

Abstract
The biological function of multiple repetitions of single amino acids, or homo-repeats, is largely unknown, but their occurrence in proteins has been associated with more than 20 hereditary diseases. Analysing 122 bacterial and eukaryotic genomes, we observed that the number of proteins containing homo-repeats is significantly larger than expected from theoretical estimates. Analysis of statistical significance indicates that the minimal size of homo-repeats varies with amino acid type and proteome. In an attempt to characterize proteins harbouring long homo-repeats, we found that those containing polar or small amino acids S, P, H, E, D, K, Q and N are enriched in structural disorder as well as protein- and RNA-interactions. We observed that E, S, Q, G, L, P, D, A and H homo-repeats are strongly linked with occurrence in human diseases. Moreover, S, E, P, A, Q, D and T homo-repeats are significantly enriched in neuronal proteins associated with autism and other disorders. We release a webserver for further exploration of homo-repeats occurrence in human pathology at http://bioinfo.protres.ru/hradis/.
Eukaryotic and bacterial genomes harbour proteins containing multiple repetitions of specific amino acids called homo-repeats. The functional role of homo-repeats is still unclear1, although a tight link with disease exists2.
Homo-repeat sizes vary from proteome to proteome3,4 and are associated with low complexity regions in eukaryotes5. Indeed, comparing H. sapiens and E. coli proteins, we previously reported a significant enrichment of homo-repeats in H. sapiens6, which can be linked to the presence of structurally disordered regions7. Some homo-repeats, such as for instance LLLLLL, occur 11 times more frequently in H. sapiens than D. melanogaster and C. elegans, while TTTTTT are 4 times more abundant in D. melanogaster and C. elegans than H. sapiens8. Similarly, the poly-N motif occurs more than 17000 times in 122 proteomes3: The NNNNN pattern is connected with fungi symbiosis and occurs 21 times in the human proteome3. Also HHHHHH repeats are particularly frequent, especially at the N- or C-terminus of polypeptide chains, but their abundance in crystal and NMR structures is often due to biochemical procedures (histidine-tags are useful for purification at a nickel-containing column)9. Yet, poly-H are highly frequent in the human proteome and linked to a number of functional roles6. For instance, protein kinase DYRK1A (poly-H length of 13) and FAM76B (poly-H length of 10) uses histidine expansions to mediate nuclear speckle trafficking10,11,12.
What functions of poly-repeats are known? For instance, poly-L expansions, especially when located at the N-terminal end of proteins13, act as signal peptides and are abundant in membrane proteins14. By contrast, poly-Q and poly-A occur more often in transcription factors and poly-K are enriched in a number of metabolic pathways3. Similarly, poly-M are connected with voltage-gated calcium channel activity3, while poly-P are associated with central nervous system, morphogenesis and through actin cytoskeleton organization, cell morphogenesis, tropomyosin binding and stereocilium3. The poly-A tract in the HOXD13 human protein (15 residues in length) is essential for limb development11.
Not all homo-repeats are associated with specific roles and investigation of their biological functions is complicated by the widespread occurrence in low-complexity regions of higher eukaryotes15. Here, we studied the distribution of homo-repeats in eukaryotic and bacterial proteomes and quantified the difference between expected and real occurrences in 1.5 million sequences. As presence of low complexity regions can cause cellular toxicity by promoting promiscuous interactions16, we investigated the relationships between homo-repeat occurrence, number of protein interactions and diseases. We release a dataset at http://bioinfo.protres.ru/hradis/ for further exploration of homo-repeats occurrence in human diseases.
Results and Discussion
In this study, we focused on the occurrence of homo-repeats in eukaryotic and bacterial proteomes. Previous analyses indicated that homo-repeats of 5 amino acids occur non-randomly14,17,18.
How large is the difference between the expected occurrences of homo-repeats with real occurrences in 122 proteomes?
How many proteins are expected to contain a homo-repeat of a certain length? If we compute the expected number of proteins <N(M)> harbouring a homo-repeat of M residues in a database containing 1 million protein sequences with average length of 500 residues and uniform amino acid frequency of 1/20, we have:
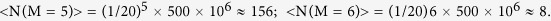 |
In the case of the human proteome our estimates indicate <N(M = 5)> ≈ 7 and <N(M = 6)> ≈ 0.3.
Can this example be expanded into a more general model to study the occurrence of homo-repeats? To this aim, we have derived a recursive equation (Materials and Methods) that estimates the probability of homo-repeats to occur in the central or terminal parts of a protein sequence (Fig. 1A and Materials and Methods). We used the equation to investigate the frequency of the longest homo-repeat M in a protein sequence of length L (Fig. 1B). Using 122 proteomes (Supplementary Table S1), we studied the length distribution of protein sequences (Fig. 2) and their amino acid frequencies (Supplementary Fig. S1) to measure the expected number of proteins N(M, L) carrying a specific motif [see Materials and Methods, Eq. 1].
Figure 1. Theoretical estimate of homo-repeat frequencies.

Given the length of the sequence (L) and the sizes of the central (M) and C-terminal (K) motifs, it is possible to compute the probability p that a homo-repeat occurs using the recursive formula presented in Eq. 2. (A) The longest homo-repeat is in the central part of the sequence. (B) The longest homo-repeat is at the C-terminal.
Figure 2. Protein length and expected homo-repeat frequencies.
