

Abstract
Background
Early detection of oral squamous cell carcinomas (OSCCs) is urgently needed to improve the prognosis and quality of life (QOL) of patients. Oral leukoplakias (OLs), known as the most common premalignant lesions in the oral cavity, often precede OSCCs. Especially, OLs with dysplasia are known to have a high risk of malignant transformation. Here, we searched for the promoter methylation characteristic of high-risk OLs.
Methods
To identify methylation-silenced genes, a combined analysis of methylated DNA immunoprecipitation (MeDIP) − CpG island (CGI) microarray analysis and expression microarray analysis after treatment with a demethylating agent was performed in two OSCC cell lines (Ca9–22 and HSC-2). The methylation statuses of each gene were examined by methylation-specific PCR.
Results
A total of 52 genes were identified as candidates for methylation-silenced genes in Ca9-22 or HSC-2. The promoter regions of 13 genes among the 15 genes randomly selected for further analysis were confirmed to be methylated in one or more of five cell lines. In OSCC tissues (n = 26), 8 of the 13 genes, TSPYL5, EGFLAM, CLDN11, NKX2-3, RBP4, CMTM3, TRPC4, and MAP6, were methylated. In OL tissues (n = 24), seven of the eight genes, except for EGFLAM, were found to be methylated in their promoter regions. There were significantly greater numbers of methylated genes in OLs with dysplasia than in those without dysplasia (p < 0.0001).
Conclusions
OLs at high risk for malignant transformation were associated with aberrant promoter methylation of multiple genes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12885-016-2371-5) contains supplementary material, which is available to authorized users.
Keywords: Methylation, Promoter methylation, Gene silencing, Oral squamous cell carcinoma, Oral leukoplakia
Background
Oral cancer is a major public health problem worldwide, and OSCC is the most common type of oral cancer. The survival rates of patients with OSCCs have remained largely unchanged for decades, with a 5-year survival rate of around 50 % despite advances in therapeutics [1–4]. In addition to that, even when patients with advanced OSCCs survive after surgery, large tissue defects of the maxillofacial region pose a serious problem. Therefore, early and accurate detection of OSCCs is important not only to improve the survival rate of patients with OSCCs but also to maintain good QOL of the patients.
For the early detection of OSCCs, a finding of oral premalignant lesions with high-risk malignant transformation is important. OL is the most common premalignant lesion in the oral cavity, and OLs often precede OSCCs. The transition frequency from OLs into OSCCs ranges widely, from 0.13 % to 36.4 % [5]. Histologically, the presence of dysplasia is often associated with the development of OSCCs [6–8]. However, the molecular mechanism underlying malignant transformation of OLs has not been elucidated yet, and molecular markers to identify patients at higher risk of developing OSCC have not been isolated [9].
As a molecular marker to identify lesions with a higher risk of malignant transformation, DNA methylation might be useful [10–14]. The accumulation of aberrant methylation in non-cancerous lesions, such as gastric mucosae with Helicobacter pylori infection, produces epigenetic field defects leading to malignant transformation [15, 16]. In the field of oral malignancy, although many reports describe methylation silencing in OSCCs [17–21], few reports focus on methylation in OLs, especially OLs with a high-risk of malignant transformation [18, 22–26].
In this study, we aimed to identify aberrant promoter methylation in OLs at high risk of malignant transformation.
Methods
Cell lines, tissue samples, and DNA extraction
Human OSCC cell lines (Ca9–22, HSC-2, HO-1-N-1, HSC-3 and SCC-4) were purchased from the Human Science Research Resources Bank (HSRRB, Osaka, Japan). A total of 24 OL tissues (average age, 64.0 years [range, 38–84 years]; 10 male and 14 female) and a total of 26 OSCC tissues (average age, 64.6 years [range, 42–89 years]; 17 male and 9 female) were obtained from patients who underwent biopsies or operations at the University of Tokyo Hospital between Dec. 2009 and Nov. 2011. The OSCCs were graded according to the Union for International Cancer Control (UICC)’s TNM classification. OL was defined as “a predominantly white lesion of the oral mucosa that can not be characterized as any other definable lesion”[27]. The presence or absence of dysplasia in OLs is determined by the degree of cellular abnormality above the epithelial basement membrane as originally defined by the World Health Organisation (WHO) [28]. Normal oral mucosae were obtained from 16 healthy volunteers. Samples were stored in RNAlater (Applied Biosystems, Foster City, CA, USA) at -80 °C until the extraction of genomic DNA. Genomic DNA was extracted by the phenol-chloroform method. This research was approved by the research ethics committee of Graduate School of Medicine and Faculty of Medicine, The University of Tokyo, approval #2819-(1), and informed consent was obtained from all patients and volunteers. Each patient’s tobacco smoking history was obtained in an interview.
