

Abstract
Transforming growth factor-beta (TGFβ) signaling in cancer is context dependent and acts either as a tumor suppressor or a tumor promoter. Loss of function mutation in TGFβ type II receptor (TβRII) is a frequent event in oral cavity squamous cell carcinoma (SCC). Recently, heterogeneity of TGFβ response has been described at the leading edge of SCC and this heterogeneity has been shown to influence stem cell renewal and drug resistance. Because exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways we investigated whether exosomes contain components of the TGFβ signaling pathway and whether exosome transfer between stromal fibroblasts and tumor cells can influence TGFβ signaling in SCC. We demonstrate that exosomes purified from stromal fibroblasts isolated from patients with oral SCC contains TβRII. We also demonstrate that transfer of fibroblast exosomes increases TGFβ signaling in SCC keratinocytes devoid of TβRII which remain non-responsive to TGFβ ligand in the absence of exosome transfer. Overall our data show that stromal communication with tumor cells can direct TGFβ signaling in SCC.
Keywords: Exosomes, transforming growth factor-beta, squamous cell carcinoma, tumor microenvironment
Introduction
Squamous cell carcinoma (SCC) arises in tissues that provide a barrier between an organism and the environment, such as the skin, oral cavity, esophagus and lung. SCC comprises over 90% of cancers of the head and neck and can occur in skin and the squamous lining of the mucosal surfaces of the upper aerodigestive tract, including the oral cavity, pharynx, larynx, and sinonasal tract [1]. Head and neck SCC (HNSCC) is the sixth most common cancer in the world, and it is anticipated that over 52,000 people are diagnosed in US each year [2,3]. Despite new therapies and improved risk stratification, overall survival in subjects with HNSCC is poor and only 50-60% of patients diagnosed with HNSCC are alive after 5 years [4,5]. Thus, HNSCC is a major clinical problem.
Transforming growth factor-beta (TGFβ) receptors participate in a signaling pathway that controls many aspects of mammalian development and tissue homeostasis [6]. The so called canonical TGFβ signaling pathway begins with binding of ligand (TGFβ1-3) to the type II receptor (TβRII) which in turn recruits the type I receptor (TβRI) leading to transphosphorylation of the resulting heterotetramer. This activated complex then recruits SMAD2 and SMAD3 which are themselves phosphorylated, leading to nuclear translocation and subsequent gene expression [7].
The role of TGFβ in SCC is controversial [8]; in murine studies of skin SCC a paradox emerges whereby TGFβ expression in the epidermis inhibits papilloma formation yet accelerates malignant conversion [9]. Furthermore, conditional ablation of TβRII induces cancer raising important questions about the timing and response to TGFβ signaling in tumor development [10,11]. In humans, mutation in TβRI leads to Ferguson-Smith syndrome, a familial disorder characterized by multiple, regressing SCC or keratoacanthomas of the skin [12]. Mutations in TβRI have also been identified in rapidly arising keratoacanthomas and skin SCC in patients receiving the broad kinase inhibitor sorafenib [13] and patients receiving the anti-TGFβ antibody, fresolimumab also frequently develop keratoacanthomas and SCC [14]. Moreover, The Cancer Genome Atlas Network identified sporadic inactivating mutations and deletions in the gene encoding TβRII in HNSCC, primarily oral cavity tumors [15]. Collectively these human clinical observations support the notion that inactivation of TGFβ leads to SCC initiation and yet TGFβ remains an important signaling pathway for cancer progression [8,9].
Recently it has been shown that response to TGFβ at the tumor stroma interface contributes to SCC progression and drug resistance [16] raising the possibility that TGFβ signaling might be influenced by intercellular communication between tumor and the microenvironment, a mechanism shown to promote therapy resistance in other cancers [17]. Local or systemic cell-cell communications have been shown to be mediated by transfer of DNA, microRNA, mRNA, lipids and proteins via exosomes, which are 30-150 nm vesicles derived from the intraluminal membrane budding of multivesicular bodies [18,19]. These vesicles are defined by enrichment of specific lipid-raft proteins, such as tetraspanins (CD9, CD63, CD81) and flotillin-1 (FLOT1) [20,21-24]. Two general mechanisms have been hypothesized to explain the transfer of exosomal content between cells; both mechanisms propose that exosomes incorporate transmembrane proteins into the plasma membrane of the recipient cell and release their lumen content into the cytoplasm [25,26].
In this study we show that exosomes isolated from stromal fibroblasts contain components of the TGFβ signaling pathway and that exosome transfer between stromal fibroblasts and tumor cells can influence TGFβ signaling in SCC.
