

Abstract
In the endometrium transforming growth factor-betas (TGF-βs) are involved mainly in menstruation and endometriosis. After binding of the ligands to the high-affinity receptors, TGF-β receptors (TBR1 and TBR2), TGF-βs activate Smad signaling to modulate gene expression and cellular functions. However, recently also Smad-independent pathways have been studied in more details. To evaluate both pathways, we have analyzed TGF-β signaling in human endometrial and endometriotic cells. Although endometrial and endometriotic cells secrete TGF-β1, secretion by stromal cells was higher compared to epithelial cells. In contrast, secretion of TGF-β2 was higher in endometriotic stromal and endometriotic epithelial cells compared to normal endometrial cells. Treatment of endometrial and endometriotic stromal and epithelial cells with TGF-β1 or TGF-β2 increased Smad-dependent secretion of plasminogen activator inhibitor-1 (PAI-1) dramatically in all three cell lines. Of note, endometriotic cells secreted clearly higher levels of PAI-1 compared to endometrial cells. Whereas a TBR1 kinase inhibitor completely blocked the TGF-β1 or TGF-β2-induced PAI-1 secretion, an ERK1/2 inhibitor only partially reduced PAI-1 secretion. This inhibition was not dependent on epidermal growth factor receptor (EGFR) activation by phosphorylation but on kinase activity of the TBR1. Finally, treatment of endometrial and endometriotic cell lines with recombinant PAI-1 showed reduced cell adhesion, especially of the endometrial cells. In summary, our results demonstrate that both Smad-dependent and TBR1-dependent ERK1/2 pathways are necessary for TGF-β-dependent high level secretion of PAI-1, which might increase cellular deadhesion.
Keywords: PAI-1, ERK, SMAD, TGF-betas, endometriosis
Introduction
TGF-βs are one of the most essential growth factors involved in cell differentiation, proliferation, motility, cancer and apoptosis in various cell types [1]. The Smad-dependent pathway is the main pathway of TGF-β signaling and is activated by phosphorylation of TBR1 which in turn phosphorylates Smad2/3 [2]. Additionally also Smad-independent pathways modulate signal transduction of TGF-βs (e.g. [3,4]).
In the human endometrium TGF-βs are differentially and stage-specifically expressed [5]. The highest expression of TGF-βs was detected during menses [6] and TGF-β1 is also elevated in serum and peritoneal fluid of women with endometriosis [7,8]. Furthermore, TGF-β1 is also differentially expressed in ovarian endometriosis and might play a role as an activator [9].
Endometriosis affects 10% of the female population in their reproductive ages [10] and is characterized by endometrial-like tissue growing outside the uterine cavity, mostly in the ovary and the peritoneum [11]. Besides animal models to study endometriosis (e.g. [12]), endometriotic cell lines with characteristics comparable to primary endometriotic cells have been established [13,14]. Of note, these cell lines offer the possibility to study epithelial cells, which are otherwise rarely analyzed in the research of endometriosis in vitro.
In endometrial cells, TGF-βs showed different effects on cell proliferation. TGF-β1 stimulated proliferation of low epithelial cell numbers, but inhibited it at high cell numbers in women with and without endometriosis [15]. However, all three TGF-β isoforms inhibited proliferation of endometrial stromal cells in vitro [16].
In a nude mouse model, preincubation of endometrial tissue with TGF-β1 together with progesterone before xenografting suppressed endometriosis-like lesion formation [17]. Possibly, TGF-β1 restored the ability of progesterone to suppress matrix metalloproteinases (MMPs) and thus prevented the establishment of endometriosis. However, TGF-β1 knockout mice on a background of SCID showed reduced lesion development of xenotransplanted human endometriotic tissue [18].
Remarkably, TGF-βs, especially TBR1, are also involved in myometrial development [19,20]. Furthermore, TGF-βs induced contraction of endometrial stromal cells in vitro [16], which might contribute to abnormal myometrial contractions found in women with endometriosis [21,22] probably resulting in increased dissemination of endometrial fragments.
In this study we aimed to investigate the TGF-β signaling pathways in endometrial and endometriotic cells to identify possible targets which might be involved in the pathology of endometriosis.
Materials and methods
Cell lines
The stromal T-HESC cells ([23] ATCC CRL-4003) have been isolated from normal endometrium and demonstrate typical endometrial characteristics [24]. The stromal cells 22B and epithelial cells 12Z have been isolated from active peritoneal endometriotic lesions ([14] generously provided by Dr. Starzinski-Powitz, Frankfurt, Germany). The cell lines show characteristics of the active phase of endometriosis and thus are suitable for studying cellular and molecular behaviour of endometriosis [24].
