

Abstract
Background
Omics approaches have significantly increased our understanding of biological systems. However, they have had limited success in explaining the dramatically increased productivity of commercially important natural products by industrial high-producing strains, such as the erythromycin-producing actinomycete Saccharopolyspora erythraea. Further yield increase is of great importance but requires a better understanding of the underlying physiological processes.
Results
To reveal the mechanisms related to erythromycin yield increase, we have undertaken an integrated study of the genomic, transcriptomic, and proteomic differences between the wild type strain NRRL2338 (WT) and the industrial high-producing strain ABE1441 (HP) of S. erythraea at multiple time points of a simulated industrial bioprocess. 165 observed mutations lead to differences in gene expression profiles and protein abundance between the two strains, which were most prominent in the initial stages of erythromycin production. Enzymes involved in erythromycin biosynthesis, metabolism of branched chain amino acids and proteolysis were most strongly upregulated in the HP strain. Interestingly, genes related to TCA cycle and DNA-repair were downregulated. Additionally, comprehensive data analysis uncovered significant correlations in expression profiles of the erythromycin-biosynthetic genes, other biosynthetic gene clusters and previously unidentified putative regulatory genes. Based on this information, we demonstrated that overexpression of several genes involved in amino acid metabolism can contribute to increased yield of erythromycin, confirming the validity of our systems biology approach.
Conclusions
Our comprehensive omics approach, carried out in industrially relevant conditions, enabled the identification of key pathways affecting erythromycin yield and suggests strategies for rapid increase in the production of secondary metabolites in industrial environment.
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-016-0496-5) contains supplementary material, which is available to authorized users.
Keywords: Saccharopolyspora erythraea, Erythromycin, Systems biology, Metabolic engineering, Transcriptomics, Proteomics, Polyketide, Substrate supply
Background
Metabolic engineering of microorganisms for industrial production of valuable compounds has been influenced strongly in recent years by the growing availability of “omics” techniques. Genomics and transcriptomics, but also proteomics and metabolomics have importantly increased our systems-wide understanding of cell physiology, from transcriptional and translational regulation to morphogenesis, stress response and many other processes in the cell [1]. In many cases, particularly for production of metabolites with well-characterized biosynthetic pathways in well-known microbes, results of omics analyses have inspired novel metabolic engineering strategies which led to significant improvements of titres of industrially important metabolites such as l-lysine, l-threonine or xylitol [2–5]. In addition, integration of different types of omics data through systems biology approaches is a very promising tool in process development which will further accelerate the development of efficient bioprocesses for production of natural products as well as non-natural chemicals [6].
Soil-dwelling actinomycete bacteria are particularly prolific producers of diverse biologically active secondary metabolites, which have found widespread use in human and veterinary medicine, as well as in agriculture [7]. These substances include antibiotics, antifungal, anti-cancer, immunosuppressive, insecticide, and other classes of bioactive substances of immense value for human health and the global economy. In natural environments these compounds are produced by bacterial cells in very small amounts as non-essential secondary metabolites. Decades of research have been devoted into improvement of diverse strains and fermentation bioprocesses used to produce these metabolites at industrial scale. Predominantly, strains were subjected to random mutagenesis treatments followed by intensive strain selection aiming at improved yields of target products [8, 9]. As a result, many industrially used strains now have drastically, up to 1000 fold, improved secondary metabolite yields. However, they are also full of uncharacterized mutations that disrupt their normal developmental cycle, make them more sensitive to environmental conditions, and hinder further yield optimization.
