

Abstract
Background
Schistocephalus solidus is a well-established model organism for studying the complex life cycle of cestodes and the mechanisms underlying host-parasite interactions. However, very few large-scale genetic resources for this species are available. We have sequenced and de novo-assembled the transcriptome of S. solidus using tissues from whole worms at three key developmental states - non-infective plerocercoid, infective plerocercoid and adult plerocercoid - to provide a resource for studying the evolution of complex life cycles and, more specifically, how parasites modulate their interactions with their hosts during development.
Findings
The de novo transcriptome assembly reconstructed the coding sequence of 10,285 high-confidence unigenes from which 24,765 non-redundant transcripts were derived. 7,920 (77 %) of these unigenes were annotated with a protein name and 7,323 (71 %) were assigned at least one Gene Ontology term. Our raw transcriptome assembly (unfiltered transcripts) covers 92 % of the predicted transcriptome derived from the S. solidus draft genome assembly currently available on WormBase. It also provides new ecological information and orthology relationships to further annotate the current WormBase transcriptome and genome.
Conclusion
This large-scale transcriptomic dataset provides a foundation for studies on how parasitic species with complex life cycles modulate their response to changes in biotic and abiotic conditions experienced inside their various hosts, which is a fundamental objective of parasitology. Furthermore, this resource will help in the validation of the S solidus gene features that have been predicted based on genomic sequence.
Electronic supplementary material
The online version of this article (doi:10.1186/s13742-016-0128-3) contains supplementary material, which is available to authorized users.
Keywords: Transcriptome, RNA-seq, de novo assembly, Schistocephalus solidus, Parasite, Cestode, Flatworm, Threespine stickleback, Gasterosteus aculeatus
Data description
Background
Parasites are increasingly recognized as critically important ecological agents that play a key role in nutrient cycling, influence inter-specific interactions and engineer the physicochemical properties of ecosystems [1]. Increased connectivity between trophic levels due to parasitic infections has been systematically investigated for more than 200 years. Peter Christian Abildgaard, a Danish veterinarian, was the first to identify a parasite being transmitted from one species to another via predation [2]. Abildgaard discovered the existence of complex parasite life cycles after observing that threespine sticklebacks (Gasterosteus aculeatus) and various fish-eating birds seemed to be infected by different forms of the same flatworm, named by Abildgaard as Taenia gasterostei. In a classic experiment, Abildgaard showed that ducks could acquire and maintain healthy T. gasterostei following the ingestion of infected threespine sticklebacks. This was the first demonstration of a complex life cycle, in which a parasite is transmitted from one host species to another [2]. After 155 years, in 1945, J.D. Smyth utilized T. gasterostei, by then named Schistocephalus solidus, as an experimental model. In an influential publication, Smyth described how S. solidus could be cultivated in vitro in the laboratory using specific experimental conditions to mimic the conditions within different hosts [3]. The infection of threespine stickleback by S. solidus has now become a model system in various research areas, including evolution, physiology, immunology, ecology, behavior (reviewed in [4]) and genomics of host parasite interactions, as fully sequenced genomes for G. aculeatus [5] and S. solidus [6, 7] are now available.
The life cycle of S. solidus can be simulated in vivo and in vitro using Smyth’s techniques [4, 8, 9]. In brief, eggs hatch in the water to release the coracidium larvae. Any species of cyclopoid copepod can eat the coracidium, after which the larva develops into the procercoid stage in the body cavity of its crustacean host. Threespine sticklebacks feed on infected copepods, allowing the procercoid to migrate into the coelom of the fish. The procercoid then undergoes a transformation to become an early, immature plerocercoid entering in a growth phase that will last 8–16 weeks. This growth period results in gains of up to 300 times its initial mass [10, 11]. Once the plerocercoid reaches a critical body mass (50 mg or more [12]) it is able to infect its final host, which is typically any species of piscivorous bird [13]. Trophic transmission of the competent (‘infective’) plerocercoid allows the parasite to complete its sexual maturation and reproduction, either by self- or cross-fertilization, in the digestive tract of the bird [14, 15]. The eggs are finally released in the water through the bird’s faeces.
Despite complex parasite life cycles being first described more than 200 years ago, the question of why and how some parasites evolved to acquire this complex strategy still remains elusive [16]. One approach to understanding the evolution of these strategies involves characterizing the molecular mechanisms that allow the parasite to transition from one stage to another as it transfers through several different hosts. The transcriptome of the parasite, consisting of all of the mRNA molecules that can be produced by the organism, represents a critical level of biological organization. It plays a key role in modulating the concentration of proteins at the interface of the molecular interactions between the parasite and its host [17, 18]. Changes in gene expression represent a major mechanism by which phenotypic traits can be ‘fine-tuned’ to achieve success in variable environments [19], including those experienced by parasites as they transit successive host species.
Understanding how parasites interact with their host environments and how they respond to changes in the biotic and abiotic conditions present at each stage is a fundamental objective of parasitology. Understanding these interactions at a molecular level requires the development of genetic resources. Here we present a comprehensive de novo transcriptome sequence that covers three key developmental life cycle stages of S. solidus that occur in vertebrate hosts, namely the non-infective plerocercoid, the infective plerocercoid and the adult. This experimental host-parasite system also represents a unique opportunity to collect valuable empirical data that will increase our knowledge of how parasites impact ecological and evolutionary processes, through effects on host behavior, sexual development and physiology [20–25]. Finally, this first large-scale transcriptomic dataset will help in the validation of the S. solidus gene features that have been predicted based on genome sequence.