Cell culture
3x105 cells were seeded into 6-well plates (TPP, Switzerland) in DMEM high glucose or DMEM/F12 media (+ 10% FCS). After culturing overnight (37°C, 5% CO2), cells were starved in fresh medium (+ 1% FCS) for 6 hours. After removal of the old medium, fresh medium containing 10 ng/ml recombinant human (rh)-TGF-β1 or rh-TGF-β2 (Promokine, Germany), respectively was added. In the untreated controls only 1x PBS was added to the medium. Cells were cultured (37°C, 5% CO2) for up to three days.
To investigate the pathways that might be involved in TGF-β signaling with respect to PAI-1 secretion, several inhibitors targeting different pathways were used: the TGF-β receptor type I kinase inhibitor: 5 µM LY364947 (Sigma-Aldrich, USA [25]) and 5 µM of the ERK inhibitor II (Merck, Germany [26]) dissolved in DMSO. The optimal dose was determined in prerun experiments. Inhibitors for other pathways like p38 MAPK (SB203580), PI3K (LY 294002), pKA (H-89) or JNK (JNK inhibitor II) were less effective on PAI-1 secretion.
Fresh media (+ 1% FCS) in 6-well with or without inhibitor(s) was added. The untreated controls were done with DMSO as vehicle. After an incubation of 2 hours (37°C, 5% CO2), cells were stimulated with 10 ng/ml TGF-β1 or TGF-β2 as described above.
Supernatants were collected and mixed with a Protease Inhibitor cocktail (Sigma-Aldrich, USA). After centrifugation (5000x g, 10 min, 4°C) the supernatants were aliquoted and stored at -20°C until use in the ELISAs. Then cell numbers were determined as described below.
Cell numbers
After removal of the medium, cells were washed two times with 1x Dulbecco’s PBS with Ca2+ and Mg2+. Then accutase was added at 37°C until all cells were detached. After adding fresh medium, 10 μl of the cell suspension was transferred to a CASY tube with 10 ml CASY ton solution and mixed thoroughly. Then the cell numbers and cell viability were measured with a CASY-counter (Schaerfe System, Germany).
Cell adhesion assay
1×106 cells/ml were seeded in 6-well plates and treated with active recombinant human-PAI-1 (final concentrations of 40 nM, 20 nM, 10 nM or 5 nM). The buffer (50 mM NaH2PO4, 150 mM NaCl, 1 mM EDTA, pH 6.6) used to dissolve PAI-1 was used for the untreated control. After 4 hours (37°C, 5% CO2) the cell numbers were quantitated with the CASY Counter.
ELISAs
Quantitation of protein secretion was performed with the following ELISAs: PAI-1 Antigen ELISA Kit (Technoclone), and TGF-β1 and TGF-β2 DuoSets (both from R&D Systems). Latent TGF-β isoforms were activated with HCl followed by neutralization with NaOH as indicated by the supplier. Each ELISA was performed according to the manufacturer’s instructions and quantitated with the Benchmark Reader infinite M2000 (Tecan). Cell numbers were used for standardization.
Statistical analysis
Each experiment was repeated independently at least three times in duplicate. Values from all experiments were used to calculate the means and the respective errors of the mean (SEM). Comparison between groups was done with the ANOVA followed by the post-hoc tests of Tukey and Dunnett by using GraphPad Instat 3 (GraphPad). p values less than 0.05 were considered statistically significant.
Results
To study endometrial and endometriotic cells in vitro, three cell lines from the human eutopic and ectopic endometrium were used. All cell lines secreted TGF-β1 (Figure 1A) as well as TGF-β2 (Figure 1B). The stromal cell lines (T-HESC and 22B) secreted higher levels compared to the epithelial cell line 12Z (Figure 1A). Interestingly, the endometriotic cell lines 22B and 12Z secreted more TGF-β2 than the normal endometrial cells (Figure 1B). All cell lines only secreted very low levels of TGF-β3 (data not shown). In the next step, we investigated secretion of the TGF-β-regulated protein PAI-1. In contrast to the epithelial cell line 12Z the stromal cell lines produced high amounts of PAI-1 (Figure 1C). The endometriotic stromal cells 22B secreted approx. 4-fold higher levels compared to the endometrial stromal T-HESC. In contrast the endometriotic epithelial 12Z cells secreted only modest amounts of PAI-1. Taken together, PAI-1 secretion of the stromal cells is much higher compared to the epithelial cells, whereas endometriotic cells secrete higher amounts of TGF-βs than the normal endometrial cells.
Figure 1.