Omics methods are clearly opening new possibilities to, more rapidly than before, turn actinomycetes into sophisticated and efficient cell factories. Better understanding of their physiology will enable researchers to selectively remove the metabolic bottlenecks of secondary metabolite biosynthesis without introducing unnecessary or deleterious changes. A “reverse engineering” approach has been proposed, whereby factors important for production could be identified by examining the differences between natural strains and industrial high-producing strains obtained by random mutagenesis and selection. Impressive insight into actinomycete biology has already been achieved by omics techniques [10]. For example, genome sequencing revealed an unimagined potential of actinomycete strains to synthesize a large number of different bioactive compounds [10]. However, mechanisms responsible for increasing the yields of valuable bioactive compounds are still poorly understood and omics approaches have shown relatively limited success in improving production of important natural products at industrial scale. This is likely due to several reasons: (1) actinomycetes have large genomes, up to 12 Mbp, which is reflected in complex networks of global and pathway-specific regulatory genes and morphological differentiation [1, 11]; (2) the interrelations between primary and secondary metabolic pathways during the production stage of the bioprocess are complex and poorly understood [12]; (3) laboratory media, in which transcriptomic and proteomic analyses are usually carried out, poorly reflect industrially relevant bioprocess conditions, whereas omics analyses in rich industrial media are hampered by difficulties in isolating high quality RNA and/or proteins; (4) most industrially used actinomycete strains have likely been “over-mutagenized” during the strain improvement process, resulting in a large number of observed genomic variants (SNPs), many of which represent neutral or even negative mutations, often resulting in morphologically and physiological unstable strains, which are difficult to manage reproducibly in industrial environment [13]. All these factors make application of omics analyses and their interpretation even more complex for industrial actinomycetes than for most other microbes.
Saccharopolyspora erythraea is a particularly interesting organism to study, as the producer of erythromycin, an industrially and clinically extremely valuable antibiotic, but also as a model representative of actinomycetes [14]. Since publication of the S. erythraea genome [15], several studies have investigated the changes in gene expression throughout the erythromycin production bioprocess, in both wild type and industrial strains [16–21]. Despite the progress made in these studies, much work remains to be done in assigning functional roles to mutations found in industrial S. erythraea strains, identifying key mechanisms influencing erythromycin yield and clarifying the connections between erythromycin biosynthesis and the rest of cellular metabolism. In order to enable rapid increase of erythromycin yield by metabolic engineering/synthetic biology approaches, much deeper understanding of these aspects is of great importance.
In an effort to address some of these questions, we have undertaken a comprehensive comparative study of the genomic, transcriptomic, and proteomic differences between the wild type S. erythraea NRRL2338 (WT) and an industrial high-producing (HP) strain of S. erythraea ABE1441, which had been subjected to mutagenesis and selection for many decades. Importantly, cultivation of both strains was carried out at bioreactor scale, using industrially relevant growth media and bioprocess parameters. Using various data analysis and integration approaches we identified several novel mechanisms that could contribute to higher erythromycin yield in the HP strain. We observed the overexpression of several genes related to branched-chain amino acid metabolism, potentially representing a novel methylmalonyl-CoA building block feeder pathway. Significant increase in final erythromycin yield was observed, when several of these genes were constitutively overexpressed in the WT strain. Our work shows that omics approaches can rapidly provide new strategies for the improvement of actinomycete based production strains, provided that the analyses are carried out with optimised methodology in industrially relevant conditions.
Results
Genome of the high-producing S. erythraea strain ABE1441
The order-of-magnitude increase in erythromycin yield, displayed in industrial cultivation conditions by the HP strain [22] compared to WT, as well as all differences in the metabolism between the two strains ultimately stem from genomic mutations. These mutations accumulated in the HP strain in numerous rounds of “classical strain” improvement by random mutagenesis and selection. Therefore, we initially sequenced the genome of the HP strain and compared it to the genome of the publicly available WT S. erythraea strain. Importantly, before comparison with the HP sequence, the originally deposited WT sequence [15] was screened for potential sequencing errors by comparing it with recently published RNA-seq data of the same NRRL 2338 strain [19]. Using this approach 40 putative sequencing errors were identified in the original WT genome of approx. 8.2 Mbp (Additional file 1). Since these sequencing errors would falsely appear as “mutations” when comparing the HP strain to the published WT genome, they were excluded from our comparative genomic analysis. Next generation sequencing of the HP strain revealed 165 genuine mutations compared to WT, affecting 147 genes (i.e. present inside the ORFs or in putative promoter or terminator regions). Out of these, 139 were single nucleotide variations (SNVs), 23 multiple nucleotide variations (MNVs), two deletions (in terminator of SACE_5310 and in SACE_6447) and one insertion (in SACE_4589). Seven genes had two mutations while five and seven mutations were identified in two transposase genes SACE_3579 and SACE_4072, respectively. Mutations were found to be distributed evenly over the whole genome. Particularly noteworthy are mutations in three genes involved in the TCA cycle (SACE_1170, SACE_6584 and SACE_6636), three genes belonging to two-component regulatory systems (SACE_4067, SACE_6086, and SACE_6447) and seven genes involved in amino acid metabolism (SACE numbers 3033, 3126, 3584, 4093, 4116, 6565 and 7125). Locations and nature of all observed mutations are presented in Additional file 2, columns H-M.