Specimen collection and laboratory infections
Parasites used in this study were obtained from experimentally-infected, laboratory-raised threespine sticklebacks at the University of Leicester (Leicester, England) according to previously described protocols [3, 26, 27]. Culturing and RNA extraction protocols are also available via the protocols.io repository [28]. Parasite eggs utilized in these experimental infections were previously produced from adult worms following the in vitro culture of plerocercoids [3] extracted from wild-caught threespine sticklebacks collected from Clatworthy Reservoir in Somerset, England (51°06’86”N, 3°35’39”W). Experimentally-infected fish were the F1 progeny of adult parents collected from the same lake as the parasite population, and from two other locations in the United Kingdom: Carsington Water in Derbyshire, England (53°06’05.09”N, 1°64’36.58”W) and Inverleith Pond in Edinburgh, Scotland (55°96’78.57”N, 3°21’67.21”W).
In brief, parasite eggs were placed in Petri dishes filled with tap water for two weeks and exposed to light to stimulate hatching. Hatched larvae were fed to laboratory-cultured copepods (Cyclops strennus Fischer) and three to four weeks later they were fed to the laboratory-raised threespine sticklebacks. Fish exposed to infected copepods were randomly selected and killed in a benzocaine solution (15 mM) between 10 and 17 weeks post-exposure. The timing of the sampling and subsequent dissection allowed both ‘non-infective’ plerocercoids (<50 mg) and ‘infective’ plerocercoids (>50 mg) to be recovered aseptically from the coelom of infected fish [12]. Non-infective worms were collected between 10 and 13 weeks post-infection, while infective worms were collected between 16 and 17 weeks post-infection. Additional infective plerocercoids extracted aseptically from naturally infected threespine sticklebacks caught from Clatworthy Reservoir were cultured in vitro to simulate the avian digestive tract environment [3] in order to obtain samples of the sexually mature adult worm life stage.
RNA extraction and library preparation
All of the worms were washed carefully with UltraPure RNase free water (Ambion Inc., Austin, TX, USA) immediately after being extracted aseptically from the fish coelom (non-infective and infective plerocercoids) or collected from incubated test tubes (adults), then quickly cut with a scalpel into square pieces of five millimeters by five millimeters, placed into RNAlater (Ambion Inc., Austin, TX, USA) at 4 °C overnight, then transferred the next morning to −80 °C until RNA extraction. Total RNA was extracted from S. solidus worms following the method developed by Chomczynski & Sacchi [29], based on acid guanidinium thiocyanate-phenol-chloroform (Trizol® reagent, Invitrogen, Carlsbad, CA, USA).
Total RNA quality assessment using an Agilent 2100 Bioanalyzer® (Agilent, Santa Clara, CA, USA) revealed profiles similar to sub-optimal, or potentially degraded sample (see [30] for sample-specific profiles). However, this profile has been consistently observed across multiple independent cestode extractions (unpublished data) and has also been documented in other taxa from the Platyhelminthes phylum (classes Trematoda, Tricladida), Nematoda phylum (classes Chromadorea, Adenophorea) and other taxonomic groups including Arthropoda and therefore is not likely to indicate degradation [31]. These profiles are most likely the result of thermal conversion producing gap-deletion patterns in the 28S rRNA, ultimately leading to its fragmentation [31]. All of our RNA samples exhibited this same gap-deletion pattern.
Total RNA samples from 14 individual worms were used to produce individual TruSeq cDNA Illumina sequencing libraries (San Diego, CA, USA) according to the manufacturer’s protocol (see Table 1). Libraries were evenly and randomly distributed into three Illumina HiSeq 2000 lanes so that each lane contained samples from all three developmental stages. Sequencing was performed on the Illumina HiSeq 2000 system at Centre de Recherche du CHU de Québec (Québec, QC, Canada) to generate a total of 375 million 100 bp paired-end reads. RNA from one adult worm was used to prepare an additional Illumina TruSeq sequencing library used to perform preliminary optimization tests on assembly parameters (see Table 1, sample cltw.A.01). This library was sequenced on the MiSeq system at Plateforme d’analyses génomiques (Institut de Biologie Intégrative et des Systèmes, Université Laval, Québec) and yielded 19.4 million 300 bp paired-end sequences (deposited into the NCBI Sequence Read Archive (SRA) with accession number SAMN04296611 associated with BioProject PRJNA304161).
Table 1.