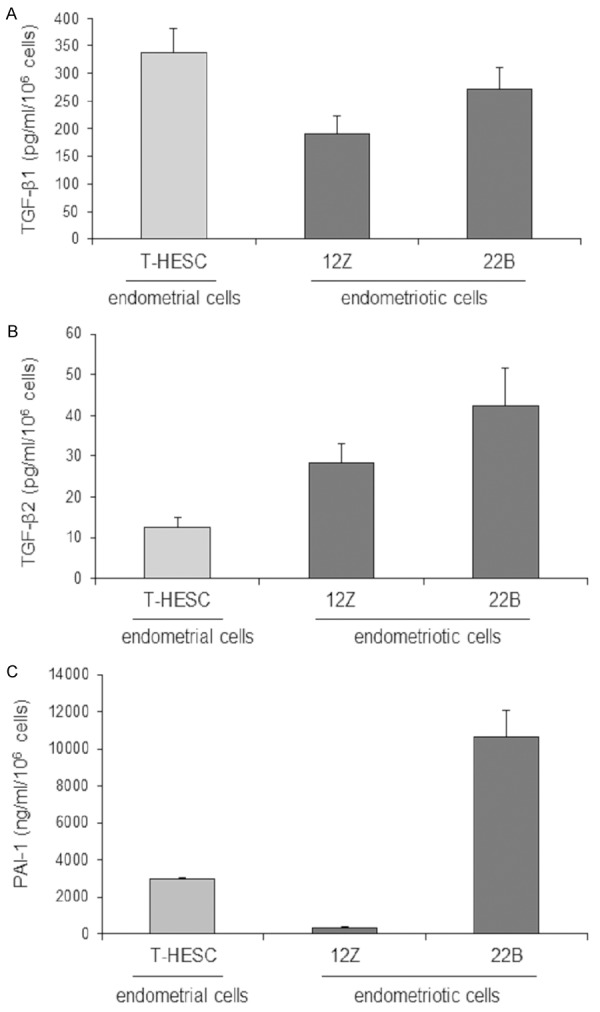
Quantification of TGF-β1, TGF-β2 and PAI-1 secretion by all cell lines. Stromal cell lines T-HESC and 22B secreted higher levels of TGF-β1 compared to epithelial cells 12Z (A). Endometriotic cell lines 22B and 12Z secreted more TGF-β2 compared to endometrial cells T-HESC (B). Endometriotic stromal cells 22B secreted 4-fold higher levels compared to endometrial stromal cells T-HESC. Endometriotic epithelial cells 12Z secreted modest amounts of PAI-1 (C). Each experiment was repeated three times in duplicates (A+B); each experiment was repeated nine times in duplicates (C).
In order to elucidate the signaling pathways involved in secretion of PAI-1, we analyzed Smad-dependent and Smad-independent pathways with pharmacological inhibitors. Our results showed that both TGF-β1 and TGF-β2 strongly increased secretion of PAI-1 in all cell lines studied (Figure 2). The inhibitor LY364947 was described to inhibit phosphorylation of Smad2 and Smad3 by blocking the kinase activity of TBR1 [27]. Addition of LY364947 to the cells together with TGF-β1 or TGF-β2 strongly decreased PAI-1 secretion to levels comparable to the untreated controls (Figure 2), demonstrating that mainly the Smad pathway is responsible for TGF-β-induced PAI-1 secretion in endometrial cells.
Figure 2.
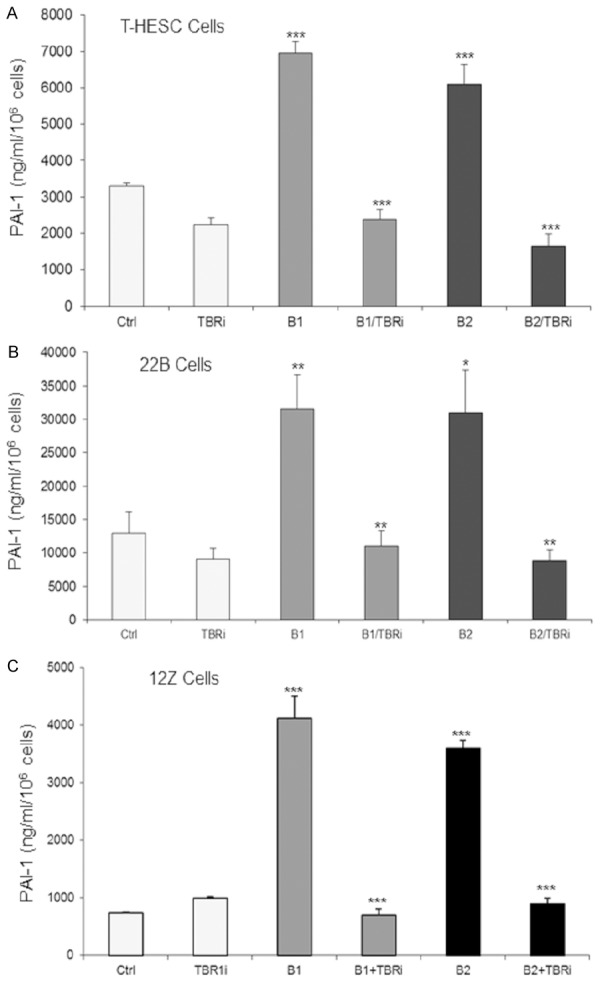
Treatment of cells with TGF-β1 or TGF-β2 (10 ng/ml), respectively, induced PAI-1 secretion in endometrial stromal T-HESC cells (A), endometriotic stromal 22B cells (B) and endometriotic epithelial 12Z cells (C). The TBR1 inhibitor (LY364947) blocked TGF-β-induced PAI-1 secretion of all cell lines studied completely to control levels (***P<0.001; **P<0.01; *P≤0.05). Each experiment was repeated three times in duplicates. Ctrl, control; TBRi; TBR1 inhibitor; B1, TGF-β1; B2, TGF-β2.