Adapted industrial-like bioprocess for erythromycin production
In order to obtain industrially relevant transcriptomic and proteomic data, three independent bioprocesses with WT and HP strains were carried out in 5 L bioreactors and samples were taken at specific time points, characterized by rates of biomass growth, erythromycin titre, and expression of erythromycin biosynthetic genes (Fig. 1; Additional file 3). Briefly, the fermentation process consists of an initial phase without erythromycin production, lasting around 24 h (time point t1—initial increase in expression of the erythromycin biosynthetic genes eryAI and eryK; see Additional file 3). The initial phase is followed by a period of rapid biomass formation and rapid consumption of glucose, when erythromycin production is most pronounced (time point t2—start of rapid growth phase and detectable erythromycin production, transient decline of erythromycin biosynthetic gene expression). Once all the glucose in the medium is consumed and the cells enter the stationary growth phase (time point t3—glucose depleted, a second increase in expression of eryA1 and eryK genes), there is an additional increase in erythromycin titre. However, the biosynthesis of erythromycin stops shortly afterwards in the WT strain (time point t4–stationary phase, end of erythromycin production in WT). In the HP strain, in contrast, erythromycin production is not only more intense during the rapid growth phase, but it also continues into the stationary phase, resulting in a more than ten-fold higher yield compared to the WT strain. Accordingly, prolonged expression of eryAI gene, encoding the erythromycin polyketide synthase, was also observed in the HP strain. Interestingly, the rate of glucose consumption is almost identical for both strains.
Fig. 1.
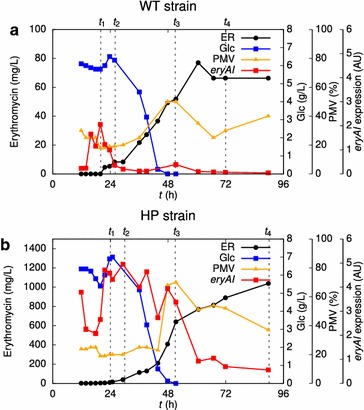
Key bioprocess parameters of cultivation of WT (a) and HP (b) strains in bioreactor scale. Erythromycin (ER) and glucose (Glc) concentrations, the packed mycelium volume (PMV), and relative expression of key erythromycin biosynthetic gene eryAI are plotted against total bioprocess time for a representative replicate of the bioprocess. The dashed lines denote the time points selected for transcriptomic and proteomic analysis. In the WT strain, erythromycin is produced primarily during the period of intense cell growth and rapid glucose consumption. In the HP strain, final erythromycin yield is much higher due to an increased rate of production during the rapid growth phase and a prolonged period of production in the late stages of the bioprocess. Expression profiles of eryAI and eryK erythromycin biosynthesis marker genes in both bioprocesses are shown in Additional file 3
Comparative gene expression analysis
The samples obtained at defined time points of the adapted industrial bioprocess allowed us to conduct a thorough comparative analysis between WT and HP strains at the gene expression level. Firstly, we identified differentially expressed genes at individual time points. Out of 7217 genes featured on the microarray, 55 were excluded from subsequent data analysis due to low signal in most samples and 1198 genes were found to be significantly differentially expressed (DE) between HP and WT in at least one time point. The number of DE genes increased over time (Fig. 2). Microarray data was validated by performing qPCR analyses, based on seven selected genes related to erythromycin biosynthesis (Additional file 4). The high correlation between the results of the two methods (r = 0.84) confirms that the microarray gene expression data is reliable.