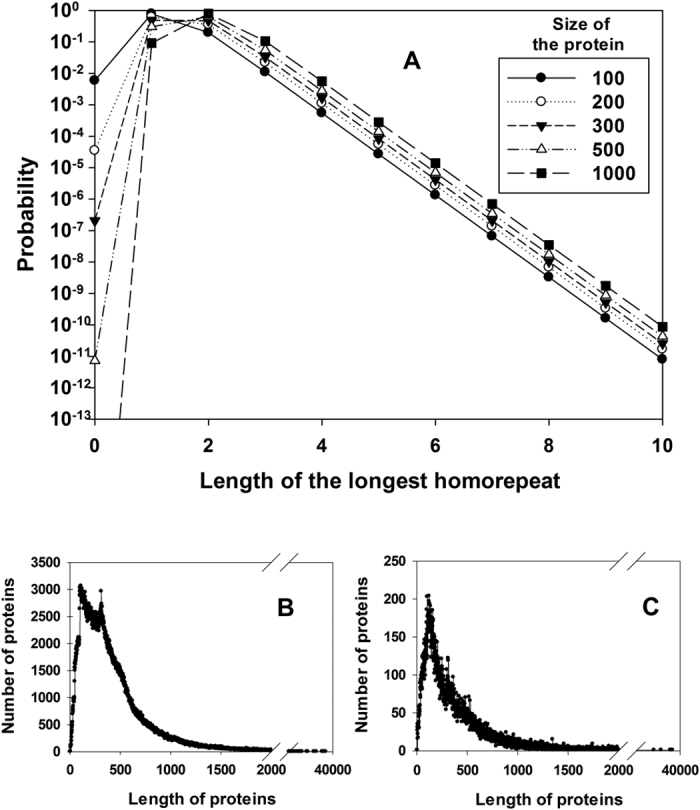
(A) Predicted frequencies of the longest homo-repeat at different proteins lengths. Protein length distribution of (B) 122 proteomes (average length is 435 ± 425 amino acids) and (C) human proteome (average length is 395 ± 530 amino acids).
The expected frequencies of motif repeats such as poly-Q, poly-L, and poly-C, differ substantially from those observed in real proteomes (Fig. 3; Supplementary Materials): the length of homo-repeats in natural proteomes is much larger than the estimate based on amino acid frequencies and protein length distribution (Fig. 2 and Supplementary Table S1). We report in Table 1 the lengths of homo-repeats whose occurrences in real proteomes have a 10-fold difference from theoretical estimates.
Figure 3. Theoretical vs observed homo-repeat frequencies.

For poly-L, poly-Q and poly-C, we report the difference expected and the measured numbers of proteins harbouring the repeats (122 proteomes and human proteome are shown).
Table 1. Lengths of homo-repeats whose frequencies in real proteomes have a 10-fold difference from theoretical estimates.
| C | W | M | H | Y | N | K | F | D | P | Q | I | T | E | S | R | V | G | A | L | N* | N** | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Metazoa | 4 | 5 | 5 | 4 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 6 | 5 | 5 | 5 | 5 | 6 | 5 | 5 | 6 | 424 | 17 |
| Viridiplantae | 4 | 4 | 5 | 4 | 5 | 5 | 5 | 5 | 5 | 4 | 4 | 6 | 5 | 5 | 5 | 5 | 6 | 5 | 5 | 6 | 108 | 5 |
| Stramenopiles | 4 | 4 | 5 | 4 | – | 4 | 4 | 5 | 5 | 4 | 4 | – | 5 | 5 | 5 | 5 | 7 | 5 | 6 | 7 | 12 | 1 |
| Choanoflagellida | 4 | 4 | 4 | 5 | 5 | 5 | 4 | 5 | 5 | 5 | 5 | 6 | 5 | 5 | 6 | 6 | 7 | 6 | 6 | 7 | 9 | 1 |
| Euglenozoa | 4 | 4 | 5 | 4 | 5 | 4 | 4 | 4 | 5 | 5 | 4 | 5 | 5 | 5 | 5 | 6 | 7 | 5 | 5 | 6 | 44 | 4 |
| Alveolata | 5 | 4 | 5 | 5 | 6 | 6 | 6 | 5 | 5 | 4 | 5 | 8 | 5 | 5 | 5 | 4 | – | 4 | 5 | 7 | 50 | 6 |
| Amoebozoa | 4 | 4 | 4 | 4 | 5 | 5 | 5 | 5 | 5 | 4 | 4 | 8 | 5 | 5 | 5 | 5 | 5 | 4 | 4 | 7 | 25 | 2 |
| Diplomonadida | – | – | – | – | – | 6 | – | – | 6 | 5 | 5 | – | 6 | 6 | 7 | 6 | 5 | 6 | 7 | – | 17 | 3 |
| Fungi | 4 | 5 | 5 | 4 | 5 | 5 | 5 | 5 | 5 | 5 | 4 | 7 | 5 | 5 | 6 | 5 | 6 | 5 | 6 | 8 | 551 | 58 |
| Bacteria | 5 | – | – | 5 | – | 6 | 5 | 6 | 6 | 5 | 5 | – | 6 | 7 | 6 | 6 | 8 | 6 | 7 | 9 | 210 | 25 |
N* is the number of proteins (×104), N** is the number of proteomes.
Although previous genome analyses indicated that the minimal homo-repeat length is between 5 and 7 residues14,17,18,19, our results indicate the size varies with the amino acid type. For polar and soluble residues20 such as H, D, N, K and P, the minimal size is 4, while W, M, Y, F, Q and T, which are often found in amyloid regions21, show lengths ≥5. Residues occurring in loops (E, S and G) have lengths ≥5, whereas those containing hydrophobic elements in their side chains (I, R and A) are associated with sizes ≥6 with exception of V and L that have lengths ≥7 and 8. In general, N, D, and K homo-repeats show shorter sizes than for Q, E, and R, although the motif length slightly depends on the kingdom (Table 1). In the case of the human proteome, all the homo-repeats show lengths ≥5 (Table 2), with exception of V, S, A, L, I and M (size: 6) and C (size: 4).
How many partners do proteins with long homo-repeats have?
Our results indicate that homo-repeats are more frequent than expected from theoretical estimates. To investigate what common characteristics have the genes harbouring homo-repeats, we analysed their protein networks using BIOGRID (version 3.4.134)22. Using 3514 human proteins carrying homo-repeats with size more than 10 fold larger than expected (Table 2), we found an increase in the number of physical partners of R, A, T, G, S, P, H, E, D, K, Q and N repeats (Fig. 4). Out of 320000 interactions reported in the human proteome, we found that 94000 physical associations involve homo-repeats. The largest number of binding partners was observed for D, K, Q, and N, while I, W and Y are not associated with any interaction (Fig. 4). Thus homo-repeat lengths can be connected with the number of physical associations. While hydrophobic homo-repeats are depleted in partners, hydrophilic ones have a larger number of interactions, which is in agreement with previous literature reporting enrichment of binding partners in polar regions with high structural disorder content15,16.