Materials and methods
Cells
Ethical approval for this investigation (#15D.548) was obtained from the Jefferson Institutional Review Board. All patients participating in this study provided written, informed consent. Primary keratinocytes and fibroblasts were isolated from fresh tissue obtained from patients undergoing SCC surgery, and cultured in media containing serum and, for keratinocytes only, supplemented with growth factors as previously described [27].
Antibodies
The antibodies used in this study were: P-SMAD2 rabbit monoclonal (Ser465/467 138D4 #3108, Cell Signaling Technology, Danvers, MA); P-SMAD3 rabbit monoclonal (Ser423/425 EP823Y ab52903, Abcam, Cambridge, MA); total-SMAD2/3 rabbit polyclonal (#3102, Cell Signaling Technology); GAPDH mouse monoclonal (G8795, Sigma Aldrich, St. Louis, MO); TβRII (L-21) rabbit polyclonal (sc-400, Santa Cruz Biotechnology Inc., Dallas, TX); TSG101 rabbit monoclonal (ab-125011, Abcam); CD9 mouse monoclonal (sc-13118, Santa Cruz Biotechnology, Inc.), calnexin (H-70) rabbit polyclonal (sc-11397, Santa-Cruz Biotechnology, Inc.).
Exosome isolation and characterization
Cells were washed with PBS and grown in serum-free medium for 48 h and exosomes were isolated as described [20,28,29].
Exosome uptake
SCC keratinocytes were serum starved for 24 h and then incubated with 20 or 30 µg/mL of fibroblasts exosomes. After 24 h, cells were washed twice with PBS and incubated with TGFβ1 (5 ng/mL dilution from 100 mg/mL stock dissolved in 20 mM Sodium Citrate pH 3.0; Cell signaling Technology) for 30 min at 37°C. Cells were then washed again and lysed using RIPA buffer. Protein quantification was performed using the BCA protein assay kit. P-SMAD3, P-SMAD2, Total SMAD2/3, TβRII expression was analyzed using immunoblotting. GAPDH was used as a loading control.
Immunoblotting
Whole cell extracts and exosome lysates were collected using RIPA buffer and protein concentration determined by BCA protein assay kit (Thermo Fisher Scientific). Proteins were separated by electrophoresis using 10% SDS/PAGE gels. Proteins were transferred to a nitrocellulose membrane using the Trans-Blot® Turbo™ system (Bio-Rad, Hercules, CA). For exosome characterization, equal amounts of total cell lysates and exosome lysates were resolved using 12.5% SDS/PAGE gels. Proteins were transferred to a PVDF membrane and analysis was performed as described before [29].
Results
Primary SCC fibroblasts respond to TGFβ
Primary keratinocytes cultured from SCC patient specimens exhibited variation in TGFβ response. Of the 14 populations of SCC keratinocytes tested, 3 were unresponsive to TGFβ1 stimulation, one of which did not express detectable levels of TβRII (Figure 1 and data not shown). In contrast 12/12 primary fibroblast populations cultured from SCC patient resected tissue robustly signaled after TGFβ1 stimulation, as determined by phospho-specific antibodies against SMAD2 and SMAD3 (Figure 1 and data not shown).
Figure 1.
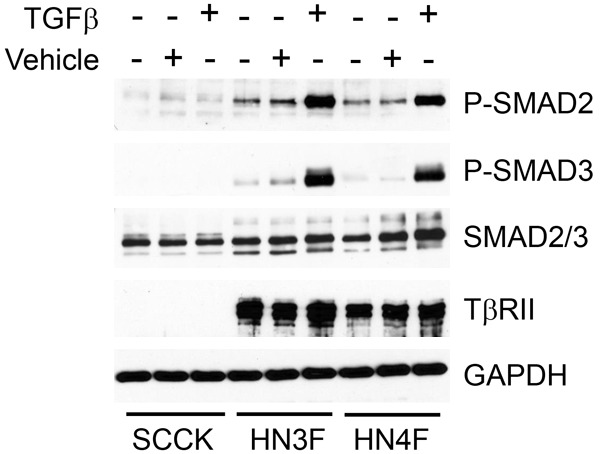
Lack of TβRII in SCC tumor keratinocytes abrogates SMAD phosphorylation response to TGFβ ligand. Of 14 populations of keratinocytes cultured from SCC patient resected tissue, 3 were unresponsive to TGFβ1 stimulation (5 ng/ml for 30 min), one of which did not express detectable levels of TβRII (SCCK, shown here). 12/12 fibroblast populations cultured from SCC patient resected tissue robustly signaled via SMAD phosphorylation (P-SMAD) after TGFβ1 stimulation. Examples of fibroblast populations, HN3 and HN4, are shown (HN3F and HN4F).