5-Aza-2′-deoxycytidine treatment
Ca9–22 and HSC-2 cells were seeded at a density of 2 × 105 cells ⁄ 10 cm plate on day 0. For 5-aza-2′-deoxycytidine (5-aza-dC; Sigma, St Louis, MO, USA) treatment, the cells were exposed to medium containing 3-μM 5-aza-dC or control medium for 24 h on days 1 and 3, and then harvested on day 5. The doses of 5-aza-dC were adjusted so that the growth of the treated cells was suppressed to 40–80 % that of nontreated cells.
Methylated DNA immunoprecipitation (MeDIP) − CpG island (CGI) microarray analysis
MeDIP − CGI microarray analysis was performed as previously described [29, 30]. Briefly, 5 μg of genomic DNA was immunoprecipitated with an anti-5-methylcytidine antibody (Diagnode, Liége, Belgium), and the precipitated DNA and input DNA were labeled with Cy5 and Cy3, respectively. A human CGI oligonucleotide microarray (Agilent Technologies, Santa Clara, CA, USA) was hybridized with the labeled probes and scanned with a G2565BA microarray scanner (Agilent Technologies). Scanned data were processed with Feature Extraction 9.1 and ChIP Analytics 1.3 software (Agilent Technologies). The signal of the probe was converted into a “Me value,” which represents the methylation level as a value from 0 (unmethylated) to 1 (methylated) [29]. Differentially methylated regions were detected by a comparison of the Me values of the two samples. When three or more consecutive probes in a locus showed differences in the Me value larger than 0.6, the locus was considered to have different methylation statuses. Promoter regions of three genes (HOXA11, NPY, and UCHL1) reported as frequently methylated in multiple cancers, including OSCCs, were used as a methylated control [31–33]. Promoter regions of three genes (ACTB, B2M, and GAPDH) known as housekeeping genes were used as unmethylated control.
Gene expression analysis by oligonucleotide microarray
Expression microarray analysis was performed by a GeneChip Human Genome U133 Plus 2.0 expression microarray (Affymetrix, Santa Clara, CA, USA). From 8 μg of total RNA, first-strand cDNA was synthesized with SuperScript III reverse transcriptase (Invitrogen) and T7-(dT)24 primer (Amersham Biosciences, Little Chalfont, UK). Double-stranded cDNA was then synthesized, and biotin-labeled cRNA was synthesized using a Bio-Array HighYield RNA transcript-labeling kit (Enzo Life Sciences, Farmingdale, NY, USA). Twenty micrograms of labeled cRNA was fragmented and hybridized to the GeneChip oligonucleotide microarray with a GeneChip hybridization control kit. The microarray was stained and scanned according to the Affymetrix protocol. The scanned data were processed using GeneChip operating software 1.4. The signal intensity of each probe was normalized so that the average signal intensity of all the probes on a microarray would be 500. The average signal intensity of all the probes for a gene was used as its transcription level. Genes were classified into those with high (>1000), moderate (250–1000), or low (<250) transcriptions according to their signal intensities [30].
Sodium bisulfite modification and methylation-specific PCR (MSP)
Sodium bisulfite treatment was performed as described previously [29] using 500 ng of DNA digested with BamHI (Toyobo, Tokyo, Japan) and suspended in 20 μl of Tris-EDTA (TE) buffer. For MSP, 1 μl of solution was used for PCR reaction with primers specific to methylated (Additional file 1: Table S1) and with primers that targeted the Alu repeat sequence; the latter were used as a control of the amount of bisulfite-treated DNA [34]. Fully methylated DNA was prepared by methylating genomic DNA using SssI-methylase (New England Biolabs, Beverly, MA, USA). Fully unmethylated DNA was prepared by amplifying genomic DNA with phi29 DNA polymerase (GenomiPhi DNA Amplification kit; GE Healthcare UK, Buckinghamshire, UK).
Statistical analysis
Associations between methylation status and various clinical parameters were evaluated by Fisher’s exact test (two-sided). SPSS Statistics (IBM Corporation, Somers, NY, USA) software version 21.0 (SPSS Inc., Chicago, IL, USA) was used for analysis. P values < 0.05 were considered to indicate significance.