Fig. 2.
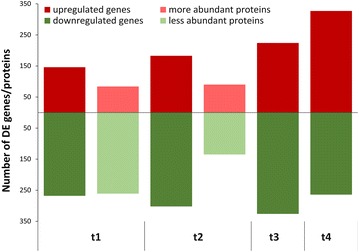
Numbers of differentially expressed genes and differentially abundant proteins at the four bioprocess time points. The numbers of up-regulated genes/proteins in HP compared to WT strain at the time points t1 to t4 of the fermentation process are shown in red and down-regulated in green. Darker colour is used for microarray analyses of gene expression and lighter colour for LC–MS/MS analyses of protein abundance. The cut-off for differential expression was |logFC| > 1 and p < 0.01 and for differential protein abundance was |logFC| > 1.5, respectively. Due to extensive proteolysis in the HP strain samples differential protein abundance was not calculated at t3 and t4
In the next step we performed a gene set enrichment analysis (GSEA) [23] to identify the functional gene groups and metabolic pathways (as defined by KEGG pathway or Gene Ontology) that were over- or under-expressed in the HP strain at selected time points during the fermentation (Table 1; Additional file 5). While some differences in gene expression between the strains can already be observed at t1, the switch to secondary metabolism at t2 is more pronounced in the HP strain, suggesting that in the HP secondary metabolism is turned on earlier than in the WT strain. This is seen as enrichment of upregulated genes in several secondary metabolic pathways and in pathways related to the metabolism of amino acids that can act as polyketide precursors, as well as by the enrichment of downregulated genes in primary metabolic pathways such as the TCA cycle and nucleotide and nitrogen metabolism. The HP strain then maintains the significantly higher expression of erythromycin biosynthetic genes as well as other polyketide-related pathways throughout the fermentation. Interestingly, the analysis shows that at t3, genes involved in DNA repair mechanisms during replication are expressed at lower level in HP compared to WT strain. Moreover, increased proteolysis, observed in proteomics, is also evident on the transcriptome level (Additional file 5). Overall, in comparison to WT, the HP strain shows increased expression of genes involved in secondary metabolism and decreased expression of several key primary metabolic pathways since the start of the exponential growth phase (t2). This becomes even more pronounced once the glucose in the medium is fully consumed (t3) (Fig. 1).
Table 1.
Transcriptional differences in HP compared to the WT strain at the level of biological processes
| Gene sets | HP vs WT | ||||||
|---|---|---|---|---|---|---|---|
| Ontology | Name | Code | Size | t1 | t2 | t3 | t4 |
| KEGG Pathway | Translation | 145 | + | + | |||
| Ribosome | sen03010 | 64 | + | + | |||
| Metabolism of terpenoids and polyketides | 128 | + | + | + | |||
| Biosynthesis of ansamycins | sen01051 | 13 | + | + | + | ||
| Biosynthesis of 12-, 14- and 16-membered macrolides | sen00522 | 8 | + | + | + | ||
| Type I polyketide structures | sen01052 | 9 | + | + | + | ||
| Nitrogen metabolism | sen00910 | 56 | − | ||||
| C5-branched dibasic acid metabolism | sen00660 | 10 | + | ||||
| Valine, leucine and isoleucine biosynthesis | sen00290 | 23 | + | ||||
| Tyrosine metabolism | sen00350 | 38 | − | ||||
| Tryptophan metabolism | sen00380 | 39 | + | + | |||
| Glycolysis/gluconeogenesis | sen00010 | 74 | − | ||||
| Citrate cycle (TCA cycle) | sen00020 | 48 | − | ||||
| Pyruvate metabolism | sen00620 | 73 | − | ||||
| Metabolism of cofactors and vitamins | 192 | + | |||||
| Lipid metabolism | 169 | − | |||||
| Fatty acid biosynthesis | sen00061 | 29 | − | ||||
| Xenobiotics biodegradation and metabolism | 175 | − | |||||
| Styrene degradation | sen00643 | 13 | + | ||||
| Atrazine degradation | sen01052 | 9 | − | − | |||
| Purine metabolism | sen00230 | 88 | − | − | |||
| Nucleotide metabolism | 118 | − | − | − | |||
| Oxidative phosphorylation | sen00190 | 49 | + | ||||