TβRII is present in exosomes isolated from SCC fibroblasts
We analyzed components of the TGFβ signaling pathway in exosomes isolated from SCC fibroblasts. In each case we demonstrated that the TGFβ receptor, TβRII was present in exosomes (Figure 2), whereas we did not detect the presence of SMAD2 or SMAD3 in exosomes isolated from SCC fibroblasts (data not shown).
Figure 2.
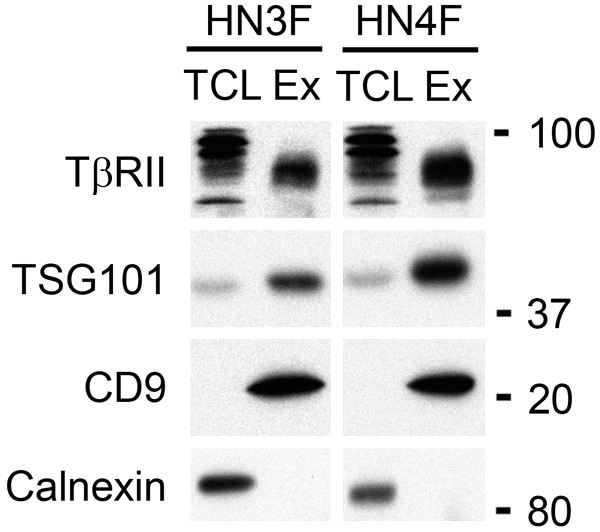
TβRII is expressed in exosomes released from fibroblasts. Exosomes were purified via ultracentrifugation from fibroblast culture media (HN3F and HN4F shown). 15 µg of exosome lysates (Ex) and total cells lysates (TCL) were loaded on a 12.5% SDS-PAGE gel. Immunoblotting analysis shows that TβRII is expressed in exosomes. Each exosome preparation was characterized by detection of TSG101 and CD9, markers that are enriched in exosomes compared to cell lysates. Calnexin, an endoplasmic reticulum protein is not detected in exosomes.
Exosome transfer from fibroblasts increases TGFβ signaling in SCC keratinocytes
Next, we investigated whether exosome transfer from TGFβ signaling-competent fibroblasts was able to confer TGFβ signaling to SCC keratinocytes deficient in TGFβ ligand response. We demonstrated that exosome transfer increased TBRII levels and SMAD2 phosphorylation in SCC keratinocytes (Figure 3) while SMAD3 phosphorylation did not consistently change (data not shown). An increase in P-SMAD2 was observed in four separate transfer experiments using either a pool of exosome preparations or exosomes isolated from a single fibroblast population. Furthermore, stimulation of SCC keratinocytes with recombinant TGFβ1 after exosome transfer resulted in a small, but further increase of phosphorylated SMAD2 (Figure 3).
Figure 3.
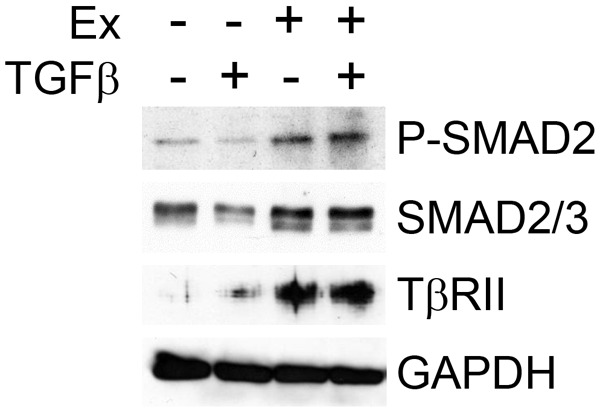
Exosomal transfer increases phosphorylation of SMAD2 and levels of TβRII in SCC tumor keratinocytes. Exosomes (Ex) were isolated and pooled from 4 separate populations of fibroblasts derived from patients with SCC. SCC keratinocytes were serum starved for 24 h and then incubated with 20 µg/mL of fibroblast exosomes (Ex +) or PBS without exosomes (Ex -). After 24 h, cells were washed twice with PBS and incubated with (TGFβ +) or without (TGFβ -) TGFβ1 (5 ng/mL) for 30 min at 37°C. Cells were then washed again and lysed using RIPA buffer. Immunoblotting was performed using antibodies to phosphorylated SMAD2, SMAD2/3, TβRII or GAPDH.
