene caused by either spontaneous or experimental reasons may result in a series of autoimmune/inflammatory diseases [24,26-29]. In addition, the levels of Foxp3 might be involved in the extent of Treg suppressive activity [30]. It has been reported that Foxp3 regulates expression and function of interleukin (IL) 2, CD25, cytotoxic T lymphocyte antigen (CTLA) 4, and glucocorticoid-induced TNF receptor (GITR) which is critical for Treg [31]. Despite significant progresses have been made to reveal the mechanistic details of its contribution to Treg cells, the mechanism of Foxp3-mediated transcriptional regulationis still largely unknown.
It has been reported that the induction of Foxp3 gene is modulated by several regulatory regions including a promoter, two enhancers (Enhancer 1 and Enhancer 2), and conserved noncoding sequence (CNS) 1-3 [32]. Enhancer 1 is located in CNS1, Enhancer 2 is located in CNS2 in the intron of the Foxp3 gene, and while CNS3 is located downstream of exon1. Nuclear factor of activated T cells (NFAT) and Smad3 could interact with CNS1 to induce the expression of Foxp3 [33]. Activation of signal transducer and activator of transcription (STAT) 5 directly binds the promoter of Foxp3 to CNS2 to maintain Foxp3 expression [34]. Also, c-Relin conjunction with Foxp3 promoter or CNS3 plays a unique role in Foxp3 gene expression, leading to the open up of the Foxp3 locus in natural Treg (nTreg) precursor cells [32]. Additionally, several transcription factors such as NFAT and acuteleukemia-1 (AML1)/runt-related transcription factor 1 (Runx1) have shown to bind to Foxp3 promoter and potentially interacts with AP-1 and nuclear factor κB (NF-κB) [35,36]. Both T cell receptor (TCR) and TGF-β signals have been reported to induce the activation of AP-1 through mitogen-activated protein kinase (MAPK) pathway, transiently inducing the expression of Foxp3 [37,38]. To better elucidate the functional role of the Foxp3 gene in Treg development and differentiation, we further explore the molecular mechanisms of AP-1 on Foxp3 gene expression. In the present study, a sepsis mouse model was induced by using a CLP method. We found that a significant elevation in Treg cells following polymicrobial sepsis in the blood, which was in line with previous studies. Additionally, adenosine and pharmacological stimulation and inhibition of adenosine were performed to the purified CD4+CD+25Treg cells. In line with previous studies [39-41], the levels of Foxp3 were significantly increased by adenosine and its selective A2A receptor agonist CGS21680, but were decreased by adenosine antagonist CSC. The results implied that the existence of an AP-1 binding site in the Foxp3 promoter region was necessary to induce the expression of Foxp3. Then we explored the possible mechanism of adenosine on the expression of Foxp3 by investigation the relationships between the expression of Foxp3 and the activation of JNK or Smad3. The results confirmed that the expression of Foxp3 was associated with the activation of JNK but not associated with Smad3. To characterize the possible signaling pathway, we studied the levels of the transcription factor AP-1 after administration of different drugs. These results confirmed that the JNK pathway and AP-1 were involved in the effect of adenosine on the expression of Foxp3. Our study confirmed with previous studies. Mantel et al. identified the AP-1 binding sites in the FOXP3 promoter [37]. Xu et al. has found that AP-1 site located in a Foxp3 enhancer region plays a significant role in TCR/TGF-β induced Foxp3 gene expression [41]. Furthermore, we identified that c-Fos and c-Jun could bind to the Foxp3 promoter by using ChIP assay. Besides, the activity of c-Fos, Fra-2, c-Jun, or JunD promoted the expression of Foxp3 in CD4+CD25+Treg cells.
In conclusion, adenosine enhanced the high expression of Foxp3 by activation of JNK/AP-1, and followed by c-fos and c-Jun activation in the Foxp3 enhancer region.
Acknowledgements
This research was supported by Grant No. 81471895 and No. 81401575 from National Natural Science Foundation of China to Tao Yang and Rui Bao (Beijing, China).
Disclosure of conflict of interests
None.