Schistocephalus solidus specimens from three different developmental stages used to generate the de novo reference transcriptomeb
| Sample ID | Mass (mg) | Stagea | Library size (no. raw reads) | Platform | Ave. read quality (PHRED score) |
|---|---|---|---|---|---|
| ssol.cltw.NI.05 | 1.7 | NI | 95.7 M | HiSeq 2000 | 37 |
| ssol.cltw. NI.08 | 1.5 | NI | 57.6 M | HiSeq 2000 | 37 |
| ssol.cltw. NI.12 | 5.2 | NI | 29.5 M | HiSeq 2000 | 37 |
| ssol.cltw. NI.13 | 1.8 | NI | 69.6 M | HiSeq 2000 | 37 |
| ssol.cltw. NI.14 | 3.4 | NI | 59.8 M | HiSeq 2000 | 37 |
| ssol.cltw. NI.22 | 7.7 | NI | 49.1 M | HiSeq 2000 | 37 |
| ssol.cltw. NI.26 | 13.1 | NI | 53.7 M | HiSeq 2000 | 37 |
| ssol.cltw. NI.63 | 102 | I | 37.5 M | HiSeq 2000 | 37 |
| ssol.cltw. NI.67 | 90.1 | I | 42.3 M | HiSeq 2000 | 37 |
| ssol.cltw. I.98–1 | 101.2 | I | 36.8 M | HiSeq 2000 | 37 |
| ssol.cltw.I.98–2 | 108.1 | I | 56.3 M | HiSeq 2000 | 37 |
| ssol.cltw.A.01 | 164.5 | A | 38.8 M | MiSeq | 38 |
| ssol.cltw.A.03 | 321 | A | 61.2 M | HiSeq 2000 | 37 |
| ssol.cltw.A.07 | 329.4 | A | 50.2 M | HiSeq 2000 | 37 |
| ssol.cltw.A.12 | 356 | A | 51.7 M | HiSeq 2000 | 37 |
a NI non-infective, I infective, A adult
bWorms in bold were selected as the three representative samples to be used to perform the initial raw de novo assembly. All 14 HiSeq libraries were then used to eliminate redundancy in the final dataset and increase assembly quality, while sample cltw.a-01 (in italic) was used to perform preliminary optimization tests on assembly parameters
Transcriptome assembly
Three worm libraries of similar size, one from each life stage were used for the initial de novo assembly (Table 1, in bold). Only one individual per life stage was used to obtain an initial set of raw de novo transcripts in order to i) minimize redundancy in assembled contigs due to allele splitting, ii) obtain the best possible balance between true transcript detection and false positives, and iii) obtain the maximum diversity of transcripts spanning all three life stages, i.e. characterize a maximum number of stage-specific genes [32–34]. In brief, reads from these pre-defined “representative worms” were combined for the initial assembly, then all 14 HiSeq libraries were used to reduce the final assembly and reduce contig redundancy as much as possible. The complete assembly pipeline is summarized in Fig. 1 and is available for download on Github [35].
Fig. 1.
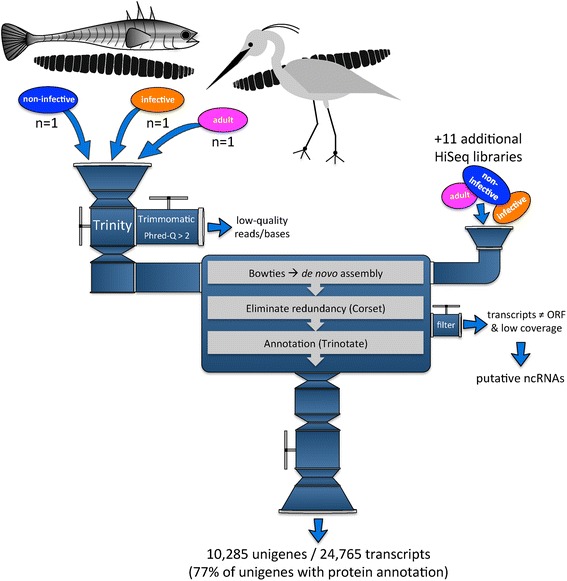
De novo assembly method used in the construction of a reference transcriptome for Schistocephalus solidus. Sequencing libraries from three developmental stages of S. solidus, non-infective plerocercoid, infective plerocercoid and adult, were trimmed (PHRED > 2, read length > 60 nucl.), concatenated and assembled de novo (1 library per stage). Next, the three libraries initially used to produce the de novo assembly, in addition to 11 libraries spanning the same three developmental stages (non-infective plerocercoid = 6 libraries, infective plerocercoid = 3 libraries, adult = 2 libraries) were aligned back to the de novo assembly. corset was used on the resulting alignment to eliminate redundancy by creating clusters of similar sequences, called ‘unigenes’. Transcripts were finally annotated through the Trinotate pipeline and transcripts poorly supported by protein-coding evidence were discarded, along with transcripts showing a low average coverage, i.e. CPM < 10 in 50 % of the samples in one group. The final transcriptome contains 24,765 transcripts accounting for 10,285 unigenes, of which 77 % could be annotated
Using Trimmomatic v0.33 [36], sequencing adaptors were removed from raw reads, reads were quality trimmed and then selected for minimum length (reads ≥ 60 bp were retained). As the primary goal of this study was to perform a de novo assembly, a less-stringent trimming threshold was selected (Phred score = 2) as suggested for increased de novo assembly quality [37]. Trimmed libraries for the three representative samples were then concatenated and used as the input for a de novo assembly through the Trinity pipeline v.2.0.6 with default parameters and a minimum contig length of 150 nucleotides [38]. Next, trimmed sequencing reads for the 14 HiSeq libraries (deposited into the NCBI SRA under accession number SRP066813, associated with BioProject PRJNA304161) were aligned with Bowtie 2 v.2.1.0 [39] against the de novo assembly, allowing multi-mapping for each read. Corset v.1.04 [40] was then utilized to cluster transcripts into unigenes based on sequence similarity and read counts (total of 14 sorted BAM files, i.e. one per individual library). Open reading frames (ORFs) were predicted for all transcripts with Transdecoder v.2.0.1 [38]. The raw transcriptome was finally filtered to discard transcripts that were poorly supported by protein-coding evidence (ORF length < 10 amino acids) and with low read counts (transcripts with CPM > 15 in at least three samples in one of the three developmental stages were kept). Considering the increasing evidence for key biological functions performed by non-coding RNAs [41], transcripts that did not contain ORFs that met our criteria were considered as “potentially non-coding” for further analysis and are provided as supplementary resource associated with this publication [30]. Results of the assembly and filtering steps can be found in Table 2.