Discussion
Here we show that TβRII is present in exosomes isolated from fibroblasts and that TβRII can be transferred to SCC keratinocytes via exosomes. We further show that exosome transfer increased TGFβ signaling, as determined by phosphorylation of SMAD2, and that stimulation of SCC keratinocytes with recombinant TGFβ1 after exosome transfer further increased phosphorylation of SMAD2, albeit only slightly.
Previous work by others has shown that cancer derived exosomes can increase TGFβ signaling in mesenchymal cells. Specifically, exosomes isolated from gastric, breast and ovarian carcinoma cell lines convert mesenchymal stem cells (derived from either adipocytes or umbilical cord) to a myofibroblast lineage determined by alpha smooth muscle actin expression and increased phosphorylation of SMAD2 [30-32]. No components of the TGFβ signaling pathway were reported to be present in cancer derived exosomes in these prior studies. To our knowledge our study is the first to demonstrate that TβRII is present in exosomes isolated from the tumor microenvironment, that TβRII can be transferred via exosomes, and that exosomal transfer can stimulate TGFβ signaling in tumor cells that are unresponsive to TGFβ ligand in the absence of exosomal transfer.
Based on our data we find the most likely explanation for increased TGFβ signaling after exosome transfer is TβRII transfer. Increased phosphorylated SMAD2 after exosome transfer alone is likely due to secretion of TGFβ ligand from the SCC keratinocytes thereby stimulating transferred TβRII. However, we cannot rule out the possibility that SMAD2 phosphorylation is a result of non-canonical TGFβ signaling after transfer of components other than SMAD2 or SMAD3 proteins, including micro-RNAs, present in fibroblast exosomes that we have yet to identify. Indeed, in the experiment shown in Figure 3 there is an observed increase, albeit small, in total SMAD2/3 levels which may be a result of exosome transfer. Regardless of these details the fact remains that TGFβ signaling can be stimulated in SCC tumor cells via exosome transfer from the tumor microenvironment.
It has been shown that membrane trafficking can regulate TβRII activity and degradation, and that sorting of TβRII into the early endosome is important for SMAD complex formation [33]. Further work is necessary to determine the precise nature of how exosome transfer regulates TGFβ signaling in SCC and whether TGFβ signaling itself can influence exosome secretion.
The paradox presented by TGFβ signaling in SCC, whereby loss of function results in tumor initiation and increased signaling promotes progression [9], cannot be explained solely by genetics, as loss of function mutation would preclude subsequent signaling. Our data address such a paradox and provide a mechanism for SCC heterogeneity in response to TGFβ, particularly at the leading edge of invading tumors where communication between the microenvironment is most likely [34]. In summary, we present a potent mechanism, exosomal transfer from the tumor microenvironment, for re-activation of TGFβ signaling in SCC tumors that have lost the response to TGFβ ligand.
Acknowledgements
We would like to thank all the participants involved in this study. This work was funded in part by: the Dystrophic Epidermolysis Bullosa Research Association (G.J.I. and A.P.S.), a Sidney Kimmel Cancer Center Gold Goal Line Award (A.P.S and J.M.C.), and National Institutes of Health Grants P01CA140043 and R01CA089720 (to L.R.L.).
Disclosure of conflict of interest
None.