Analysis of several Smad-independent pathway inhibitors as indicated in the Materials and Methods section revealed only for the ERK pathway a significant contribution to secretion of PAI-1 (Figure 3). Our results showed that the TGF-β1-induced or TGF-β2-induced PAI-1 secretion was reduced by a highly selective ERK1/2 inhibitor by 35-50% in the cell lines studied. Furthermore, our results showed that stimulation with TGF-β1 or TGF-β2 strongly enhanced phosphorylation of ERK1/2 in all three cell types studied. However, we found that the EGF receptor is not phosphorylated by the TGF-βs (data not shown).
Figure 3.
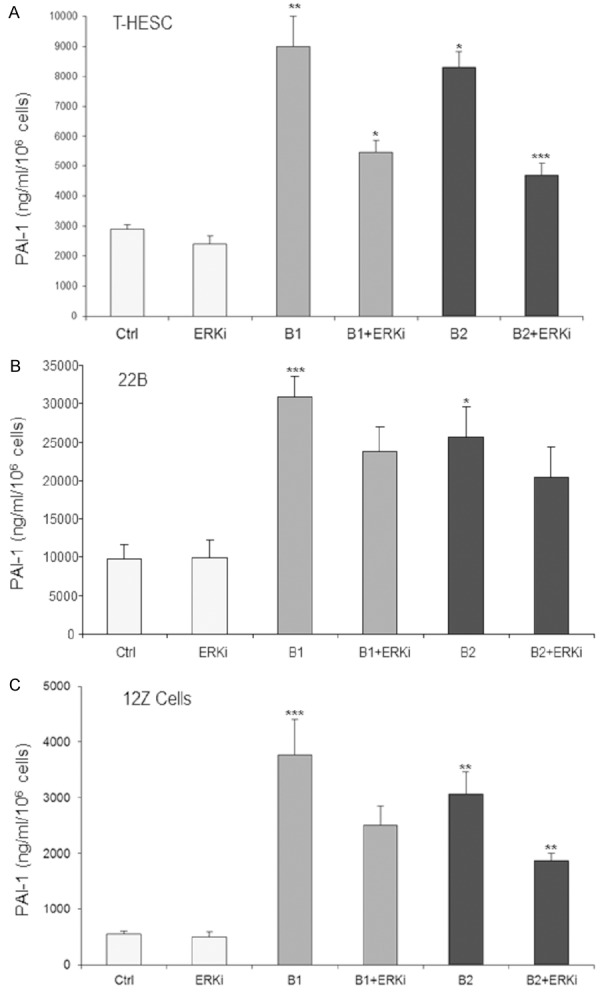
TGF-β1 or TGF-β2 induced PAI-1 secretion was moderately but significantly inhibited by the ERK inhibitor (ERKi) in endometrial stromal T-HESC cells (A), endometriotic stromal 22B cells (B), and endometriotic epithelial 12Z cells (C) by 50%, 30%, and 40%, respectively (**P<0.001, **P<0.01; *P<0.05). Each experiment was repeated three times in duplicates. Ctrl, control; B1, TGF-β1; B2, TGF-β2.
Besides effects of PAI-1 on activation of plasmin, it was also reported that PAI-1 inhibited cell attachment to the extracellular matrix (ECM [28]). In order to test this hypothesis, cells were stimulated with or without recombinant PAI-1 for four hours and then the cell numbers were counted. PAI-1 decreased the number of adherent cells in a dose-dependent manner (Figure 4). Interestingly PAI-1 demonstrated stronger inhibitory effects on the endometrial cell line T-HESC compared to the endometriotic cell lines 22B and 12Z. In addition to cell numbers, the CASY counter also measures cell vitality, which however was similar in all cell lines studies. Thus, we could exclude reduced cell adhesion by cell death.