Table 2. Lengths of homo-repeats whose occurrence differs at least 10-fold between natural and expected human proteomes.
| AA | C | W | M | H | Y | N | K | F | D | P | Q | I | T | E | S | R | V | G | A | L |
| Length | 4 | 5 | 6 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 6 | 5 | 5 | 6 | 5 | 6 | 5 | 6 | 6 |
Figure 4. Homo-repeats and protein interactions.
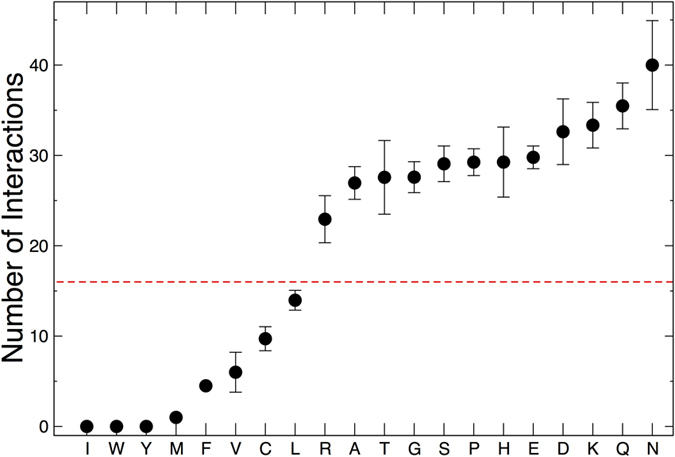
Using a total of 94000 physical associations available from BioGRID22, we found that human proteins containing poly-E, poly-D, poly-K, poly-Q, and poly-N have more interactions than the rest of the proteome (homo-repeat size is chosen according to Table 2; mean and standard error of the mean are shown). The red line indicates the average number of partners (16 interactions) in H. sapiens (total of 320000 interactions).
What physico-chemical features define human proteins with many interactions?
To understand what physico-chemical features contribute to the interaction ability of homo-repeat proteins, we used the multicleverMachine approach23,24. Based on the consensus of different predictors, multicleverMachine identifies signals in protein groups14. By directly comparing proteins that contain hydrophobic (A, G, C, V, I, L, M, F, Y and W; total of 1261 proteins) and hydrophilic (P, S, N, E, K, R, H, Q and T; total of 2672 proteins) homo-repeats, we found that the latter are enriched in RNA-binding ability and structural disorder (the analysis is based on homo-repeat sizes reported in Table 2 and is reported at the webserver link http://www.tartaglialab.com/cs_multi/confirm/1358/5f36e6e108/)25,26. As shown in Fig. 5, the enrichments are significant for both RNA-binding ability (p-value = 10−35; Kolmogorov-Smirnov test and area under the ROC curve AUC = 0.68) and structural disorder (p-value = 10−38; Kolmogorov-Smirnov test and AUC = 0.72). In agreement with the analysis reported in Fig. 4, proteins with a lower number of interacting partners (i.e., containing homo-repeats with C, F, I, L, M, V, W and Y amino acids) show a decreased amount of structural disorder, while those with a high number of partners (i.e., containing homo-repeats with E, D, G, S, Q, N, K, and H amino acids as well as the intermediate cases R and T) have increased nucleic-acid binding propensity. Thus, our findings are in agreement with previous evidence showing that structural disorder correlates with presence of small and polar amino acids27,28 and is associated with RNA-binding ability26,29. Moreover, gene ontology analysis performed with the multicleverMachine approach indicates that not only proteins containing poly-R and poly-K (Fig. 6A), but also those with negatively charged homo-repeats are able to bind RNA (Fig. 6A,B), as highlighted by recent studies30.
Figure 5. Structural disorder and RNA-binding ability of homo-repeats.

Using the multicleverMachine approach23,24, we found that proteins harboring hydrophilic (P, S, N, E, K, R, H, Q and T) homo-repeats are more prone to be RNA-binding26 and structurally disordered25 than those containing hydrophobic (A, G, C, V, I, L, M, F, Y and W) homo-repeats. (A) Box plot analysis (p-value = 10−35; Kolmogorov-Smirnov test) and (B) Receiver operating characteristic (area under the curve AUC = 0.68) indicate strong enrichments in RNA-binding abilities of hydrophilic homo-repeats. Similar results were observed for structural disorder: (C) Box plot (p-value = 10−38; Kolmogorov-Smirnov test) and (B) Receiver operating characteristic (AUC = 0.72). The analysis, as well as the original datasets can be found at the link http://www.tartaglialab.com/cs_multi/confirm/1358/5f36e6e108/.
Figure 6. Nucleic acid binding and gene ontology analysis.

multiCleverMachine analysis of AmiGO annotations51 indicate that proteins containing poly-R (A) and poly-E homo-repeats (B) reveal the increase in RNA- and DNA-binding abilities. GO labels are shown together with word-cloud visualization (p-values < 0.01 calculated with Bonferroni’s correction on whole human proteome). The analysis is available at the following links http://www.tartaglialab.com/GO_analyser/render_GO_universal/839/3158792f91/(poly-E) and http://www.tartaglialab.com/GO_analyser/render_GO_universal/840/ea98f8b320/(poly-R).
Relation of homo-repeats to human diseases
In agreement with previous literature data31,32,33,34,35, we found that Q, G, L, P, T, D, A, H and V homo-repeats have strong propensities to be coupled with pathology (Fig. 7; Table 3; Material and Methods). Indeed, a number of reports indicate that sequences containing repeats such as, for instance, poly-A are associated with diseases, including synpolydactyly type II (gene HOXD13), blepharophimosis (FOXL2), oculopharyngeal muscular dystrophy (PABPN1), infantile spasm syndrome (ARX), and holoprosencephaly (ZIC2)11. Similarly poly-Q expansions have been associated with Huntington’s disease, Dentatorubral Pallidolysian Atrophy (DRPLA), and Spinocerebellar Ataxias (SCA)36.