Results
Chemical genomic screening of methylation-silenced genes in OSCC cell lines
To identify methylation-silenced genes in OSCC, we took a combined approach of MeDIP–CGI microarray analysis in two OSCC cell lines (Ca9–22, HSC-2) and expression microarray analysis before and after treatment of the two cell lines with a demethylating agent, 5-aza-dC. In the Ca9–22 cell line, the MeDIP − CGI microarray showed that 797 promoter CGIs were hypermethylated, and the expression microarray data showed that the expression levels of 675 genes were upregulated three-fold or more after 5-aza-dC treatment. By integrating the MeDIP − CGI microarray data and the expression microarray data, 50 genes were indicated to be methylation-silenced in the Ca9–22 cell line (Fig. 1). In the HSC-2 cell line, the MeDIP − CGI microarray showed that 513 promoter CGIs were hypermethylated, and the expression microarray data showed that the expression levels of 212 genes were upregulated three-fold or more after 5-aza-dC treatment. By integrating the MeDIP − CGI microarray data and the expression microarray data, eight genes were indicated to be methylation-silenced in the HSC-2 cell line (Fig. 1). After duplicates were removed, 52 genes were indicated to be methylation-silenced in either OSCC cell line. Of these 52 genes, 15 genes were “randomly” selected for further analysis (Fig. 1).
Fig. 1.
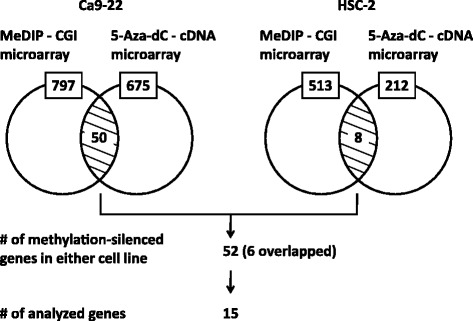
Isolation of methylation-silenced genes in two OSCC cell lines. MeDIP–CGI microarray analysis showed that 797 and 513 promoter CGIs were hypermethylated in Ca9–22 and HSC-2, respectively. 5-aza-dC – cDNA microarray analysis showed that the expression levels of 675 and 212 genes were upregulated in Ca9–22 and HSC-2, respectively. By integrating these data, 50 and 8 genes were indicated methylation-silenced in Ca9–22 and HSC-2, respectively. After duplicates were removed, 52 genes were indicated to be methylation-silenced in either cell line. Of these 52 genes, 15 genes were randomly selected for further analysis
Methylation profiles of the 15 promoter CGIs in OSCC cell lines
The selected 15 genes were analyzed in five OSCC cell lines by MSP. Of the 15 genes, 13 were methylated in one or more of these cell lines (Fig. 2a) and selected for further analysis. Representatively, a promoter CGI of TSPYL5 (Testis-specific protein, Y-encoded-like 5) was methylated in all five cell lines (Fig. 2b). A promoter CGI of TRPC4 (Transient receptor potential cation channel, subfamily C, member 4) was methylated in three cell lines.
Fig. 2.
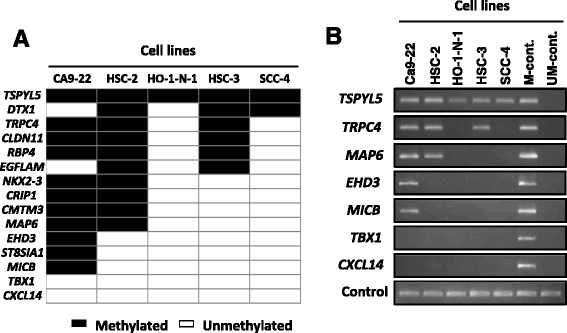
Methylation profiles of 15 promoter CGIs in five OSCC cell lines. a Promoter CGIs of 13 of the 15 genes were methylated in one or more of the five OSCC cell lines (Ca9–22, HSC-2, HO-1-N-1, HSC-3 and SCC-4). Closed box, methylated DNA detected; open box, methylated DNA not detected. b Representative results of MSP in the five OSCC cell lines are shown. Methylated DNA-specific primer sets were used to detect aberrant DNA methylation. Primer sets that target the Alu repeat sequence were used as a control of the amount of bisulfite-treated DNA. M-cont and UM-cont are fully methylated and fully unmethylated DNA, respectively
Methylation profile of the 13 promoter CGIs in OSCC tissues
Promoter methylation of genes expressed in normal oral mucosae can affect gene function, and is potentially important for malignant transformation. On the other hand, promoter methylation of genes unexpressed in normal oral mucosae is considered to be passenger methylation during OSCC carcinogenesis [34]. Therefore, the expression levels of the 13 methylated genes in OSCC cell lines were investigated in normal oral mucosae using the GEO database (GEO database; http://www.ncbi.nlm.nih.gov/geo/). Eight of those genes − CMTM3 (CKLF-like MARVEL transmembrane domain containing 3), RBP4 (Retinol-binding protein 4), NKX2–3 (NK2 homeobox 3), TSPYL5, CLDN11 (Claudin 11), EGFLAM (EGF-like, fibronectin type III and laminin G domain), CRIP1 (Cysteine-rich protein 1), and EHD3 (EH-domain containing 3) − were expressed in normal oral mucosae (GSM447398, GSM447399, GSM447408, GSM447404, GSM447406 and GSM447407 in the GEO database). Five of the 13 genes − TRPC4, MAP6 (Microtubule-associated protein 6), DTX1 (Deltex homolog 1), ST8SIA1 (ST8 alpha-N-acetyl-neuraminide alpha-2, 8-sialyltransferase 1), and MICB (MHC class I polypeptide-related sequence B) − were unexpressed in normal oral mucosae (Fig. 3).