Figure 4.
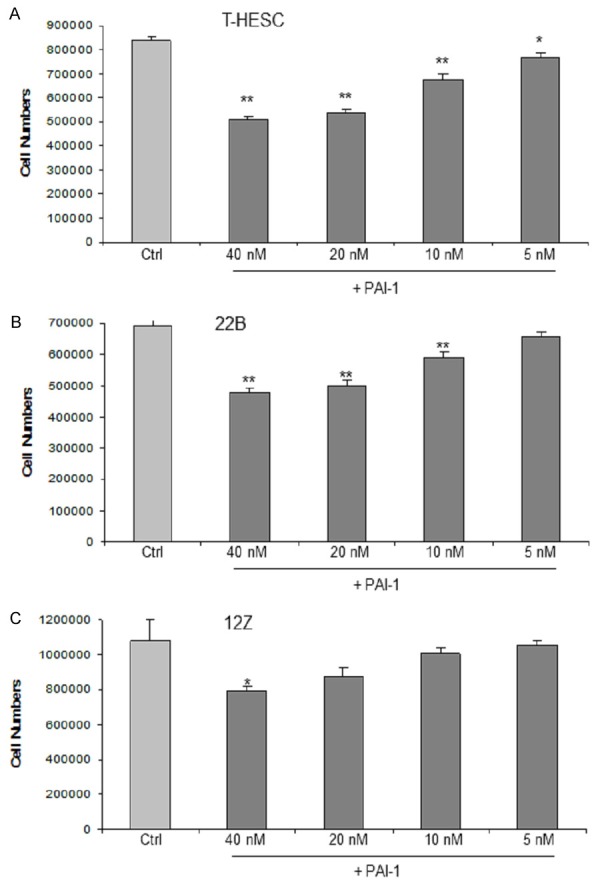
PAI-1 dose dependently inhibited cell adhesion of endometrial stromal T-HESC cells (A), endometriotic stromal 22B cells (B), and endometriotic epithelial 12Z cells (C). The numbers of attached cells decreased in a range of 2-40% after treatment for four hours with various concentrations of recombinant PAI-1 (***P<0.001, **P<0.01, *P<0.05). Each experiment was repeated three times in triplicates.
Discussion
In our study, we quantified secretion of TGF-β1 and TGF-β2 by endometrial and endometriotic cell lines and demonstrated that TGF-β1 secretion was higher in stromal cells in comparison to the epithelial cells. Of note, endometriotic cell lines secreted considerably higher levels of TGF-β2 compared to normal endometrial cell lines. Our results concur with those of Pizzo et al. [7] who reported increased TGF-β2 secretion by endometrial cells at advanced stages of endometriosis. Similarily, Chegini et al. [29] observed increased TGF-β2 secretion in surgically induced endometriosis.
Besides the main function of PAI-1 to inhibit the tissue/urokinase plasminogen activator (tPA/uPA) induced fibrinolysis, PAI-1 is important also in adhesion, migration, signal transduction and anti-apoptosis [30,31]. In endometrial and endometriotic tissues, Bruse et al. [32,33] showed that expression of PAI-1 and uPA is much higher in endometriotic and endometrial tissue of women with endometriosis, compared to women without endometriosis. Also PAI-1 levels were higher in ectopic tissues than in eutopic tissues of the same woman [33].
Although there are many genes targeted by TGF-βs, PAI-1 is one of the most highly upregulated [34]. Accordingly we found a Smad-dependent increased secretion of PAI-1 in endometrial and endometriotic cells upon stimulation with TGF-β1 or TGF-β2 in vitro. Our results showed clearly that endometriotic cells secreted more PAI-1 than the endometrial cells, and stromal cells (endometrial and endometriotic) secreted considerably more PAI-1 compared to epithelial cells. Our results concur with those of Bruse et al. [33] who showed that PAI-1 is mainly produced by endometrial endothelial and stromal cells.