Figure 7. Fraction of proteins linked to disease.
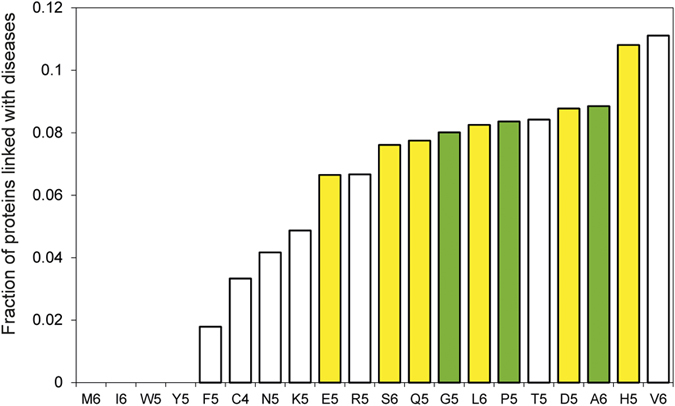
Using the OMIM database available at http://www.omim.org/, we found that poly-G, poly-A, and poly-P are strongly associated with disease (standardized Z-score > 5; Material and Methods), followed by poly-E, poly-S, poly-Q, poly-L, poly-D and poly-H. Green colour corresponds to homo-repeats with Z-score > 5, yellow to 3 < Z-score < 5, and white with Z-score < 3 (homo-repeat size is chosen according to Table 2).
Table 3. Number of proteins with homo-repeats larger than 4 associated with disease according to the OMIM database http://www.omim.org/ (bold characters correspond to standardized Z-score > 5).
| Proteins | C | M | F | I | L | V | W | Y | A | G | T | S | Q | N | E | D | H | R | K | P | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | 59053 | 38 | 3 | 56 | 25 | 1503 | 49 | 1 | 7 | 1300 | 836 | 190 | 1175 | 529 | 24 | 1625 | 262 | 148 | 270 | 554 | 1363 |
| Disease | 2501 | 2 | 0 | 1 | 1 | 125 | 7 | 0 | 0 | 105 | 67 | 16 | 86 | 41 | 1 | 108 | 23 | 16 | 18 | 27 | 114 |
Recently, Manuel Irimia and colleagues identified a number of neuron-specific micro-exons (i.e., 27 nt in length) that are switched on during neural differentiation to enhance specific protein-protein interactions. Most of the micro-exon containing proteins are enriched in structurally disordered regions37 and about 30% of them are misregulated in the brains of individuals with autism spectrum disorder37.
We studied the occurrence of homo-repeats in proteins harbouring micro-exons (895 cases)37 comparing their frequencies with expected values calculated on 20 random extractions of the human proteome (Table 4). Increasing the motif length from 4 to 9 amino acids, we found that the following homo-repeats are significantly enriched: 4 – S, E, P, A, Q and T; 5 – S, E, P, A, Q, D and T; 6 – S, E, P, Q, D and T; 7 – S, E, P, Q, T and H; 8 – S, E, P, A, Q, T and H; 9 – S, E, P, Q and T (Table 4; Figs 6 and 8; S, E, P, Q and T are enriched in all considered cases). Interestingly, the enrichments involve polar (D, E, H, Q, S and T) as well as small (A, P and G) amino acids, which can be connected to patterns occurring in proteins with a large number of interactions (i.e., S, P, H, G, D and Q; Fig. 4).
Table 4. Homo-repeat enrichments in neuronal proteins harboring micro-exons.
| C (inclusions) |
R (proteome) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 5 | 6 | 7 | 8 | 9 | 4 | 5 | 6 | 7 | 8 | 9 | |
| S | 174 ± 13 | 63 ± 8 | 26 ± 5 | 14 ± 4 | 12 ± 3 | 10 ± 3 | 64.5 ± 15.0 | 15.3 ± 6.0 | 6.0 ± 2.4 | 3.1 ± 1.7 | 1.9 ± 1.4 | 1.4 ± 1.0 |