References
- 1.Marur S, Forastiere AA. Head and neck cancer: changing epidemiology, diagnosis, and treatment. Mayo Clin Proc. 2008;83:489–501. doi: 10.4065/83.4.489. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Bose P, Brockton NT, Dort JC. Head and neck cancer: from anatomy to biology. Int J Cancer. 2013;133:2013–2023. doi: 10.1002/ijc.28112. [DOI] [PubMed] [Google Scholar]
- 4.Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, Lin C, Leach C, Cannady RS, Cho H, Scoppa S, Hachey M, Kirch R, Jemal A, Ward E. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62:220–241. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- 5.Pulte D, Brenner H. Changes in survival in head and neck cancers in the late 20th and early 21st century: a period analysis. Oncologist. 2010;15:994–1001. doi: 10.1634/theoncologist.2009-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 7.Ross S, Hill CS. How the Smads regulate transcription. Int J Biochem Cell Biol. 2008;40:383–408. doi: 10.1016/j.biocel.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Connolly EC, Akhurst RJ. The complexities of TGF-beta action during mammary and squamous cell carcinogenesis. Curr Pharm Biotechnol. 2011;12:2138–2149. doi: 10.2174/138920111798808284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, Akhurst RJ. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531–542. doi: 10.1016/s0092-8674(00)80127-0. [DOI] [PubMed] [Google Scholar]
- 10.Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007;12:313–327. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, Li AG, Tang CF, Siddiqui Y, Nord J, Andersen P, Corless CL, Wang XJ. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goudie DR, D’Alessandro M, Merriman B, Lee H, Szeverenyi I, Avery S, O’Connor BD, Nelson SF, Coats SE, Stewart A, Christie L, Pichert G, Friedel J, Hayes I, Burrows N, Whittaker S, Gerdes AM, Broesby-Olsen S, Ferguson-Smith MA, Verma C, Lunny DP, Reversade B, Lane EB. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat Genet. 2011;43:365–369. doi: 10.1038/ng.780. [DOI] [PubMed] [Google Scholar]
- 13.Arnault JP, Mateus C, Escudier B, Tomasic G, Wechsler J, Hollville E, Soria JC, Malka D, Sarasin A, Larcher M, Andre J, Kamsu-Kom N, Boussemart L, Lacroix L, Spatz A, Eggermont AM, Druillennec S, Vagner S, Eychene A, Dumaz N, Robert C. Skin tumors induced by sorafenib; paradoxic RAS-RAF pathway activation and oncogenic mutations of HRAS, TP53, and TGFBR1. Clin Cancer Res. 2012;18:263–272. doi: 10.1158/1078-0432.CCR-11-1344. [DOI] [PubMed] [Google Scholar]
- 14.Lacouture ME, Morris JC, Lawrence DP, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Berzofsky JA, Hsu FJ, Guitart J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor beta by the monoclonal antibody fresolimumab (GC1008) Cancer Immunol Immunother. 2015;64:437–446. doi: 10.1007/s00262-015-1653-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oshimori N, Oristian D, Fuchs E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160:963–976. doi: 10.1016/j.cell.2015.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H, Ter Brugge PJ, Jonkers J, Slingerland J, Minn AJ. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159:499–513. doi: 10.1016/j.cell.2014.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 19.Simons M, Raposo G. Exosomes--vesicular carriers for intercellular communication. Curr Opin Cell Biol. 2009;21:575–581. doi: 10.1016/j.ceb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 doi: 10.1002/0471143030.cb0322s30. Chapter 3: Unit 3 22. [DOI] [PubMed] [Google Scholar]
- 21.Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 24.Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, Zhang J, Weitz J, Chin L, Futreal A, Kalluri R. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014;289:3869–3875. doi: 10.1074/jbc.C113.532267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 26.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purdie KJ, Pourreyron C, South AP. Isolation and culture of squamous cell carcinoma lines. Methods Mol Biol. 2011;731:151–159. doi: 10.1007/978-1-61779-080-5_14. [DOI] [PubMed] [Google Scholar]
- 28.Trerotola M, Jernigan DL, Liu Q, Siddiqui J, Fatatis A, Languino LR. Trop-2 promotes prostate cancer metastasis by modulating beta(1) integrin functions. Cancer Res. 2013;73:3155–3167. doi: 10.1158/0008-5472.CAN-12-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fedele C, Singh A, Zerlanko BJ, Iozzo RV, Languino LR. The alphavbeta6 integrin is transferred intercellularly via exosomes. J Biol Chem. 2015;290:4545–4551. doi: 10.1074/jbc.C114.617662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho JA, Park H, Lim EH, Lee KW. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int J Oncol. 2012;40:130–138. doi: 10.3892/ijo.2011.1193. [DOI] [PubMed] [Google Scholar]
- 31.Cho JA, Park H, Lim EH, Kim KH, Choi JS, Lee JH, Shin JW, Lee KW. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol Oncol. 2011;123:379–386. doi: 10.1016/j.ygyno.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Gu J, Qian H, Shen L, Zhang X, Zhu W, Huang L, Yan Y, Mao F, Zhao C, Shi Y, Xu W. Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-beta/Smad pathway. PLoS One. 2012;7:e52465. doi: 10.1371/journal.pone.0052465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang F, Chen YG. Regulation of TGF-beta receptor activity. Cell Biosci. 2012;2:9. doi: 10.1186/2045-3701-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curry JM, Sprandio J, Cognetti D, Luginbuhl A, Bar-ad V, Pribitkin E, Tuluc M. Tumor microenvironment in head and neck squamous cell carcinoma. Semin Oncol. 2014;41:217–234. doi: 10.1053/j.seminoncol.2014.03.003. [DOI] [PubMed] [Google Scholar]