To investigate possible Smad-independent pathways, we used several inhibitors, but found only for ERK a moderate but significant downregulation in TGF-β1- and TGF-β2-induced secretion of PAI-1 in all cell lines. Thus, besides Smad signaling, the ERK pathway contributes to high level secretion of PAI-1 in endometrial and endometriotic cells. Interestingly, there are several connections between the TGF-β-induced Smad pathway and the ERK pathway [35], for example the small GTPase Ras and the ERKs are implicated in TGF-β signaling [3]. Similarly, TGF-β phosphorylated ERK Ras-dependently and through activation of p21 activated kinase-2 [36]. Furthermore activated TBR1 directly phosphorylated ShcA (Src homology 2 domain containing) inducing association with GRB2 (EGF receptor-bound protein-2) and SOS (son of sevenless), which then leads to a sequential activation of Ras, Raf, MEK1/2 and ERK1/2 [37]. These data suggest that ERKs can be activated directly by the TBR1 without involvement of the Smad pathway or EGF receptor phosphorylation, the latter is normally required for phosphorylation of ERKs [38]. In this study, we did not observe phosphorylation of EGF receptors after TGF-β stimulation of endometrial and endometriotic cells (data not shown).
Czekay et al. [39] showed that PAI-1 lowered attachment of cells to the ECM through inhibition of uPAR-vitronectin interaction, suggesting that PAI-1 negatively affects the cell-to-ECM connection. We found that recombinant PAI-1 reduced cell attachment in all cell lines studied. This effect is not due to increased apoptosis, because PAI-1 is nearly exclusively described as an anti-apoptotic protein [31]. Of note, in our study PAI-1 demonstrated stronger inhibitory effects on endometrial compared to endometriotic cells. Thus, we suggest that PAI-1 might play a role in endometriosis by facilitating detachment of endometrial cells.
In conclusion, we showed in this study that mainly stromal cells secrete TGF-βs and PAI-1, which in the case of PAI-1 is regulated Smad-dependently and Smad-independently by ERKs. However, we could also show that epithelial cells contribute to secretion of TGF-βs and PAI-1 albeit only moderately. The connection of the ERK pathway to TGF-β signaling as suggested in this study seems to depend only on the kinase activity of the TBR1. Moreover, it is striking that we found no apparent differences in cell signaling between endometrial and endometriotic cells. However, we hypothesize that increased TGF-β and PAI-1 levels as observed in women with endometriosis might possibly contribute to endometriosis via enhanced cell shedding of endometrial cells by increased PAI-1 levels, a hypothesis which warrants further investigations.
Acknowledgements
We thank Dorina Zoltan for excellent technical assistance.
Disclosure of conflict of interest
None.
References
- 1.Ikushima H, Miyazono K. Biology of transforming growth factor-β signalling. Curr Pharm Biotechnol. 2011;12:2099–2107. doi: 10.2174/138920111798808419. [DOI] [PubMed] [Google Scholar]
- 2.Massagué J, Wotton D. Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yue J, Mulder KM. Requirement of Ras/MAPK pathway activation by transforming growth factor β for transforming growth factor β1 production in a Smad-dependent pathway. J Biol Chem. 2000;275:30765–30773. doi: 10.1074/jbc.M000039200. [DOI] [PubMed] [Google Scholar]
- 4.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Omwandho CO, Konrad L, Halis G, Oehmke F, Tinneberg HR. Role of TGF-βs in normal human endometrium and endometriosis. Hum Reprod. 2010;25:101–109. doi: 10.1093/humrep/dep382. [DOI] [PubMed] [Google Scholar]
- 6.Gaide Chevronnay HP, Cornet PB, Delvaux D, Lemoine P, Courtoy PJ, Henriet P, Marbaix E. Opposite regulation of transforming growth factors-β2 and -β3 expression in the human endometrium. Endocrinology. 2008;149:1015–1025. doi: 10.1210/en.2007-0849. [DOI] [PubMed] [Google Scholar]
- 7.Pizzo A, Salmeri FM, Ardita FV, Sofo V, Tripepi M, Marsico S. Behaviour of cytokine levels in serum and peritoneal fluid of women with endometriosis. Gynecol Obstet Invest. 2002;54:82–87. doi: 10.1159/000067717. [DOI] [PubMed] [Google Scholar]