Fig. 3.

Methylation profiles of the 13 promoter CGIs in OSCC tissues. The methylation statuses of the 13 genes showing promoter methylation in the OSCC cell lines were examined in 16 normal oral mucosae and 26 OSCC tissues. Eight genes were expressed in normal oral mucosae, six of which showed promoter methylation in the OSCC tissues. Five genes were unexpressed in normal oral mucosae, two of which were methylated in their promoter regions in the OSCC tissues. Closed box, methylated DNA detected; open box, methylated DNA not detected
Of the eight expressed genes, six (CMTM3, RBP4, NKX2–3, TSPYL5, CLDN11, and EGFLAM) showed promoter methylation in 10, 9, 8, 8, 7, and 5 of 26 OSCC tissues, respectively. Two of the five unexpressed genes (TRPC4 and MAP6) showed promoter methylation in 11 and 8 of the 26 OSCC tissues, respectively (Fig. 3).
Clinicopathological analysis was performed to examine the association between the methylation status of the eight identified genes and the clinical parameters. The categorizations of cases for each parameter are shown in Additional file 2: Table S2. The methylation status of TSPYL5 was inversely associated with the differentiation levels of OSCCs (p < 0.01) and was prone to be methylated in the tongue rather than in the other sites (p < 0.05). However, none of the other genes showed an association between methylation status and age (above average or below average), sex (male or female), stage (early or advanced), or smoking history (presence or absence).
Methylation profile of the eight promoter CGIs in OL tissues and identification of aberrant methylation in OLs with dysplasia
The methylation statuses of the eight (six expressed and two unexpressed) genes that showed promoter methylation in OSCC tissues were examined in 13 and 11 OLs with- and without dysplasia, respectively (Fig. 4a). Of the six expressed genes, five (CMTM3, RBP4, NKX2–3, TSPYL5, and CLDN11) were methylated in 6, 3, 3, 1, and 1 OL(s), respectively. The two unexpressed genes (TRPC4 and MAP6) were methylated in four and three OLs, respectively.
Fig. 4.
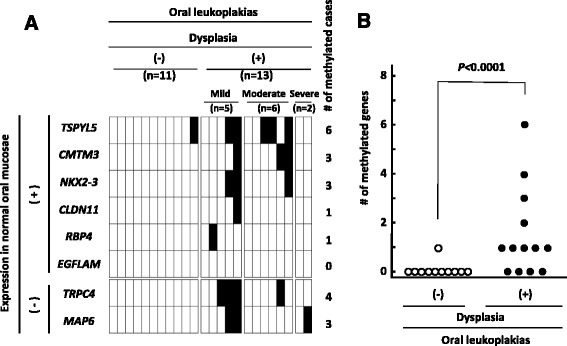
Methylation profiles of the eight promoter CGIs in OL tissues and identification of aberrant methylation in OLs with dysplasia. a The methylation statuses of the eight genes showing promoter methylation in OSCC tissues were examined in 24 OL tissues. Six and two of the eight genes were expressed and unexpressed, respectively, in normal oral mucosae. Thirteen and 11 cases were OLs with and without dysplasia, respectively. The OLs with dysplasia were categorized into mild (n = 5), moderate (n = 6), and severe grade (n = 2). Five of the six expressed genes and both of the unexpressed genes showed promoter methylation in OL(s) with dysplasia. b When the number of methylated genes was summed and compared between OLs with and without dysplasia, the methylation status showed a significant association with the presence of dysplasia (p < 0.0001)
When the number of methylated genes in an OL was compared between OLs with and without dysplasia, it was found that OLs with dysplasia had significantly more methylated genes (Fig. 4b, p < 0.0001). Only one OL tissue without dysplasia, diagnosed as an acanthosis, showed an aberrant promoter methylation. As for individual genes, TRPC4 methylation was associated with the presence of dysplasia in OLs (p = 0.04). The OLs with dysplasia were categorized into mild (n = 5), moderate (n = 6), and severe grade (n = 2), however any association between the grade of dysplasia and methylation status of the identified genes was not observed (Fig. 4a).