Table 2.
De novo assembly and annotations metrics for the transcriptome of the cestode Schistocephalus solidus
| De novo assembly | |
| Assembled bases | 195 089 904 |
| Assembled transcripts | 293 731 |
| Unigenes – Unfiltered | 115 318 |
| Unigenes – Expression filter | 12 291 |
| Unigenes – Expression & ORF filters (transcripts) | 10 285 (24 765) |
| Average transcripts per filtered unigene | 2.41 |
| Sum of filtered transcripts (Mbp) | 367.83 |
| Average length (bp) – Filtered transcripts (min - max : median) | 2 684 (174–25 376 : 2168) |
| Annotation | |
| Unigenes with protein name | 7 920 (77 %) |
| Unigenes with Gene Ontology (% of unigenes) | 7 323 (71 %) |
| Proteins with complete ORF (% of unigenes) | 20 335 (82 %) |
| Unigenes/transcripts with KEGG ID (% of unigenes) | 4 270/7 798 (35 %) |
Annotation
Annotation was performed on predicted protein amino acid sequences using Trinotate v.2.0.2 [38] to assign a protein name and GO terms to each transcript. Predicted proteins were analyzed by several methods for functional annotation, starting with a sequence homology search on UniProtKB/Swissprot (downloaded 11 May 2015). Protein sequences were then mined for functional domains through HMMER v3.1 [42] and Pfam v.28.0 [43]. Signal peptides and transmembrane domains were assigned to coding sequences according to hidden Markov model-based prediction algorithms implemented in SignalP v.4.1 [44] and tmHMM v.2.0 [45], respectively. Transcripts were finally compared to curated annotation databases including EMBL Uniprot [46], KEGG [47], eggNOG v.3.0 [48] and GO pathways [49]. The steps and scripts built to implement this annotation pipeline are available on Github [50]. Transvestigator [51] was finally used to prepare the data for submission to NCBI Transcriptome Shotgun Assembly (TSA), by confirming that ORFs were on the positive strand and that each transcript was associated with at least one ORF. Annotation information based on the results obtained with Trinotate was also included in the TSA submission (accessible through GigaDB accession publication associated with this publication, see [30]).
Comparison with gene-prediction models
A predicted transcriptome for S. solidus is currently available on WormBase v1.5.4 [6]. These gene predictions were generated by the Parasite Genomics group at the Wellcome Trust Sanger Institute from the genome by a combination of programs such as MAKER [52] and Augustus [53], as well as protein sequence homology searches against the taxonomically nearest reference helminth genome. Although gene prediction models can generate informative data when working with genome sequences, an essential task in characterizing gene features in a newly sequenced genome is to confirm and validate predicted coding sequences with empirical mRNA data [54]. The de novo transcriptome generated here was thus compared to the predicted version of the transcriptome and the complete genome from WormBase using two complementary approaches. First, a reciprocal best-hit analysis was performed, and second, our mRNA sequencing reads were aligned to the reference genome. We expected that only a partial representation of the predicted coding sequences on WormBase would be observed in the data presented here. This prediction stems from the fact that our de novo transcriptome was assembled using mRNA sequences for parasite stages in the fish and the bird hosts only. The developing embryo, free-swimming larval and procercoid stages were not considered in this assembly.
Results of a standard BLAST approach showed that 7,399 (72 %) of de novo unigenes (i.e. unigenes with valid ORF(s) and evidence of expression) give significant blast results (e < 1e-10, minimum 50 % overlap) when compared to the WormBase predicted transcriptome. Using reciprocal best hit BLAST [55] reduces the number of de novo unigenes with significant matches on the predicted transcriptome to 5,176 (50 %). Using the genome as a target, 9,877 (96 %) of our de novo unigenes return a significant blastn match (e-value < 1e-10, minimum 50 % overlap) on the WormBase genome with an average and a median sequence similarity of 92 % (range = 62–100 %, mode = 100 %, see Additional file 1). Recent work on the landscape genetics of S. solidus in Alaska (USA) revealed significant genetic differences among populations from several lakes along a gradient of isolation by distance [56]. The strong population structure and low admixture levels found in these lakes are indicative of low migration rates among populations. This could help explain why we obtain a median similarity of 92 % and not higher when comparing populations from the UK (de novo transcriptome generated in this study) with populations from Germany (WormBase genome).