| Inositol phosphate metabolism | sen00562 | 20 | + | ||||
| Membrane transport | 138 | − | |||||
| ABC transporters | sen02010 | 117 | − | ||||
| GO biological process | DNA biosynthetic process | GO:0071897 | 18 | − | − | ||
| DNA replication | GO:0006260 | 29 | − | − | |||
| DNA integration | GO:0015074 | 48 | − | ||||
| DNA recombination | GO:0006310 | 44 | + | − | |||
| DNA repair | GO:0006281 | 45 | − | ||||
| Transposition, DNA-mediated | GO:0006313 | 62 | − | − | − | ||
| DNA-templated transcription, initiation | GO:0006352 | 36 | − | − | − | ||
| Negative regulation of transcription, DNA-templated | GO:0045892 | 19 | − | − | |||
| Translation | GO:0006412 | 68 | + | + | + | ||
| Methylation | GO:0032259 | 126 | + | ||||
| Dephosphorylation | GO:0016311 | 26 | + | ||||
| Biosynthetic process | GO:0009058 | 59 | + | + | |||
| Fatty acid biosynthetic process | GO:0006633 | 23 | − | ||||
| Antibiotic biosynthetic process | GO:0017000 | 16 | + | + | + | ||
| Proteolysis | GO:0006508 | 152 | + | + | + | ||
| Amino acid transmembrane transport | GO:0003333 | 33 | − | ||||
| Carbohydrate transport | GO:0008643 | 25 | − | ||||
For each ontology used, KEGG pathways or GO Biological process, gene set names, codes and sizes (total numbers of S. erythraea genes associated with the gene set) are shown. Significant enrichment (p < 0.01 for KEGG and p < 0.05 for GO) of the individual gene sets in up- or down-regulated genes at individual time points (t1–t4) is represented by ‘+’ or ‘−’, respectively. Pathways with missing codes are the top KEGG ontology terms
Complementary to analyses described above where only differences in individual time points were assessed, time-course profile analysis was performed that identified 6 clusters of genes with differential time-course profiles between the two strains (Additional file 6). In two clusters the gene expression was higher and in 4 lower in WT compared to HP throughout the fermentation process.
Co-expression analysis
In the next step we also investigated the correlations between gene expression levels [24, 25]. For each gene in the genome we calculated Pearson correlation coefficients between the expression levels of that gene and all other genes across all experimental samples. The same analysis was also performed on subsets of data limited to the WT strain and the HP strain, respectively. Our analysis revealed that the ery cluster is highly correlated to a wide range of genes encoding diverse functions (Fig. 3). At least two distinct groups of genes are consistently correlated with the expression of the ery cluster in both WT and HP strains while also having a strong connection to at least one mutated gene (Fig. 3a). The first group contains a number of genes of the TCA cycle and fatty acid metabolism, as well as mutB (SACE_5639, a member of the mcm operon involved in converting between succinyl- and methylmalonyl-CoA). These genes are expected to be involved in the supply of precursors for erythromycin biosynthesis from the core carbon metabolism. The second group is mostly composed of genes related to the phosphate metabolism or to the biosynthesis of secondary metabolites. The latter include a pair of apparent polyketide synthase (PKS) clusters, pks2 (SACE_2593-635) and pks8 (SACE_5532-45). Interestingly, even though their expression is much lower in HP than in WT, these PKS genes maintain a positive correlation with the ery cluster across the time points of WT as well as HP fermentation. This suggests they may be subject to dual regulation where one system strives to coordinate their expression to that of ery, while a separate mechanism strongly downregulates their expression in HP. Our analysis also identified other highly internally connected groups of genes corresponding to ribosomal proteins and several transposons (Fig. 3a), confirming that the expression correlation approach can successfully identify co-regulated genes.