| L | 94 ± 10 | 36 ± 6 | 18 ± 4 | 2±1 | – | – | 61.2 ± 10.2 | 18.7 ± 6.0 | 7.1 ± 3.4 | 4.0 ± 1.5 | 1.5 ± 1.0 | 1.0 ± 1.0 |
| E | 182 ± 13 | 66 ± 8 | 45 ± 7 | 30 ± 5 | 19 ± 4 | 14 ± 4 | 55.9 ± 10.9 | 20.2 ± 6.3 | 8.9 ± 3.2 | 6.2 ± 3.2 | 3.4 ± 2.0 | 2.2 ± 1.9 |
| P | 136 ± 12 | 67 ± 8 | 39 ± 6 | 28 ± 5 | 22 ± 5 | 13 ± 4 | 50.3 ± 12.7 | 17.8 ± 5.8 | 8.5 ± 2.9 | 5.3 ± 3.2 | 3.0 ± 1.4 | 1.7 ± 1.2 |
| A | 98 ± 10 | 45 ± 7 | 25 ± 5 | 12 ± 3 | 11 ± 3 | 4 ± 2 | 48.3 ± 8.2 | 15.4 ± 3.6 | 8.1 ± 3.4 | 4.4 ± 1.8 | 2.7 ± 1.6 | 2.0 ± 1.4 |
| G | 75 ± 9 | 43 ± 7 | 21 ± 5 | 11 ± 3 | 5 ± 2 | 1 ± 1 | 34.8 ± 8.5 | 12.6 ± 4.0 | 5.8 ± 2.9 | 3.7 ± 2.2 | 1.7 ± 1.1 | 0.9 ± 1.1 |
| K | 47 ± 7 | 17 ± 4 | 5 ± 2 | 1 ± 1 | – | – | 24.0 ± 7.3 | 5.8 ± 3.1 | 2.5 ± 1.5 | 0.9 ± 1.1 | 1.0 ± 1.5 | 0.4 ± 0.6 |
| R | 31 ± 6 | 10 ± 3 | – | – | – | – | 23.6 ± 4.6 | 4.9 ± 2.2 | 1.2 ± 0.9 | 0.3 ± 0.5 | – | 0.1 ± 0.3 |
| Q | 42 ± 6 | 29 ± 5 | 24 ± 5 | 22 ± 5 | 21 ± 5 | 11 ± 3 | 13.6 ± 4.3 | 4.7 ± 1.0 | 2.9 ± 2.2 | 1.5 ± 1.0 | 1.3 ± 1.2 | 0.4 ± 0.7 |
| V | 12 ± 3 | 1 ± 1 | – | – | – | – | 10.1 ± 4.0 | 1.1 ± 1.0 | 0.2 ± 0.4 | – | – | – |
| D | 25 ± 5 | 15 ± 4 | 9 ± 3 | 4 ± 2 | – | – | 9.6 ± 3.4 | 3.2 ± 2.2 | 1.2 ± 0.9 | 0.7 ± 0.7 | 0.5 ± 0.8 | 0.1 ± 0.3 |
| T | 38 ± 6 | 24 ± 5 | 17 ± 4 | 6 ± 2 | 4 ± 2 | 4 ± 2 | 9.0 ± 3.5 | 2.3 ± 1.7 | 0.9 ± 1.1 | 0.3 ± 0.4 | 0.2 ± 0.5 | 0.1 ± 0.3 |
| I | 1 ± 1 | – | – | – | – | – | 2.8 ± 1.5 | 0.3 ± 0.5 | – | – | – | – |
| H | 5 ± 2 | 5 ± 2 | 5 ± 2 | 5 ± 2 | 4 ± 2 | 3 ± 2 | 2.5 ± 1.3 | 1.1 ± 0.8 | 0.9 ± 1.0 | 0.9 ± 0.8 | 0.5 ± 0.7 | 0.4 ± 0.7 |
| F | 4 ± 2 | – | – | – | – | – | 2.4 ± 2.3 | 0.3 ± 0.7 | – | – | – | – |
| N | 8 ± 3 | 2 ± 1 | – | – | – | – | 2.2 ± 1.3 | 0.5 ± 0.7 | – | – | 0.1 ± 0.2 | – |
| C | 3 ± 2 | – | – | – | – | – | 1.8 ± 1.6 | 0.6 ± 0.7 | 0.4 ± 0.5 | 0.1 ± 0.2 | – | – |
| W | – | – | – | – | – | – | 1.1 ± 0.9 | 0.1 ± 0.2 | – | – | – | – |
| M | – | – | – | – | – | – | 0.3 ± 0.5 | – | – | – | – | – |
| Y | – | – | – | – | – | – | 0.1 ± 0.2 | – | – | – | – | – |
C indicates the number of cases associated with an amino acid motif of length between 4 and 9 (895 cases) and R indicates the average motif counts measured on 20 random extractions from human proteome (each sample contains 895 cases)37. The standard deviation associated with 20 extractions is reported. Homo-repeats with standardized Z-score > 5 are given in bold.
Figure 8. Examples of homo-repeat occurrences in different datasets.
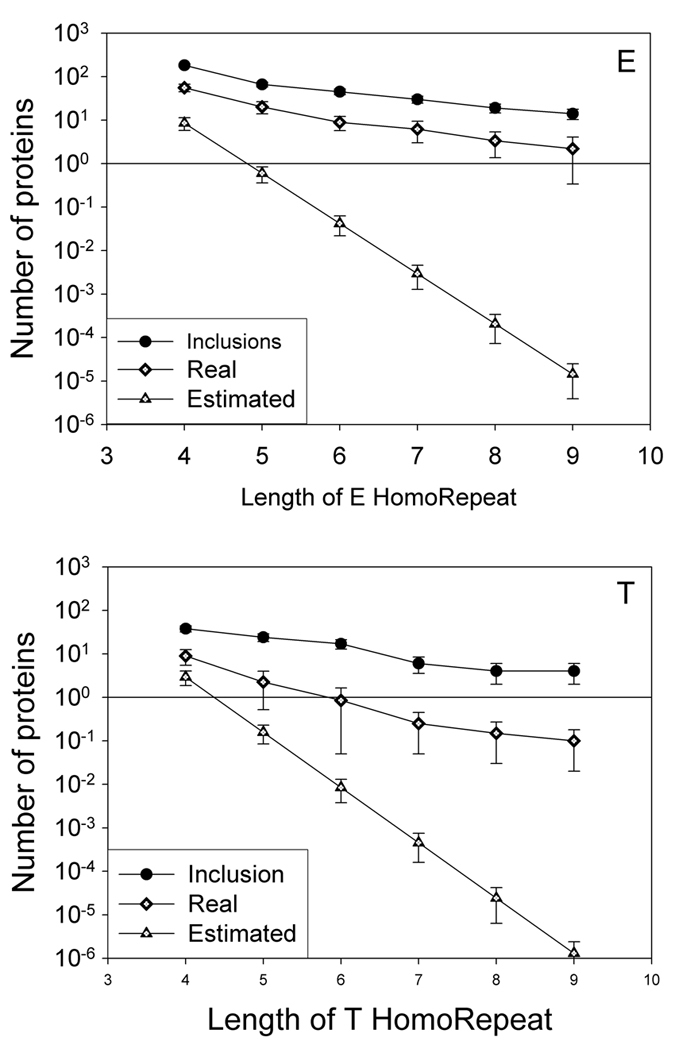
For E and T homo-repeats, we report occurrences in protein harbouring micro-exons inclusion (895 neuronal proteins)37, human proteome and theoretical estimates based on the occurrence of amino acids in 122 proteomes.