Clinicopathological analysis was performed to examine the association between the methylation status of each of the seven genes and the clinical parameters. The categorizations of cases for each parameter are shown in Additional file 2: Table S2. None of the genes showed an association with age, sex, stage, site, or smoking history.
Discussion
We identified seven genes aberrantly methylated in their promoter regions not only in OSCCs but also in OLs. The number of methylated genes was significantly associated with the presence of dysplasia in OLs, which is known to be associated with a high risk of malignant transformation into OSCCs [7, 8]. This result indicates that accumulation of aberrant methylation might be associated with the malignant transformation of OLs, Aberrant promoter methylation is known to accumulate also in other organs, in high-risk tissues such as gastric mucosae with Helicobacter pylori infection, in liver tissue at the precancerous stage, in colonic mucosae with ulcerative colitis, and in esophageal mucosae [15, 16, 34–38]. These previous reports support the hypothesis that the accumulation of aberrant methylation in OLs produces epigenetic field defects leading to malignant transformation.
In addition to the methylation of multiple genes, the methylation silencing of a specific gene may be functionally involved in malignant transformation. The five genes methylation-silenced in OLs (TSPYL5, CMTM3, NKX2–3, CLDN11, and RBP4) were expressed in normal oral mucosae, which indicates the functional importance of these genes [34]. Especially, TSPYL5 was most frequently methylated in OLs and was associated with differentiation levels of OSCCs (p < 0.01). The methylation silencing of TSPYL5 has been reported in esophageal cancers, gastric cancers and malignant gliomas [33, 39, 40], and TSPYL5 has been suggested to be a tumor suppressor gene [40]. Furthermore, TSPYL5 is located on chromosome 8q22, which shows loss of heterozygosity (LOH) in OSCCs with high frequency [41].
In histopathological diagnosis, the presence of dysplasia, remains the golden standard for predicting the risk of cancerization in oral premalignant lesions [42]. However, this invasive approach cannot be repeated frequently because of its poor acceptance by patients. Furthermore, a diagnosis of dysplasia is also subject to the experience of a pathologist, and the consensus among pathologists is still poor [43]. On the other hand, quantitative DNA methylation analysis is currently available and is considered to be objective. Moreover, sufficient numbers of cells for methylation analysis can be obtained by non-invasive procedures [44], such as scraping of the oral mucosae. Thus, the risk characterization using aberrant DNA methylation in patients with OLs is considered clinically feasible. Accordingly, the accumulation of identified aberrant methylation is potentially useful as a risk marker of malignant transformation from OLs to OSCCs.
We identified a novel promoter methylation associated with the risk of malignant transformation of OLs. However, the number of OL samples was limited here. Therefore, it is necessary to validate the association by another larger population.
Conclusions
Here, we identified aberrant promoter methylation of multiple genes in high-risk OLs. This result demonstrates that the accumulation of aberrant methylation in oral premalignant lesions produces an epigenetic field of cancerization.
Abbreviations
5-aza-dC, 5-aza-2′-deoxycytidine; CGI, CpG island; LOH, loss of heterozygosity; MeDIP, methylated DNA immunoprecipitation; MSP, methylation-specific PCR; OL, oral leukoplakia; OSCC, oral squamous cell carcinoma; QOL, quality of life; TE, Tris-EDTA; UICC, Union for International Cancer Control; WHO, World Health Organisation.
Acknowledgment
The authors are grateful to Dr. T. Ushiku for his assistance of pathological diagnosis. This study was supported by a Grant-in-Aid for Scientific Research (KAKENHI).
Funding
No funding.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Authors’ contributions
MA, YM, and TU conceived and designed this study. MA performed the experiments, collected and analyzed the data, and wrote the manuscript. TU and SY contributed with critical review of data analyses, interpretation of findings and critical edit of the manuscript. YM, TA, HS, and KH reviewed the manuscript. TU and TT supervised the study. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This research was approved by the research ethics committee of Graduate School of Medicine and Faculty of Medicine, The University of Tokyo, approval #2819-(1), and informed consent was obtained from all patients and volunteers.