Fig. 3.
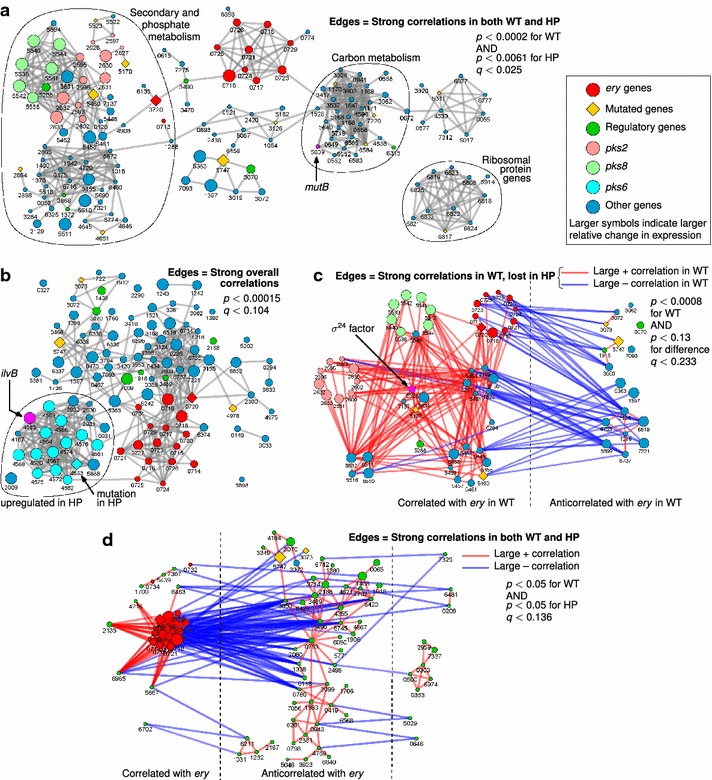
Graph representations of networks of genes with correlated expression profiles. Nodes represent genes, identified by their SACE numbers, and edges are drawn between genes whose expression levels show a strong (positive or negative) correlation in different contexts. a Edges are drawn between genes whose expression levels show a strong correlation in the WT strain and maintain the same correlation in the HP strain. Only genes closely correlated to at least one mutated gene (p < 0.00017 in WT) are shown. The ery cluster is connected to both the primary carbon metabolism (including mutB/SACE_5639, highlighted in magenta) and secondary metabolic genes. A separate cluster of ribosomal proteins was also identified using this approach, but it is not correlated to ery genes. b Edges are drawn between genes whose expression is strongly correlated in the full dataset, across both strains. Only genes with a strong correlation to the ery cluster (top 6 % of all genes) and a large change in expression between WT and HP strains (top 3.7 %) are shown. Genes of the pks6 cluster (SACE_4561-76) form a strongly interconnected group that is overexpressed in HP and includes a mutated gene (SACE_4563) and ilvB (SACE_4565, highlighted in magenta). c Edges are drawn between genes that are strongly correlated in the WT strain but whose correlation is greatly perturbed in the HP strain. Only genes with a high WT correlation to at least one mutated gene (p < 0.00016) and with a large change in expression between HP and WT (top 3.7 %) are shown. The putative sigma24 factor (SACE_5521, highlighted in magenta) loses many positive correlations to pks2 and pks8 genes. d Correlations between the ery cluster and regulatory genes. Edges are drawn between genes that maintain strong correlations in both strains. Genes consistently correlated with ery represent potential activators and those anticorrelated represent potential repressors
We also examined genes that are globally regulated (across both strains and all time points) in concert with ery and, simultaneously, are dramatically over-or under-expressed in the HP strain (Fig. 3b). This includes a tightly interconnected cluster of genes (SACE_4561-76) whose expression does not correlate very strongly with the ery cluster in WT, but closely mirrors ery overexpression in the HP strain. These genes were also identified in the same cluster as ery genes in the regression-based time-course profile analysis (Additional file 6). They are annotated as encoding an “alternative” polyketide synthase system, pks6, along with an acetolactate synthase (ilvB1, SACE_4565) and a valine-pyruvate transaminase (avtA, SACE_4564) that presumably ensure a steady supply of precursor metabolites. Importantly, one of the pks6 genes (SACE_4563, unknown function) is mutated in the HP strain, which may block the actual synthesis of the corresponding polyketide product.