As a complement to the BLAST approach, reads used to construct the de novo assembly were also aligned on the genome and predicted transcriptome using the bwa-mem algorithm [57] with default parameters. Mapping results indicate that 84 % of the reads successfully align on the genome (MAPQ ≥ 15), and 51 % of the reads successfully align on the predicted transcriptome. In total, 15,840 (78 %) predicted transcripts show some evidence of expression. The partial correspondence between the de novo assembly and the predicted transcriptome, as shown by the two complementary approaches, confirms our initial prediction that only a subset of all the possible genes in S. solidus would be represented in the stages assessed in this de novo transcriptome. On the other hand, 21 % of the de novo assembled transcripts exhibiting a valid ORF and evidence of expression across several individual worms were not represented in the predicted transcriptome. These transcripts were however detected in the genome, highlighting the importance of using RNA-seq data to further improve genome assemblies and annotations based on gene prediction models [58]. As only 84 % of the reads from the de novo transcriptome align on the genome (and not 100 %) may indicate i) regions missing from the current reference genome, ii) reads not mapping properly due to low complexity sequences, or iii) that polymorphisms between the individuals prevents mapping. Globally, these results call for a collaborative strategy taking advantage of multiple sources of information of a genomic and transcriptomic nature, towards an integrated and complete characterization of the genome structure of S. solidus.
An un-gapped blastn analysis of our raw transcriptome (unfiltered transcripts) against the WormBase predicted transcriptome revealed that it covers 92 % (18,608 transcripts) of the predicted transcripts, suggesting that some of the transcripts discarded in our pipeline based on protein-coding evidence and expression levels might in fact be true transcripts encoded in the genome. We consider these transcripts as ‘putative ncRNAs’. They are available in the public repository associated with this publication [30].
Novel resource for phylogenomic analyses
Establishing the evolutionary relationships among parasitic species represents a fundamental step towards understanding how parasitism evolved and how complex life cycles were acquired by certain taxa [59–62]. To date, very few genomes from parasitic worm species belonging to the Pseudophyllidea order (Phylum: Platyhelminthes, Class: Cestoda) have been fully sequenced [63] and currently there are no empirical transcriptomic resources available for any species belonging to the Schistocephalidae family, apart from the work on Schistocephalus solidus presented here. This new resource gives us an opportunity to fill this knowledge gap about evolutionary relationships. We assessed sequence homology between our de novo transcriptome and transcriptomes from seven other parasitic worm species by using OrthoMCL v.2.0.9 [64] according to the protocol developed by Fischer et al. [65]. Specifically, we included coding sequences from Wormbase ParaSite [66] for Hymenolepis microstoma (cestode, Hymenolepiditae family), Taenia solium, Echinococcus granulosus, Echinococcus multilocularis (cestodes, Taenidae family, described in [67]), Spirometra erinaceieuropaei (cestode, Diphyllobothriidae family, described in [63]) in addition to Schistosoma mansonii (trematode, Schistosomatidae family) and Clonorchis sinensis (tremadote, Opisthorchiidae) as outgroups in our analysis. Phylogenetic relationships among these eight species were built using a set of 565 groups of orthologous genes identified with OrthoMCL, each containing one single-copy gene per worm species (“single-copy orthologs”). Per species, single-copy orthologs were concatenated and aligned against one another using MAFFT v.7.245 [68]. Well-aligned regions were extracted using Gblocks 0.91b [69], which resulted in 80,865 aligned amino acid positions in 1,607 selected blocks. This final alignment was used to construct a phylogenetic tree with RAxML v.8.2.0 [70], following a gamma model rate of heterogeneity, combined with a WAG substitution matrix and a maximum likelihood search of 100 bootstraps. The resulting tree presented in Fig. 2 was visualized with Dendroscope v.3.4.4 [71]. Orthologous amino acid sequences for each species are provided in the GigaDB accession associated with this publication, as well as a full list of all potential orthologs between the species. In total, 8,329 unigenes (80 %) were identified as putative orthologs by OrthoMCL, meaning they show a sufficiently high sequence similarity to be aligned with at least one of the worm species included in the analysis. The results also suggest that 1,637 of the filtered unigenes (16 %) could potentially be specific to S. solidus since OrthoMCL identified them as singletons (no orthologous hit to any species included in the analysis). These specific unigenes may hold important species-specific information about this species and will be important to explore further. Of this subset, 43 were annotated with putative protein names, and 57 % remained as unknowns.
Fig. 2.
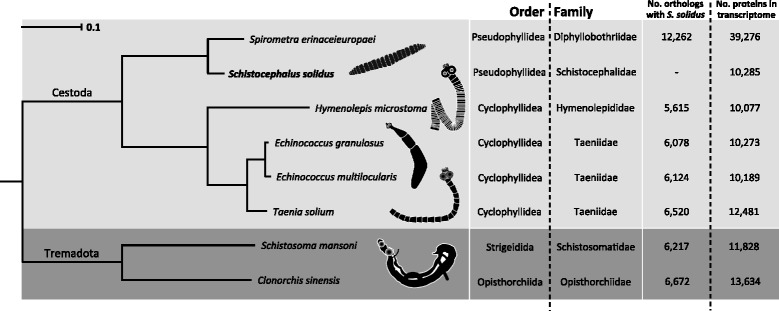
Phylogenetic relationships between Schistocephalus solidus (Schistocephalidae) and seven other parasitic worm species. These other species include five cestodes from the Cyclophyllidea and Pseudophyllidea orders and two trematodes (outgroups). More specifically, species from the cestode phylum include Hymenolepis microstoma (rodent tapeworm), Taenia solium (pork tapeworm), Echinococcus multilocularis (fox tapeworm), Echinococcus granulosus (dog tapeworm) and Spirometra erinaceieuropaei (responsible for the sparganosis infection), while the trematode outgroups are represented by Schistosoma mansoni (responsible for diseases such as schistosomiatis) and Clonorchis sinensis (Chinese liver fluke). Bootstrap values were all 100. Total number of single-copy orthologs used to produce the phylogenetic tree = 4 520 (distributed in 565 core orthologous groups). The number of orthologs shared with S. solidus is defined as the number of amino acid sequences in a given species that are part of an orthologous group identified by orthoMCL that also contains sequences from S. solidus
In conclusion, despite its position as a historical model system for the development of laboratory techniques now widely used in parasitology [72], S. solidus still remains an understudied species in terms of genomics. With novel resources such as the de novo transcriptome described here, S. solidus may additionally be a model for the study of conserved functions among parasitic worms, as well as offering species-specific genomic traits, among which may provide insight on key components of the complex life cycle of this model parasite.