Additional files
List of primers for MSP. (XLSX 37 kb)
Clinicopathological information of OL and OSCC cases. (XLSB 33 kb)
References
- 1.Marsh D, Suchak K, Moutasim KA, Vallath S, Hopper C, Jerjes W, Upile T, Kalavrezos N, Violette SM, Weinreb PH, et al. Stromal features are predictive of disease mortality in oral cancer patients. J Pathol. 2011;223(4):470–481. doi: 10.1002/path.2830. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka T, Tanaka M. Oral carcinogenesis and oral cancer chemoprevention: a review. Patholog Res Int. 2011;2011:431246. doi: 10.4061/2011/431246. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45(4-5):309–316. doi: 10.1016/j.oraloncology.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Wang WL, Lee CT, Lee YC, Hwang TZ, Wang CC, Hwang JC, Wang CC, Hwang JC, Tai CM, Chang CY, Tsai SS, Wang CP, et al. Risk factors for developing synchronous esophageal neoplasia in patients with head and neck cancer. Head & neck. 2011;33(1):77–81. [DOI] [PubMed]
- 5.Arduino PG, Bagan J, El-Naggar AK, Carrozzo M. Urban legends series: oral leukoplakia. Oral Dis. 2013;19(7):642–659. doi: 10.1111/odi.12065. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi T, Maruyama S, Abe T, Cheng J, Takagi R, Saito C, Kobayashi T, Maruyama S, Abe T, Cheng J, Takagi R, Saito C, Saku T. Keratin 10-positive orthokeratotic dysplasia: a new leucoplakia-type precancerous entity of the oral mucosa. Histopathology. 2012;61(5):910–20. [DOI] [PubMed]
- 7.Silverman S., Jr Demographics and occurrence of oral and pharyngeal cancers. The outcomes, the trends, the challenge. J Am Dent Assoc. 2001;132(Suppl):7S–11S. doi: 10.14219/jada.archive.2001.0382. [DOI] [PubMed] [Google Scholar]
- 8.Stelow EB, Mills SE. Squamous cell carcinoma variants of the upper aerodigestive tract. Am J Clin Pathol. 2005;124(Suppl):S96–109. doi: 10.1309/CR5JXUY3J2YGTC1D. [DOI] [PubMed] [Google Scholar]
- 9.Mithani SK, Mydlarz WK, Grumbine FL, Smith IM, Califano JA. Molecular genetics of premalignant oral lesions. Oral Dis. 2007;13(2):126–133. doi: 10.1111/j.1601-0825.2006.01349.x. [DOI] [PubMed] [Google Scholar]
- 10.Ushijima T. Detection and interpretation of altered methylation patterns in cancer cells. Nat Rev Cancer. 2005;5(3):223–231. doi: 10.1038/nrc1571. [DOI] [PubMed] [Google Scholar]
- 11.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abe M, Watanabe N, McDonell N, Takato T, Ohira M, Nakagawara A, Ushijima T. Identification of genes targeted by CpG island methylator phenotype in neuroblastomas, and their possible integrative involvement in poor prognosis. Oncology. 2008;74(1-2):50–60. doi: 10.1159/000139124. [DOI] [PubMed] [Google Scholar]
- 13.Abe M, Ohira M, Kaneda A, Yagi Y, Yamamoto S, Kitano Y, Takato T, Nakagawara A, Ushijima T. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res. 2005;65(3):828–834. [PubMed] [Google Scholar]
- 14.Abe M, Westermann F, Nakagawara A, Takato T, Schwab M, Ushijima T. Marked and independent prognostic significance of the CpG island methylator phenotype in neuroblastomas. Cancer Lett. 2007;247(2):253–258. doi: 10.1016/j.canlet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(3 Pt 1):989–995. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 16.Nakajima T, Yamashita S, Maekita T, Niwa T, Nakazawa K, Ushijima T. The presence of a methylation fingerprint of Helicobacter pylori infection in human gastric mucosae. International journal of cancer Journal international du cancer. 2009;124(4):905–910. doi: 10.1002/ijc.24018. [DOI] [PubMed] [Google Scholar]