Finally, we focused on genes that have both their expression levels as well as their coordination/correlations with other genes strongly perturbed in the HP strain (Fig. 3c). This group includes several genes of the PKS clusters pks2 and pks8, as well as a putative sigma24 factor (SACE_5521) with strong correlations to both of these PKS clusters in the WT strain. All three are strongly downregulated in the HP strain. In addition, the most prominent regulatory proteins that correlate strongly with the ery cluster in either or both strains are examined further in Fig. 3d and Additional file 7. Several regulators are co-expressed with ery and others show the opposite expression pattern (negative correlation). Although such correlation does not imply a direct functional relation, the two groups of regulatory genes would be good targets for identification of activators and repressors of ery expression, respectively.
Proteomic analysis
In order to complement the results of comparative genomic and transcriptomic analysis with data on protein concentration levels we sampled the WT and HP fermentation broths at the four time points, t1– t4, and subjected the samples to quantitative proteomic analysis. In the first approach, cell lysates were separated by SDS-PAGE, analysed by LC–MS/MS, and quantified by spectral counting. 1000–1400 proteins were identified per sample at each time point. In the second approach, 2-D gel electrophoresis analysis was carried out. Reliable data for protein profiles of the samples could only be obtained for the time points t1 and t2, whereas in time points t3 and t4 extensive proteolytic degradation of the samples from the HP strain was observed in spite of using different available protease inhibitors and increasing their concentrations. Therefore, proteomic data from time points t3 and t4 were not included in data analysis. A decrease in protein abundance and in the number of detected protein spots in the later phases of the bioprocess was also confirmed by 2-D PAGE (Additional file 8).
The observed differences in protein abundance between the WT and HP strains (LC–MS/MS analysis) for time points t1 and t2 (Fig. 2) agree well with the results of the transcriptome analysis, with the exception that fewer proteins displayed significant changes in abundance at t2 than at t1. As shown in Fig. 4 and Additional file 9, the erythromycin-producing PKS is strongly upregulated in the HP, as are some of the enzymes involved in the branched-chain amino acid synthesis pathway. On the other hand, enzymes of the TCA cycle, ribosomal proteins, and most other proteins with significant abundance changes are downregulated in the HP strain. Some of the most striking changes in abundance were observed with proteins belonging to biosynthetic pathways of secondary metabolites: the pks2 and pks8 systems are strongly downregulated in the HP strain while ery, pks6 and a cluster of terpenoid-quinone metabolic enzymes (tpc4, SACE_4645-51) are upregulated.
Fig. 4.