References
- 1.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med. 2001;29:S109–116. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 3.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 4.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;15:496–497. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD 2nd, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306:2594–2605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busse M, Traeger T, Potschke C, Billing A, Dummer A, Friebe E, Kiank C, Grunwald U, Jack RS, Schutt C, Heidecke CD, Maier S, Broker BM. Detrimental role for CD4+ T lymphocytes in murine diffuse peritonitis due to inhibition of local bacterial elimination. Gut. 2008;57:188–195. doi: 10.1136/gut.2007.121616. [DOI] [PubMed] [Google Scholar]
- 7.Stromberg PE, Woolsey CA, Clark AT, Clark JA, Turnbull IR, McConnell KW, Chang KC, Chung CS, Ayala A, Buchman TG, Hotchkiss RS, Coopersmith CM. CD4+ lymphocytes control gut epithelial apoptosis and mediate survival in sepsis. FASEB J. 2009;23:1817–1825. doi: 10.1096/fj.08-119024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 9.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 10.Venet F, Chung CS, Monneret G, Huang X, Horner B, Garber M, Ayala A. Regulatory T cell populations in sepsis and trauma. J Leukoc Biol. 2008;83:523–535. doi: 10.1189/jlb.0607371. [DOI] [PubMed] [Google Scholar]
- 11.Bacchetta R, Gambineri E, Roncarolo MG. Role of regulatory T cells and FOXP3 in human diseases. J Allergy Clin Immunol. 2007;120:227–235. doi: 10.1016/j.jaci.2007.06.023. quiz 236-227. [DOI] [PubMed] [Google Scholar]
- 12.Chatila TA. Role of regulatory T cells in human diseases. J Allergy Clin Immunol. 2005;116:949–959. doi: 10.1016/j.jaci.2005.08.047. quiz 960. [DOI] [PubMed] [Google Scholar]
- 13.Ramsdell F. Foxp3 and natural regulatory T cells: key to a cell lineage? Immunity. 2003;19:165–168. doi: 10.1016/s1074-7613(03)00207-3. [DOI] [PubMed] [Google Scholar]
- 14.Leng FY, Liu JL, Liu ZJ, Yin JY, Qu HP. Increased proportion of CD4(+)CD25(+)Foxp3(+) regulatory T cells during early-stage sepsis in ICU patients. J Microbiol Immunol Infect. 2013;46:338–344. doi: 10.1016/j.jmii.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 15.Nascimento DC, Alves-Filho JC, Sonego F, Fukada SY, Pereira MS, Benjamim C, Zamboni DS, Silva JS, Cunha FQ. Role of regulatory T cells in long-term immune dysfunction associated with severe sepsis. Crit Care Med. 2010;38:1718–1725. doi: 10.1097/CCM.0b013e3181e78ad0. [DOI] [PubMed] [Google Scholar]
- 16.Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 17.Scumpia PO, Delano MJ, Kelly KM, O’Malley KA, Efron PA, McAuliffe PF, Brusko T, Ungaro R, Barker T, Wynn JL, Atkinson MA, Reeves WH, Salzler MJ, Moldawer LL. Increased natural CD4+CD25+ regulatory T cells and their suppressor activity do not contribute to mortality in murine polymicrobial sepsis. J Immunol. 2006;177:7943–7949. doi: 10.4049/jimmunol.177.11.7943. [DOI] [PubMed] [Google Scholar]
- 18.Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock--a review of laboratory models and a proposal. J Surg Res. 1980;29:189–201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 19.Munford RS, Pugin J. Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am J Respir Crit Care Med. 2001;163:316–321. doi: 10.1164/ajrccm.163.2.2007102. [DOI] [PubMed] [Google Scholar]
- 20.Kessel A, Bamberger E, Masalha M, Toubi E. The role of T regulatory cells in human sepsis. J Autoimmun. 2009;32:211–215. doi: 10.1016/j.jaut.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 21.Cao C, Ma T, Chai YF, Shou ST. The role of regulatory T cells in immune dysfunction during sepsis. World J Emerg Med. 2015;6:5–9. doi: 10.5847/wjem.j.1920-8642.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, Monneret G. Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25- lymphocytes. Crit Care Med. 2004;32:2329–2331. doi: 10.1097/01.ccm.0000145999.42971.4b. [DOI] [PubMed] [Google Scholar]
- 23.Ziegler SF, Buckner JH. Influence of FOXP3 on CD4+CD25+ regulatory T cells. Expert Rev Clin Immunol. 2006;2:639–647. doi: 10.1586/1744666X.2.4.639. [DOI] [PubMed] [Google Scholar]
- 24.Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 25.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 26.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 27.Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- 28.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 29.Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006;24:209–226. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
- 30.Campbell DJ, Ziegler SF. FOXP3 modifies the phenotypic and functional properties of regulatory T cells. Nat Rev Immunol. 2007;7:305–310. doi: 10.1038/nri2061. [DOI] [PubMed] [Google Scholar]
- 31.Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, Lang S, Jackson EK, Gorelik E, Whiteside TL. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem. 2010;285:7176–7186. doi: 10.1074/jbc.M109.047423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 34.Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, Laurence A, Robinson GW, Shevach EM, Moriggl R, Hennighausen L, Wu C, O’Shea JJ. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 36.Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- 37.Mantel PY, Ouaked N, Ruckert B, Karagiannidis C, Welz R, Blaser K, Schmidt-Weber CB. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol. 2006;176:3593–3602. doi: 10.4049/jimmunol.176.6.3593. [DOI] [PubMed] [Google Scholar]
- 38.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, Drake CG, Powell JD. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111:251–259. doi: 10.1182/blood-2007-03-081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bynoe MS, Viret C. Foxp3+CD4+ T cell-mediated immunosuppression involves extracellular nucleotide catabolism. Trends Immunol. 2008;29:99–102. doi: 10.1016/j.it.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 41.Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity. 2010;33:313–325. doi: 10.1016/j.immuni.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]