- 17.Arantes LM, de Carvalho AC, Melendez ME, Carvalho AL, Goloni-Bertollo EM. Methylation as a biomarker for head and neck cancer. Oral Oncol. 2014;50(6):587–592. doi: 10.1016/j.oraloncology.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 18.Diez-Perez R, Campo-Trapero J, Cano-Sanchez J, Lopez-Duran M, Gonzalez-Moles MA, Bascones-Ilundain J, Bascones-Martinez A. Methylation in oral cancer and pre-cancerous lesions (Review) Oncol Rep. 2011;25(5):1203–1209. doi: 10.3892/or.2011.1205. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki E, Imoto I, Pimkhaokham A, Nakagawa T, Kamata N, Kozaki KI, Amagasa T, Inazawa J. PRTFDC1, a possible tumor-suppressor gene, is frequently silenced in oral squamous-cell carcinomas by aberrant promoter hypermethylation. Oncogene. 2007;26(57):7921–7932. doi: 10.1038/sj.onc.1210589. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa M, Nelson HH, Peters E, Ringstrom E, Posner M, Kelsey KT. Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene. 2002;21(27):4231–4236. doi: 10.1038/sj.onc.1205528. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Ramirez I, Garcia-Cuellar C, Sanchez-Perez Y, Granados-Garcia M. DNA methylation in oral squamous cell carcinoma: molecular mechanisms and clinical implications. Oral Dis. 2011;17(8):771–778. doi: 10.1111/j.1601-0825.2011.01833.x. [DOI] [PubMed] [Google Scholar]
- 22.Asokan GS, Jeelani S, Gnanasundaram N. Promoter hypermethylation profile of tumour suppressor genes in oral leukoplakia and oral squamous cell carcinoma. J Clin Diagn Res. 2014;8(10):ZC09–12. doi: 10.7860/JCDR/2014/9251.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sengupta S, Chakrabarti S, Roy A, Panda CK, Roychoudhury S. Inactivation of human mutL homolog 1 and mutS homolog 2 genes in head and neck squamous cell carcinoma tumors and leukoplakia samples by promoter hypermethylation and its relation with microsatellite instability phenotype. Cancer. 2007;109(4):703–712. doi: 10.1002/cncr.22430. [DOI] [PubMed] [Google Scholar]
- 24.Youssef EM, Lotan D, Issa JP, Wakasa K, Fan YH, Mao L, Hassan K, Feng L, Lee JJ, Lippman SM, et al. Hypermethylation of the retinoic acid receptor-beta(2) gene in head and neck carcinogenesis. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10(5):1733–1742. doi: 10.1158/1078-0432.CCR-0989-3. [DOI] [PubMed] [Google Scholar]
- 25.Chan WH, Chang KP, Yang SW, Yao TC, Ko TY, Lee YS, Tsai CL, Tsai CN. Transcriptional repression of DLEC1 associates with the depth of tumor invasion in oral squamous cell carcinoma. Oral Oncol. 2010;46(12):874–879. doi: 10.1016/j.oraloncology.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Furuta H, Kondo Y, Nakahata S, Hamasaki M, Sakoda S, Morishita K. NDRG2 is a candidate tumor-suppressor for oral squamous-cell carcinoma. Biochem Biophys Res Commun. 2010;391(4):1785–1791. doi: 10.1016/j.bbrc.2009.12.156. [DOI] [PubMed] [Google Scholar]
- 27.Lodi G, Sardella A, Bez C, Demarosi F, Carrassi A. Interventions for treating oral leukoplakia. The Cochrane database of systematic reviews. 2006;4:CD001829. doi: 10.1002/14651858.CD001829.pub3. [DOI] [PubMed] [Google Scholar]
- 28.Smith J, Rattay T, McConkey C, Helliwell T, Mehanna H. Biomarkers in dysplasia of the oral cavity: a systematic review. Oral Oncol. 2009;45(8):647–653. doi: 10.1016/j.oraloncology.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Yamashita S, Hosoya K, Gyobu K, Takeshima H, Ushijima T. Development of a novel output value for quantitative assessment in methylated DNA immunoprecipitation-CpG island microarray analysis. DNA Res. 2009;16(5):275–286. doi: 10.1093/dnares/dsp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeshima H, Yamashita S, Shimazu T, Niwa T, Ushijima T. The presence of RNA polymerase II, active or stalled, predicts epigenetic fate of promoter CpG islands. Genome Res. 2009;19(11):1974–1982. doi: 10.1101/gr.093310.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang JA, Lee BB, Kim Y, Park SE, Heo K, Hong SH, Kim YH, Han J, Shim YM, Lee YS, et al. HOXA11 hypermethylation is associated with progression of non-small cell lung cancer. Oncotarget. 2013;4(12):2317–2325. doi: 10.18632/oncotarget.