Availability and requirements
Project name: Corset & Trinotate pipelines
Project home page: https://github.com/fohebert/corset_pipeline & https://github.com/fohebert/Trinotate_pipeline
Operating system(s): Unix.
Programming language: Bash.
Other requirements: TRIMMOMATIC, Trinity, Bowtie, CORSET, Samtools, limma-voom.
License: GNU GPL v3
None.
Availability of supporting data
The raw datasets supporting the results of this article are available in the GigaDB repository associated with this publication [30]. All the sequencing data are available and associated with the NCBI BioProject PRJNA304161. Culturing and RNA extraction protocols are also available via the protocols.io repository [28].
Ethical statement
Fish were captured under U.K. Environment Agency permit and with the permission of the landowner. All experiments were undertaken under a U.K. Home Office license (PPL80/2327), in accordance with local and national regulations, and in line with ABS/ASAB guidelines for the ethical treatment of animals in behavioral research.
Acknowledgements
This work was funded by a FRQ-NT Projet de Recherche en Équipe grant to NAH and CRL, a Natural Science and Engineering Research Council of Canada (NSERC) Discovery grant to NAH, a NSERC Vanier Canada Graduate Scholarship and a Ressources Aquatiques Québec (RAQ) travel fellowship to FOH, and a UK BBSCR MITBP fellowship to SG. CRL holds the Canada Research Chair in Evolutionary Cell and Systems Biology. FOH would like to thank Ben J. G. Sutherland for reading and commenting on the first version of the manuscripts and for the numerous pieces of advice on technical and theoretical aspects of this work.
Abbreviations
- CPM
counts per million
- GO
Gene Ontology
- ncRNA
non-coding RiboNucleic Acid
- ORF
open reading frame
- RNA
Ribonucleic acid
- SRA
Sequence Read Archive
- TSA
Transcriptome Shotgun Assembly
Additional file
Sequence comparison between the genome and the de novo transcriptome. Description: Distribution of sequence similarities between the reference genome from WormBase and the de novo transcriptome. (PDF 450 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FOH and SG carried out the laboratory infections and fish dissections. FOH conceived the study, participated in its design, carried out the RNA extractions, prepared the Illumina sequencing libraries, carried out the bioinformatic analyses and drafted the manuscript. IB funded the study, participated in its design and in the fish dissections in the laboratory. CRL and NAH funded the study, participated in its design and coordination and helped draft the manuscript. All authors read and approved the final manuscript.
References
- 1.Hatcher MJ, Dick JT, Dunn AM. Diverse effects of parasites in ecosystems: linking interdependent processes. Front Ecol Environ. 2012;10:186–94.
- 2.Abildgaard PC. Almindelige Betragtninger Over Indvolde-Orme, Bemaekninger Ved Hundstellens Baendelorm, Og Beskrivelse Med Figurer Af Nogel Nye Baendelorme. Skrivt Naturhist Selskab Københ. 1790;1:26–64. [Google Scholar]
- 3.Smyth DJ. Studies on tapeworm physiology, the cultivation of Schistocephalus solidus in vitro. J Exp Biol. 1946;23:47–70. doi: 10.1242/jeb.23.1.47. [DOI] [PubMed] [Google Scholar]
- 4.Barber I, Scharsack JP. The three-spined stickleback-Schistocephalus solidus system: an experimental model for investigating host-parasite interactions in fish. Parasitology. 2010;137:411. doi: 10.1017/S0031182009991466. [DOI] [PubMed] [Google Scholar]
- 5.European Bioinformatic Institute. Ensembl genomes: Gasterosteus aculeatus. http://ensembl.org/Gasterosteus_aculeatus/Info/Index. Accessed 16 Apr 2016.
- 6.WormBase ParaSite: Schistocephalus solidus genome assembly. WormBase database. http://parasite.wormbase.org/Schistocephalus_solidus_prjeb527/Info/Index/. Accessed 16 Apr 2015.