The HRaDis database
8145 out of 59053 H. sapiens proteins (reviewed and un-reviewed entries in the Uniprot database) contain homo-repeats longer than 4 amino acids, which represents a non-negligible component of the proteome (14%). By considering all the homo-repeats currently linked to disease (578 out of 2501 entries; Table 3), the fraction raises to 23%, indicating that homo-repeats are tightly linked with pathology. For instance, out of all the proteins related to neurodegenerative diseases (90 entries), 13 harbour homo-repeats: PERQ2 amino acid-rich with GYF domain-containing protein 2 (PERQ2): poly-Q (sizes: 5, 6, 7, 8 and 9) and poly-K (size: 5); Huntingtin (HD): poly-Q (size: 21) and poly-E (sizes: 5 and 6), poly-P (sizes: 10 and 11); RNA-binding protein FUS (FUS): poly-G (sizes: 7, 10 and 10); Amyloid beta A4 protein (A4) – poly-T (size: 7); Ataxin-2 (ATX2): poly-Q (size: 23); Gap junction gamma-2 protein (CXG2): poly-E (size: 7); Dynactin subunit 1 (DCTN1): poly-A size: 5); NADH-ubiquinone oxidoreductase chain 6 (NU6M): poly-V (size: 5); Pantothenate kinase 2, mitochondrial (PANK2): poly-E (size: 6); Presenilin-2 (PSN2): poly-E (size: 5); Probable helicase senataxin (SETX): poly-D (size: 5); Synphilin-1 (SNCAP): poly-N (size: 5) and Mitochondrial import inner membrane translocase subunit Tim8 A (TIM8A): poly-S (size: 6). This list expands publicly available repositories, such as for instance “PolyQ”38, in which only four proteins (ATX1, ATX2, ATX7 and IID) were associated with disease.
To better investigate the link between homo-repeat occurrence and disease, we release the HRaDis database (HomoRepeats and human Diseases, available at http://bioinfo.protres.ru/hradis/), in which human sequences are reported along with OMIM classifications and GO annotations.
Conclusions
In this work, we showed that the number of homo-repeats in eukaryotic and bacterial proteomes is significantly larger than expected from theoretical estimates. Our calculations indicate that the minimal length that is statistically significant varies with amino acid type and proteome. In H. sapiens, occurrence of homo-repeats is associated with high content of structurally disordered regions and enhanced RNA-binding potential, which is in line with recent experimental findings26,29. We also observed that protein containing homo-repeats have a large number of interactions, which can promote perturbation of protein networks and cause dysfunction39.
Although the functional roles of homo-repeats are unknown, we found that their occurrence is associated with pathology. Certain homo-repeats such as for instance the poly-A tract in Homeobox 2B protein (PHOX2B) are highly conserved in vertebrate species and might have biological function. Yet, it has been reported that poly-A is frequently linked with diseases such as synpolydactyly type II (HOXD13), blepharophimosis (FOXL2), oculopharyngeal muscular dystrophy (PABPN1) and infantile spasm syndrome (ARX)11. Similarly, poly-Q expansions are associated with neurodegeneration36 and their length is proportional to disease severity40. The link between homo-repeats and disease is particularly relevant if we consider that a recent study report involvement of low complexity regions in proteins involved in autism37.
Possible models for the evolution of homo-repeats have been proposed41,42,43,44. Yet, they are still debated, and to assess possible functions, further biological information is necessary. One interesting mechanism that links homo-repeats with protein dysfunction, is that amino acid expansions can be caused by slippage errors in DNA replication, recombination and repair45,46,47,48,49.
We hope that our work will be useful for the characterization of homo-repeats in the human proteome and that starting from direct analysis of sequences available at http://bioinfo.protres.ru/hradis/, it will be possible to build a catalogue to decipher the biological functions as well as the evolutionary patterns of these sequences.
Material and Methods
Probability of occurrence of the longest homo-repeat at different protein lengths
For a polypeptide of length L containing two amino acid types A and X (any amino acid different from A), the probability of finding A in any region of the chain is equal to p (the probability of finding X is equal to 1-p). Assuming that M is the longest homo-repeat of amino acid A (if A is absent, then M = 0) and K is the length of the homo-repeat adjacent to the C-end of the chain (if the chain terminates with X, then K = 0), we can determine the probability of a homo-repeat in an iterative way. Indeed, if A is added at the C-end, K increases by 1 (if K = M, then M is incremented by 1). The probability of adding A is P(p, M, K + 1, L) = P(p, M, K, L − 1)*p or P(p, M + 1, M + 1, L) = P(p, M, M, L − 1)*p (Fig. 1). By contrast, if X is added, then M does not change, and K becomes 0, and the probability event is P(p, M, 0, L) = P(p, M, K, L − 1)*(1 − p) (Fig. 1). Thus, knowing the joint distribution of M and K for the chain length L–1, it is possible to calculate the distribution of M and K for the chain length L. For a chain with one residue: P(p, M = 0, K = 0, 1) = p and P(p, M = 1, K = 1, 1) = 1 − p (the probability of other M and K values for a chain with one residue is equal to zero). By adding up the values for K (0 ≤ K ≤ M), we calculated the probability depending on the length of the largest homo-repeat M and the chain length L (see Results section).
If we take the distribution lengths of proteins and frequencies from the set of 122 proteomes (see Supplementary Table 1) we can measure the expected number of proteins carrying a specific motif size M:
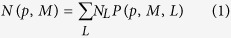 |
where 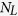 is the number of proteins with length L in the database.
is the number of proteins with length L in the database.
Calculation of the probability of homo-repeats occurence
If the probability of finding two homo-repeats with length M is small, our Eq. 1 can be approximated (M ≪ L and M ≠ 0). If the homo-repeat lies at the C-term of the protein, there will be M amino acids of type A and another amino acid X with probability of pM(1 − p). If the homo-repeat lies in the middle of the protein, there will be M amino acids of type A and two other amino acids at the edges with probability of pM(1 − p)2. Taken into account that the homo-repeat can be placed in two positions at the edges of the protein and (L − M − 1) in the middle position, the overall homo-repeat probability is:
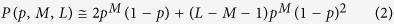 |
As natural proteins are shorter than 1000 residues, the approximation works at p ≤ 0.05 and M ≥ 4 (LpM < 0.01). We note that some amino acids, such as for instance leucine, occur with frequency p ≈ 0.1. In such cases, the approach works well if M ≥ 5.