- 8.Young VJ, Brown JK, Saunders PT, Duncan WC, Horne AW. The peritoneum is both a source and target of TGF-β in women with endometriosis. PLoS One. 2014;9:e106773. doi: 10.1371/journal.pone.0106773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vehmas AP, Muth-Pawlak D, Huhtinen K, Saloniemi-Heinonen T, Jaakkola K, Laajala TD, Kaprio H, Suvitie PA, Aittokallio T, Siitari H, Perheentupa A, Poutanen M, Corthals GL. Ovarian endometriosis signatures established through discovery and directed spectrometry analysis. J Proteome Res. 2014;13:4983–4994. doi: 10.1021/pr500384n. [DOI] [PubMed] [Google Scholar]
- 10.Bulun SE. Endometriosis. N Engl J Med. 2009;360:268–279. doi: 10.1056/NEJMra0804690. [DOI] [PubMed] [Google Scholar]
- 11.Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364:1789–1799. doi: 10.1016/S0140-6736(04)17403-5. [DOI] [PubMed] [Google Scholar]
- 12.Becker CM, Beaudry P, Funakoshi T, Benny O, Zaslavsky A, Zurakowski D, Folkman J, D’Amato RJ, Ryeom S. Circulating endothelial progenitor cells are up-regulated in a mouse model of endometriosis. Am J Pathol. 2011;178:1782–1791. doi: 10.1016/j.ajpath.2010.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starzinski-Powitz A, Gaetje R, Zeitvogel A, Kotzian S, Handrow-Metzmacher H, Herrmann G, Fanning E, Baumann R. Tracing cellular and molecular mechanisms involved in endometriosis. Hum Reprod Update. 1998;4:724–729. doi: 10.1093/humupd/4.5.724. [DOI] [PubMed] [Google Scholar]
- 14.Zeitvogel A, Baumann R, Starzinski-Powitz A. Identification of an invasive, N-cadherin-expressing epithelial cell type in endometriosis using a new cell culture model. Am J Pathol. 2001;159:1839–1852. doi: 10.1016/S0002-9440(10)63030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meresman GF, Bilotas M, Buquet RA, Baranao RI, Sueldo C, Tesone M. Gonadotropin-releasing hormone agonist induces apoptosis and reduces cell proliferation in eutopic endometrial cultures from women with endometriosis. Fertil Steril. 2003;80:702–707. doi: 10.1016/s0015-0282(03)00769-6. [DOI] [PubMed] [Google Scholar]
- 16.Nasu K, Nishida M, Matsumoto H, Bing S, Inoue C, Kawano Y, Miyakawa I. Regulation of proliferation, motility and contractivity of cultured human endometrial stromal cells by transforming growth factor-β isoforms. Fertil Steril. 2005;84(Suppl 2):1114–1123. doi: 10.1016/j.fertnstert.2005.02.055. [DOI] [PubMed] [Google Scholar]
- 17.Bruner-Tran KL, Eisenberg E, Yeaman GR, Anderson TA, McBean J, Osteen KG. Steroid regulation of matrix metalloproteinase expression in endometriosis and the establishment of experimental endometriosis in nude mice. J Clin Endocrinol Metab. 2002;87:4782–4791. doi: 10.1210/jc.2002-020418. [DOI] [PubMed] [Google Scholar]
- 18.Hull ML, Johan MZ, Hodge WL, Robertson SA, Ingman WV. Host derived TGF-β1 deficiency suppresses lesion development in a mouse model of endometriosis. Am J Pathol. 2012;180:880–887. doi: 10.1016/j.ajpath.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 19.Li Q, Agno JE, Edson MA, Nagaraja AK, Nagashima T, Matzuk MM. Transforming growth factor β receptor type 1 is essential for female reproductive tract integrity and function. PLoS Genetics. 2011;7:e1002320. doi: 10.1371/journal.pgen.1002320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Y, Bayless KJ, Li Q. TGFBR1 is required for mouse myometrial development. Mol Endocrinol. 2014;28:380–394. doi: 10.1210/me.2013-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulletti C, de Ziegler D, Polli V, Del Ferro E, Palini S, Flamigni C. Characteristics of uterine contractility during menses in women with mild to moderate endometriosis. Fertil Steril. 2002;77:1156–1161. doi: 10.1016/s0015-0282(02)03087-x. [DOI] [PubMed] [Google Scholar]
- 22.Bulletti C, de Ziegler D, Setti PL, Cicinelli E, Polli V, Flamigni C. The patterns of uterine contractility in normal menstruating women: from physiology to pathology. Ann N Y Acad Sci. 2004;1034:64–83. doi: 10.1196/annals.1335.007. [DOI] [PubMed] [Google Scholar]
- 23.Krikun G, Mor G, Alvero A, Guller S, Schatz F, Sapi E, Rahman M, Caze R, Qumsiyeh M, Lockwood CJ. A novel immortalized human endometrial stromal cell line with normal progestational response. Endocrinology. 2004;145:2291–2296. doi: 10.1210/en.2003-1606. [DOI] [PubMed] [Google Scholar]