- 7.Howe KL, Bolt BJ, Cain S, Chan J, Chen WJ, Davis P, et al. WormBase 2016: expanding to enable helminth genomic research. Nucleic Acids Res. 2016;44:D774–80. doi: 10.1093/nar/gkv1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smyth JD. In vitro cultivation of parasitic helminths. London: CRC Press; 1990. [Google Scholar]
- 9.Jakobsen PJ, Scharsack JP, Hammerschmidt K, Deines P, Kalbe M, Milinski M. In vitro transition of Schistocephalus solidus (Cestoda) from coracidium to procercoid and from procercoid to plerocercoid. Exp Parasitol. 2012;130:267–73. doi: 10.1016/j.exppara.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Hammerschmidt K, Kurtz J. Surface carbohydrate composition of a tapeworm in its consecutive intermediate hosts: individual variation and fitness consequences. Int J Parasitol. 2005;35:1499–507. doi: 10.1016/j.ijpara.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 11.Scharsack JP, Koch K, Hammerschmidt K. Who is in control of the stickleback immune system: interactions between Schistocephalus solidus and its specific vertebrate host. P R Soc B. 2007;274:3151–8. doi: 10.1098/rspb.2007.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tierney JF, Crompton DW. Infectivity of plerocercoids of Schistocephalus solidus (Cestoda: Ligulidae) and fecundity of the adults in an experimental definitive host. Gallus gallus J Parasitol. 1992;78:1049–54. [PubMed] [Google Scholar]
- 13.Clarke AS. Studies on the life cycle of the pseudophyllidean cestode Schistocephalus solidus. Proceedings of the Zoological Society of London. Blackwell Publishing Ltd. 1954;124:257–302. [Google Scholar]
- 14.Kiessling F. Ueber den Bau von Schistocephalus dimorphus Creplin und Ligula simplicissima Rudolphi. 1882. [Google Scholar]
- 15.Joyeux C, Baer JG. Faune De France 30: Cestodes. Fédération Française des sociétés de sciences naturelles - Office central de faunistique. Paris: Paul Lechevalier; 1936. [Google Scholar]
- 16.Jackson AP. Preface. The evolution of parasite genomes and the origins of parasitism. Parasitology. 2015;142(Suppl 1):S1–5. doi: 10.1017/S0031182014001516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hébert FO, Aubin-Horth N. Ecological genomics of host behavior manipulation by parasites. Adv Exp Med Biol. 2014;781:169–90. doi: 10.1007/978-94-007-7347-9_9. [DOI] [PubMed] [Google Scholar]
- 18.Jovanovic M, Rooney MS, Mertins P, Przybylski D, Chevrier N, Satija R, et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science. 2015;347:1259038. doi: 10.1126/science.1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aubin-Horth N, Renn SCP. Genomic reaction norms: using integrative biology to understand molecular mechanisms of phenotypic plasticity. Mol Ecol. 2009;18:3763–80. doi: 10.1111/j.1365-294X.2009.04313.x. [DOI] [PubMed] [Google Scholar]
- 20.Giles N. Behavioural effects of the parasite Schistocephalus solidus (Cestoda) on an intermediate host, the three-spined stickleback, Gasterosteus aculeatus L. Anim Behav. 1983;31:1192–4. doi: 10.1016/S0003-3472(83)80025-6. [DOI] [Google Scholar]
- 21.Folstad I, Hope AM, Karter A, Skorping A. Sexually selected color in male sticklebacks: A signal of both parasite exposure and parasite resistance? Oikos Nordic Society Oikos. 1994;69:511–5. [Google Scholar]
- 22.Ranta E. Schistocephalus infestation improves prey-size selection by three-spined sticklebacks, Gasterosteus aculeatus. J Fish Biol. 1995;46:156–8. doi: 10.1111/j.1095-8649.1995.tb05954.x. [DOI] [Google Scholar]
- 23.Barber I, Walker P, Svensson PA. Behavioural responses to simulated avian predation in female three spined sticklebacks: the effect of experimental schistocephalus solidus infections. Behaviour BRILL. 2004;141:1425–40. doi: 10.1163/1568539042948231. [DOI] [Google Scholar]
- 24.Heins DC, Baker JA, Toups MA, Birden EL. Evolutionary significance of fecundity reduction in threespine stickleback infected by the diphyllobothriidean cestode Schistocephalus solidus. Biol J Linn Soc. 2010;100:835–46. doi: 10.1111/j.1095-8312.2010.01486.x. [DOI] [Google Scholar]
- 25.Quinn TP, Kendall NW, Rich HB, Chasco BE. Diel vertical movements, and effects of infection by the cestode Schistocephalus solidus on daytime proximity of three-spined sticklebacks Gasterosteus aculeatus to the surface of a large Alaskan lake. Oecologia. 2012;168:43–51. doi: 10.1007/s00442-011-2071-4. [DOI] [PubMed] [Google Scholar]
- 26.Barber I, Arnott SA, Braithwaite VA, Andrew J, Huntingford FA. Indirect fitness consequences of mate choice in sticklebacks: offspring of brighter males grow slowly but resist parasitic infections. Proc Biol Sci The Royal Society. 2001;268:71–6. doi: 10.1098/rspb.2000.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barber I, Svensson PA. Effects of experimental Schistocephalus solidus infections on growth, morphology and sexual development of female three-spined sticklebacks, Gasterosteus aculeatus. Parasitology. 2003;126:359–67. doi: 10.1017/S0031182002002925. [DOI] [PubMed] [Google Scholar]
- 28.Hébert FO, Grambauer S, Barber I, Landry CR. Protocols for “Transcriptome sequences spanning key developmental states as a resource for the study of the cestode Schistocephalus solidus, a threespine stickleback parasite”. protocols.io. http://dx.doi.org/10.17504/protocols.io.ew9bfh6. [DOI] [PMC free article] [PubMed]
- 29.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- 30.Hébert FO, Grambauer S, Barber I, Landry CR, Aubin-Horth N. Resource for “Transcriptome sequences spanning key developmental states as a resource for the study of the cestode Schistocephalus solidus, a threespine stickleback parasite.” GigaScience Database. http://dx.doi.org/10.5524/100197 [DOI] [PMC free article] [PubMed]
- 31.McCarthy SD, Dugon MM, Power AM. ‘Degraded’ RNA profiles in Arthropoda and beyond. PeerJ. 2015;3:e1436. doi: 10.7717/peerj.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cahais V, Gayral P, Tsagkogeorga G, Melo-Ferreira J, Ballenghien M, Weinert L, et al. Reference-free transcriptome assembly in non-model animals from next-generation sequencing data. Mol Ecol Resour. 2012;12:834–45. doi: 10.1111/j.1755-0998.2012.03148.x. [DOI] [PubMed] [Google Scholar]
- 33.Vijay N, Poelstra JW, Künstner A, Wolf JBW. Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments. Mol Ecol. 2012;22:620–34. doi: 10.1111/mec.12014. [DOI] [PubMed] [Google Scholar]
- 34.Francis WR, Christianson LM, Kiko R, Powers ML, Shaner NCD, Haddock SH. A comparison across non-model animals suggests an optimal sequencing depth for de novo transcriptome assembly. BMC Genomics. 2013;14:167. doi: 10.1186/1471-2164-14-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hébert FO. corset_pipeline: First complete release. Zenodo. 2016; http://dx.doi.org/10.5281/zenodo.50971.