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell SM, Ross JP, Drew HR, Ho T, Brown GS, Saunders NF, Duesing KR, Buckley MJ, Dunne R, Beetson I, et al. A panel of genes methylated with high frequency in colorectal cancer. BMC Cancer. 2014;14:54. doi: 10.1186/1471-2407-14-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oka D, Yamashita S, Tomioka T, Nakanishi Y, Kato H, Kaminishi M, Ushijima T. The presence of aberrant DNA methylation in noncancerous esophageal mucosae in association with smoking history: a target for risk diagnosis and prevention of esophageal cancers. Cancer. 2009;115(15):3412–3426. doi: 10.1002/cncr.24394. [DOI] [PubMed] [Google Scholar]
- 34.Kikuyama M, Takeshima H, Kinoshita T, Okochi-Takada E, Wakabayashi M, Akashi-Tanaka S, Ogawa T, Seto Y, Ushijima T. Development of a novel approach, the epigenome-based outlier approach, to identify tumor-suppressor genes silenced by aberrant DNA methylation. Cancer Lett. 2012;322(2):204–212. doi: 10.1016/j.canlet.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 35.Nagashio R, Arai E, Ojima H, Kosuge T, Kondo Y, Kanai Y. Carcinogenetic risk estimation based on quantification of DNA methylation levels in liver tissue at the precancerous stage. International journal of cancer Journal international du cancer. 2011;129(5):1170–1179. doi: 10.1002/ijc.26061. [DOI] [PubMed] [Google Scholar]
- 36.Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61(9):3573–3577. [PubMed] [Google Scholar]
- 37.Matsuda Y, Yamashita S, Lee YC, Niwa T, Yoshida T, Gyobu K, Igaki H, Kushima R, Lee S, Wu MS, et al. Hypomethylation of Alu repetitive elements in esophageal mucosa, and its potential contribution to the epigenetic field for cancerization. Cancer causes & control : CCC. 2012;23(6):865–873. doi: 10.1007/s10552-012-9955-4. [DOI] [PubMed] [Google Scholar]
- 38.Jin Z, Cheng Y, Olaru A, Kan T, Yang J, Paun B, Ito T, Hamilton JP, David S, Agarwal R, et al. Promoter hypermethylation of CDH13 is a common, early event in human esophageal adenocarcinogenesis and correlates with clinical risk factors. International journal of cancer Journal international du cancer. 2008;123(10):2331–2336. doi: 10.1002/ijc.23804. [DOI] [PubMed] [Google Scholar]
- 39.Jung Y, Park J, Bang YJ, Kim TY. Gene silencing of TSPYL5 mediated by aberrant promoter methylation in gastric cancers. Lab Invest. 2008;88(2):153–160. doi: 10.1038/labinvest.3700706. [DOI] [PubMed] [Google Scholar]
- 40.Kim TY, Zhong S, Fields CR, Kim JH, Robertson KD. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res. 2006;66(15):7490–7501. doi: 10.1158/0008-5472.CAN-05-4552. [DOI] [PubMed] [Google Scholar]
- 41.Chien HT, Liao CT, Huang SF, Chen IH, Liu TY, Jou YS, Wang HM, Hsieh LL. Clinical significance of genome-wide minimally deleted regions in oral squamous cell carcinomas. Genes Chromosomes Cancer. 2011;50(5):358–369. doi: 10.1002/gcc.20861. [DOI] [PubMed] [Google Scholar]
- 42.Liu W, Shi LJ, Wu L, Feng JQ, Yang X, Li J, Zhou ZT, Zhang CP. Oral cancer development in patients with leukoplakia--clinicopathological factors affecting outcome. PLoS One. 2012;7(4):e34773. doi: 10.1371/journal.pone.0034773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warnakulasuriya S, Reibel J, Bouquot J, Dabelsteen E. Oral epithelial dysplasia classification systems: predictive value, utility, weaknesses and scope for improvement. J Oral Pathol Med. 2008;37(3):127–133. doi: 10.1111/j.1600-0714.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- 44.Hayashi M, Bernert H, Kagohara LT, Maldonado L, Brait M, Schoenberg M, Bivalacqua T, Netto GJ, Koch W, Sidransky D, et al. Epigenetic inactivation of VGF associated with Urothelial cell carcinoma and its potential as a non-invasive biomarker using urine. Oncotarget. 2014;5(10):3350–3361. doi: 10.18632/oncotarget.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.