- 24.Lee J, Banu SK, Rodriguez R, Starzinski-Powitz A, Arosh JA. Selective blockade of prostaglandin E2 receptors EP2 and EP4 signaling inhibits proliferation of human endometriotic epithelial cells and stromal cells through distinct cell cycle arrest. Fertil Steril. 2010;93:2498–2506. doi: 10.1016/j.fertnstert.2010.01.038. [DOI] [PubMed] [Google Scholar]
- 25.Sawyer JS, Anderson BD, Beight DW, Campbell RM, Jones ML, Herron DK, Lampe JW, McCowan JR, McMillen WT, Mort N, Parsons S, Smith EC, Vieth M, Weir LC, Yan L, Zhang F, Yingling JM. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J Med Chem. 2003;46:3953–3956. doi: 10.1021/jm0205705. [DOI] [PubMed] [Google Scholar]
- 26.Ohori M, Kinoshita T, Okubo M, Sato K, Yamazaki A, Arakawa H, Nishimura S, Inamura N, Nakajima H, Neya M, Miyake H, Fujii T. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem Biophys Res Commun. 2005;336:357–363. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]
- 27.Peng SB, Yan L, Xia Y, Watkins SA, Brooks HB, Beight D, Herron DK, Jones ML, Lampe JW, McMillen WT, Mort N, Sawyer JS, Yingling JM. Kinetic characterization of novel pyrazole TGF-β receptor I kinase inhibitors and their blockade of the epithelial-mesenchymal transition. Biochemistry. 2005;44:2293–2304. doi: 10.1021/bi048851x. [DOI] [PubMed] [Google Scholar]
- 28.Czekay RP, Aertgeerts K, Curriden SA, Loskutoff DJ. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol. 2003;160:781–791. doi: 10.1083/jcb.200208117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chegini N, Zhao Y, Williams RS, Flanders KC. Human uterine tissue throughout the menstrual cycle expresses transforming growth factor-β1 (TGFβ1), TGFβ2, TGFβ3, and TGFβ type II receptor messenger ribonucleic acid and protein and contains [125I] TGFβ1-binding sites. Endocrinology. 1994;135:439–449. doi: 10.1210/endo.135.1.8013382. [DOI] [PubMed] [Google Scholar]
- 30.Binder BR, Christ G, Gruber F, Grubic N, Hufnagl P, Krebs M, Mihaly J, Prager GW. Plasminogen activator inhibitor 1: physiological and pathophysiological roles. News Physiol Sci. 2002;17:56–61. doi: 10.1152/nips.01369.2001. [DOI] [PubMed] [Google Scholar]
- 31.Kwaan HC, McMahon B. The role of plasminogen-plasmin system in cancer. Cancer Treat Res. 2009;148:43–66. doi: 10.1007/978-0-387-79962-9_4. [DOI] [PubMed] [Google Scholar]
- 32.Bruse C, Bergqvist A, Carlström K, Fianu-Jonasson A, Lecander I, Astedt B. Fibrinolytic factors in endometriotic tissue, endometrium, peritoneal fluid, and plasma from women with endometriosis and in endometrium and peritoneal fluid from healthy women. Fertil Steril. 1998;70:821–826. doi: 10.1016/s0015-0282(98)00285-4. [DOI] [PubMed] [Google Scholar]
- 33.Bruse C, Radu D, Bergqvist A. In situ localization of mRNA for the fibrinolytic factors uPA, PAI-1 and uPAR in endometriotic and endometrial tissue. Mol Hum Reprod. 2004;10:159–166. doi: 10.1093/molehr/gah033. [DOI] [PubMed] [Google Scholar]
- 34.Ha H, Oh EY, Lee HB. The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nat Rev Nephrol. 2009;5:203–211. doi: 10.1038/nrneph.2009.15. [DOI] [PubMed] [Google Scholar]
- 35.Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the ERK pathway is required for TGF-β1-induced EMT in vitro. Neoplasia. 2004;6:603–610. doi: 10.1593/neo.04241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki K, Wilkes MC, Garamszegi N, Edens M, Leof EB. Transforming growth factor beta signaling via Ras in mesenchymal cells requires p21-activated kinase 2 for extracellular signal-regulated kinase-dependent transcriptional responses. Cancer Res. 2007;67:3673–3682. doi: 10.1158/0008-5472.CAN-06-3211. [DOI] [PubMed] [Google Scholar]
- 37.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957–3967. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samarakoon R, Higgins SP, Higgins CE, Higgins PJ. TGF-β1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60(c-src)/EGFR(Y845) and Rho/ROCK signaling. J Mol Cell Cardiol. 2008;44:527–538. doi: 10.1016/j.yjmcc.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Czekay RP, Wilkins-Port CE, Higgins SP, Freytag J, Overstreet JM, Klein RM, Higgins CE, Samarakoon R, Higgins PJ. PAI-1: An integrator of cell signaling and migration. Int J Cell Biol. 2011;231:462–481. doi: 10.1155/2011/562481. [DOI] [PMC free article] [PubMed] [Google Scholar]