- 36.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macmanes MD. On the optimal trimming of high-throughput mRNA sequence data. Front Genet. 2014;5:13. doi: 10.3389/fgene.2014.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davidson NM, Oshlack A. Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014;15:1–14. doi: 10.1186/gb-2014-15-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.St Laurent G, Wahlestedt C, Kapranov P. The Landscape of long noncoding RNA classification. Trends Genet. 2015;31:239–51. doi: 10.1016/j.tig.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–63. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 43.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 45.Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998;6:175–82. [PubMed] [Google Scholar]
- 46.UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–12. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, et al. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012;40:D284–9. doi: 10.1093/nar/gkr1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hébert FO. Trinotate_pipeline: Annotation Pipeline - Trinotate. Zenodo. 2016. http://dx.doi.org/10.5281/zenodo.50974.
- 51.DeRego T, Hall B, ben-guin, Geib S. Transvestigator early release. Zenodo. 2014. http://dx.doi.org/10.5281/zenodo.10470.
- 52.Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res Cold Spring Harbor Lab. 2008;18:188–96. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl 2):ii215–25. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 54.Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, et al. Automated eukaryotic gene structure annotation using evidence modeler and the program to assemble spliced alignments. Genome Biol BioMed Central. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC bioinformatics. BioMed Central. 2009;10:1. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sprehn CG, Blum MJ, Quinn TP, HEINS DC. Landscape Genetics of Schistocephalus solidus Parasites in Threespine Stickleback (Gasterosteus aculeatus) from Alaska. Britton R, editor. Plos One. 2015;10:e0122307–17. doi: 10.1371/journal.pone.0122307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salem M, Paneru B, Al-Tobasei R, Abdouni F, Thorgaard GH, Rexroad CE, et al. Transcriptome assembly, gene annotation and tissue gene expression atlas of the rainbow trout. Jaiswal P, editor. Plos One. 2015;10:e0121778. doi: 10.1371/journal.pone.0121778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005;6:361–75. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- 60.Davila A, Tschoeke D, Nunes G, Jardim R, Lima J, Dumaresq A, et al. The comparative genomics and phylogenomics of leishmania amazonensis. Parasite. 2014;10:131–23. doi: 10.4137/EBO.S13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korhonen PK, Pozio E, La Rosa G, Chang BCH, Koehler AV, Hoberg EP, et al. Phylogenomic and biogeographic reconstruction of the Trichinella complex. Nat Commun. 2016;7:1–8. doi: 10.1038/ncomms10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunt VL, Tsai IJ, Coghlan A, Reid AJ, Holroyd N, Foth BJ, et al. The genomic basis of parasitism in the Strongyloides clade of nematodes. Nat Genet. 2016;48:1–11. doi: 10.1038/ng.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bennett HM, Mok HP, Gkrania-Klotsas E, Tsai IJ, Stanley EJ, Antoun NM, et al. The genome of the sparganosis tapeworm Spirometra erinaceieuropaei isolated from the biopsy of a migrating brain lesion. Genome Biol. 2015;15:1–17. doi: 10.1186/s13059-014-0510-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen F, Mackey AJ, Stoeckert CJ, Roos DS. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;34:D363–8. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinformatics. 2011;Chapter 6:Unit6.12:1–19. doi: 10.1002/0471250953.bi0612s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.WormBase ParaSite. WormBase consortium. http://parasite.wormbase.org/. Accessed 5 Oct 2015.
- 67.Tsai IJ, Zarowiecki M, Holroyd N, Garciarrubio A, Sanchez-Flores A, Brooks KL, et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature. 2013;496:1–7. doi: 10.1038/nature12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 70.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol. 2012;61:1061–7. doi: 10.1093/sysbio/sys062. [DOI] [PubMed] [Google Scholar]
- 72.Smyth JD. Parasitological serendipity: from Schistocephalus to Echinococcus. Int J Parasitol. 1990;20:411–23. doi: 10.1016/0020-7519(90)90190-X. [DOI] [PubMed] [Google Scholar]