Statistical analysis of homo-repeats and link with disease
If homo-repeat and disease frequencies are independent, the distribution has an average number of proteins.
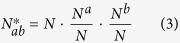 |
root-mean-square deviation
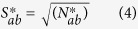 |
Z-value
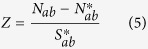 |
In Eq. 3, 4 and 5, N is the number of proteins in the human proteome, 59053. Na is the number of proteins associated with disease (2501, see Table 3), and Nb is the number of proteins with homo-repeats with the length larger or equal to 5. Nab is the number of proteins carrying both characters in our database.
cleverMachine
The cleverMachine (CM) algorithm analyzes physico-chemical properties of two protein datasets50. The tool creates profiles, or physico-chemical signatures, for each protein, utilizing a large set of features - both experimentally and statistically derived from other tools. In our analysis we used a number of physico-chemical properties (hydrophobicity, alpha-helix, beta-sheet, disorder, burial, aggregation, membrane and nucleic acid-binding propensities) and 10 propensity predictors per feature. Only differentially enriched properties (p-values < 10−5 using Fisher’s exact test) were used in the calculations. Further information can be found at http://s.tartaglialab.com/page/clever_suite.
multiCleverMachine
The multicleverMachine extends the concept of binary comparisons (CM) between protein datases by introducing signal and negative sets23,24. After submission of one or more sets for signal and one or more sets as a negative group, the multicleverMachine creates a CM run for each possible combination of elements from the signal and negative sets. The result is presented in an easy-to-read format, allowing at-a-glance interpretation of the CM submission. The multicleverMachine provides visualisation of enrichment strengths per group, enabling to see easily for which groups the various properties like disorder, alpha-helical propensity, etc. are enriched. More details about the method are available at http://www.tartaglialab.com/cs_multi/submission. In addition to the visualisation of individual enrichments, multiCM links each of the datasets to gene ontology analysis (http://www.tartaglialab.com/GO_analyser/universal and related documentation). To calculate GO enrichments, multicleverMachine uses built-in datasets containing all entries available for the proteome of interest (reference set)23,24.
Additional Information
How to cite this article: Lobanov, M. Y. et al. Non-random distribution of homo-repeats: links with biological functions and human diseases. Sci. Rep. 6, 26941; doi: 10.1038/srep26941 (2016).
Supplementary Material
Acknowledgments
The authors would like to thank R. Guigó, B. Lehner and A. Breschi for stimulating discussions. This study was supported by the Russian Science Foundation grant number 14-14-00536 for OVG and the programs “Molecular and Cellular Biology” (01201353567) for MYL and IVS. GGT received funding from the European Union Seventh Framework Programme (FP7/2007–2013), through the European Research Council, under grant agreement RIBOMYLOME_309545, and from the Spanish Ministry of Economy and Competitiveness. PK and GGT also acknowledge support from the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2013–2017’ (SEV-2012-0208). PK is recipient of a “La Caixa” studentship.
Footnotes
Author Contributions M.Y.L. and O.V.G. developed the statistical models, I.V.S. implemented the webserver. P.K., M.Y.L., O.V.G. and G.G.T. analysed the data. G.G.T. and P.V. calculated the physico-chemical features of homo-repeats. O.V.G. and G.G.T. wrote the manuscript.
References
- Siwach P. & Ganesh S. Tandem repeats in human disorders: mechanisms and evolution. Front. Biosci. J. Virtual Libr. 13, 4467–4484 (2008). [DOI] [PubMed] [Google Scholar]
- Sabate R., Rousseau F., Schymkowitz J. & Ventura S. What Makes a Protein Sequence a Prion? PLos Comput. Biol. 11, e1004013 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov M. Y., Sokolovskiy I. V. & Galzitskaya O. V. HRaP: database of occurrence of HomoRepeats and patterns in proteomes. Nucleic Acids Res. 42, D273–278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov M. Y. & Galzitskaya O. V. Occurrence of disordered patterns and homorepeats in eukaryotic and bacterial proteomes. Mol. Biosyst. 8, 327–337 (2012). [DOI] [PubMed] [Google Scholar]
- Lobanov M. Y. & Galzitskaya O. V. Disordered patterns in clustered Protein Data Bank and in eukaryotic and bacterial proteomes. PLos One 6, e27142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov M. Y., Furletova E. I., Bogatyreva N. S., Roytberg M. A. & Galzitskaya O. V. Library of disordered patterns in 3D protein structures. PLos Comput. Biol. 6, e1000958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia G. G., Pellarin R., Cavalli A. & Caflisch A. Organism complexity anti-correlates with proteomic beta-aggregation propensity. Protein Sci. 14, 2735–2740 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov M. I., Bogatyreva N. S. & Galzitskaia O. V. Occurrence of motifs with six amino acid residues in three eukaryotic proteomes. Mol. Biol. (Mosk.) 46, 184–190 (2012). [PubMed] [Google Scholar]
- Hengen P. Purification of His-Tag fusion proteins from Escherichia coli. Trends Biochem. Sci. 20, 285–286 (1995). [DOI] [PubMed] [Google Scholar]
- Alvarez M., Estivill X. & de la Luna S. DYRK1A accumulates in splicing speckles through a novel targeting signal and induces speckle disassembly. J. Cell Sci. 116, 3099–3107 (2003). [DOI] [PubMed] [Google Scholar]
- Mularoni L., Ledda A., Toll-Riera M. & Albà M. M. Natural selection drives the accumulation of amino acid tandem repeats in human proteins. Genome Res. 20, 745–754 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salichs E., Ledda A., Mularoni L., Albà M. M. & de la Luna S. Genome-wide analysis of histidine repeats reveals their role in the localization of human proteins to the nuclear speckles compartment. PLos Genet. 5, e1000397 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albà M. M. & Guigó R. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 14, 549–554 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S., Brocchieri L., Bergman A. & Mrazek J. & Gentles, A. J. Amino acid runs in eukaryotic proteomes and disease associations. Proc. Natl. Acad. Sci. USA 99, 333–338 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia G. G. & Caflisch A. Computational analysis of the S. cerevisiae proteome reveals the function and cellular localization of the least and most amyloidogenic proteins. Proteins 68, 273–278 (2007). [DOI] [PubMed] [Google Scholar]
- Vavouri T., Semple J. I., Garcia-Verdugo R. & Lehner B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 138, 198–208 (2009). [DOI] [PubMed] [Google Scholar]
- Karlin S. Statistical significance of sequence patterns in proteins. Curr. Opin. Struct. Biol. 5, 360–371 (1995). [DOI] [PubMed] [Google Scholar]
- Katti M. V., Ranjekar P. K. & Gupta V. S. Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol. Biol. Evol. 18, 1161–1167 (2001). [DOI] [PubMed] [Google Scholar]
- Faux N. G. et al. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res. 15, 537–551 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S., Levy E. D., Tartaglia G. G. & Vendruscolo M. Physicochemical principles that regulate the competition between functional and dysfunctional association of proteins. Proc. Natl. Acad. Sci. USA 106, 10159–10164 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia G. G., Cavalli A., Pellarin R. & Caflisch A. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci. Publ. Protein Soc. 14, 2723–2734 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A. et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 43, D470–478 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klus P., Cirillo D., Botta Orfila T. & Gaetano Tartaglia G. Neurodegeneration and Cancer: Where the Disorder Prevails. Sci. Rep. 5, 15390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klus P., Ponti R. D., Livi C. M. & Tartaglia G. G. Protein aggregation, structural disorder and RNA-binding ability: a new approach for physico-chemical and gene ontology classification of multiple datasets. BMC Genomics 16, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campen A. et al. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept. Lett. 15, 956–963 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A. et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406 (2012). [DOI] [PubMed] [Google Scholar]
- Lobanov M. Y., Garbuzynskiy S. O. & Galzitskaya O. V. Statistical analysis of unstructured amino acid residues in protein structures. Biochemistry (Moscow) 75, 192–200 (2010). [DOI] [PubMed] [Google Scholar]
- Uversky V. N. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 22, 693–724 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo D. et al. Constitutive patterns of gene expression regulated by RNA-binding proteins. Genome Biol. 15, R13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M. et al. Discovering RNA-protein interactome by using chemical context profiling of the RNA-protein interface. Cell Rep. 3, 1703–1713 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. E. & Rubinsztein D. C. Polyalanine and polyserine frameshift products in Huntington’s disease. J. Med. Genet. 43, 893–896 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan A. Tandem repeat polymorphisms genetic plasticity, neural diversity and disease. (Springer, 2012). [Google Scholar]
- Sudol M. The WW domain binds polyprolines and is involved in human diseases. Exp. Mol. Med. 28, 65–69 (1996). [Google Scholar]
- Todd P. K. et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eyk C. L., McLeod C. J., O’Keefe L. V. & Richards R. I. Comparative toxicity of polyglutamine, polyalanine and polyleucine tracts in Drosophila models of expanded repeat disease. Hum. Mol. Genet. 21, 536–547 (2012). [DOI] [PubMed] [Google Scholar]
- Menon R. P. et al. The Role of Interruptions in polyQ in the Pathology of SCA1. PLos Genet. 9, e1003648 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M. et al. A Highly Conserved Program of Neuronal Microexons Is Misregulated in Autistic Brains. Cell 159, 1511–1523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson A. L., Bate M. A., Androulakis S. G., Bottomley S. P. & Buckle A. M. PolyQ: a database describing the sequence and domain context of polyglutamine repeats in proteins. Nucleic Acids Res. 39, D272–276 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gsponer J., Futschik M. E., Teichmann S. A. & Babu M. M. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science 322, 1365–1368 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M. & Finkbeiner S. Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dover G. A. Slips, strings and species. Trends Genet. TIG 5, 100–102 (1989). [DOI] [PubMed] [Google Scholar]
- Cooper D. N. & Krawczak M. Mechanisms of insertional mutagenesis in human genes causing genetic disease. Hum. Genet. 87, 409–415 (1991). [DOI] [PubMed] [Google Scholar]
- Reue K. & Leete T. H. Genetic variation in mouse apolipoprotein A-IV due to insertion and deletion in a region of tandem repeats. J. Biol. Chem. 266, 12715–12721 (1991). [PubMed] [Google Scholar]
- Bliskovskiĭ V. V. & Tandem D. N. A. repeats in the vertebrate genome: structure, possible mechanisms of formation and evolution. Mol. Biol. (Mosk.) 26, 965–982 (1992). [PubMed] [Google Scholar]
- Kelly R., Bulfield G., Collick A., Gibbs M. & Jeffreys A. J. Characterization of a highly unstable mouse minisatellite locus: evidence for somatic mutation during early development. Genomics 5, 844–856 (1989). [DOI] [PubMed] [Google Scholar]
- Kelly R., Gibbs M., Collick A. & Jeffreys A. J. Spontaneous mutation at the hypervariable mouse minisatellite locus Ms6-hm: flanking DNA sequence and analysis of germline and early somatic mutation events. Proc. Biol. Sci. 245, 235–245 (1991). [DOI] [PubMed] [Google Scholar]
- Gibbs M., Collick A., Kelly R. G. & Jeffreys A. J. A tetranucleotide repeat mouse minisatellite displaying substantial somatic instability during early preimplantation development. Genomics 17, 121–128 (1993). [DOI] [PubMed] [Google Scholar]
- Imbert G., Kretz C., Johnson K. & Mandel J. L. Origin of the expansion mutation in myotonic dystrophy. Nat. Genet. 4, 72–76 (1993). [DOI] [PubMed] [Google Scholar]
- Buard J. & Vergnaud G. Complex recombination events at the hypermutable minisatellite CEB1 (D2S90). EMBO J. 13, 3203–3210 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klus P. et al. The cleverSuite approach for protein characterization: predictions of structural properties, solubility, chaperone requirements and RNA-binding abilities. Bioinformatics 30, 1601–1608 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S. et al. AmiGO: online access to ontology and annotation data. Bioinformatics 25, 288–289 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.