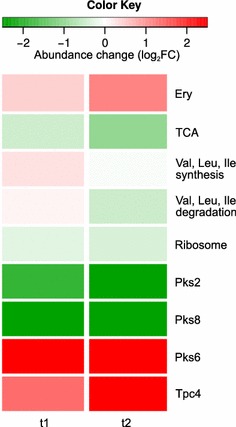
Relative changes in protein abundance (LC-MS/MS) between HP and WT strains for two time points. The changes in abundance on the level of biological processes are presented as log2 of the ratio between HP and WT with red fields representing increased abundance in HP and green fields representing decreased abundance in HP. Proteins are grouped according to their functional annotations in KEGG and abundance levels of group-wise averages are shown. Ery, proteins of the ery cluster; TCA, enzymes of the TCA cycle. Proteins of the ery cluster are strongly upregulated, as are some of the enzymes responsible for the synthesis of aliphatic amino acids. Enzymes of the TCA cycle are downregulated. Proteins of the pks6 and tpc4 secondary metabolic clusters are upregulated in HP while proteins of the pks2 and pks8 clusters are downregulated. Abundance changes for individual proteins belonging to each of these groups are presented in Additional file 9
As a complementary method for proteome analysis we used 2-D PAGE with subsequent identification of differentially abundant proteins between HP and WT samples by LC–MS analysis (Additional file 10). This method confirmed that the protein profiles of the WT and HP strains differ significantly at time point t2. Differential abundance was observed, for example, for proteins related to glycolysis, TCA cycle, erythromycin biosynthesis, nucleotide metabolism and amino acid biosynthesis. Notably, several proteins of the carbohydrate metabolism (e.g. succinyl-CoA synthetase α subunit, SACE_6668; dihydrolipoamide succinyltransferase, SACE_1638; phosphogluconate dehydratase, SACE_1740; 6-phosphofructokinase, SACE_1704; ribose-phosphate pyrophosphokinase, SACE_0816) and nucleotide metabolism (inosine-5′-monophosphate dehydrogenase, SACE_6708; phospho-2-dehydro-3-deoxyheptonate aldolase, SACE_1708; phosphoribosylaminoimidazolecarboxamide formyltransferase, SACE_6664) were observed in the WT strain, but not in the HP strain. Additionally, more than one spot on the gel was found to belong to some of these proteins (SACE_1638, SACE_6668). Three glycolytic enzymes (glucose-6-phosphate isomerase, SACE_2158; 2-phosphoglycerate dehydratase, SACE_0838; phosphoglycerate mutase, SACE_6967) were detected only in the HP strain while glyceraldehyde 3-phosphate dehydrogenase (SACE_2143) was overexpressed in the HP strain. Additionally, the ketol-acid reductoisomerase IlvC (SACE_6157), related to valine biosynthesis, was downregulated in the HP strain. The differentially abundant proteins according to 2D PAGE are listed in Additional file 10 and are also integrated with data from other approaches in Additional file 2.
Integrating genomics, transcriptomics and proteomics adds a new perspective
In general, when comparing data from transcriptomic and proteomic analyses (Additional file 2) we found good agreement between the expression levels of genes and abundance of the corresponding proteins. For the first two time points the observed correlation reached Pearson correlation coefficients (r) of 0.59 for t1 and 0.65 for t2 which is in the range of or even exceeding commonly reported values [26]. There are only a few exceptions where comparative transcriptomic and proteomic analyses show opposite patterns of transcript/protein abundance in WT and HP strains (Additional file 11).
Further, we aimed to integrate diverse types of omics data to gain a more comprehensive picture of the S.erythraea physiology. Since the core carbon metabolism and amino acid biosynthesis exhibit significant differential expression between WT and HP, we mapped our experimental data onto these metabolic pathways to visualize their connections with erythromycin biosynthesis (Fig. 5). In the HP strain, several genes of the TCA cycle are mutated and several others are significantly downregulated compared to the WT strain. On the other hand, some genes such as ilvB (SACE_4565), acd (SACE_4125 and SACE_5025), and mmsA (SACE_4672) are expressed at significantly higher levels in the HP strain. Together, these data suggest that mutations and changes in the regulation of primary metabolic pathways could result in an alternative route to supplying the erythromycin precursor methylmalonyl-CoA via the branched-chain amino acid biosynthesis/degradation pathway (Fig. 5 inset). Note that several genes have multiple paralogs present in the genome (multiple gene nodes connected to the same protein node in Fig. 5) and some of these are also known to be part of secondary metabolic gene clusters. For instance, SACE_4565 (ilvB) and SACE_5542-4 (pyruvate dehydrogenase, pdh) are part of the pks6 and pks8 cluster, respectively.
Fig. 5.
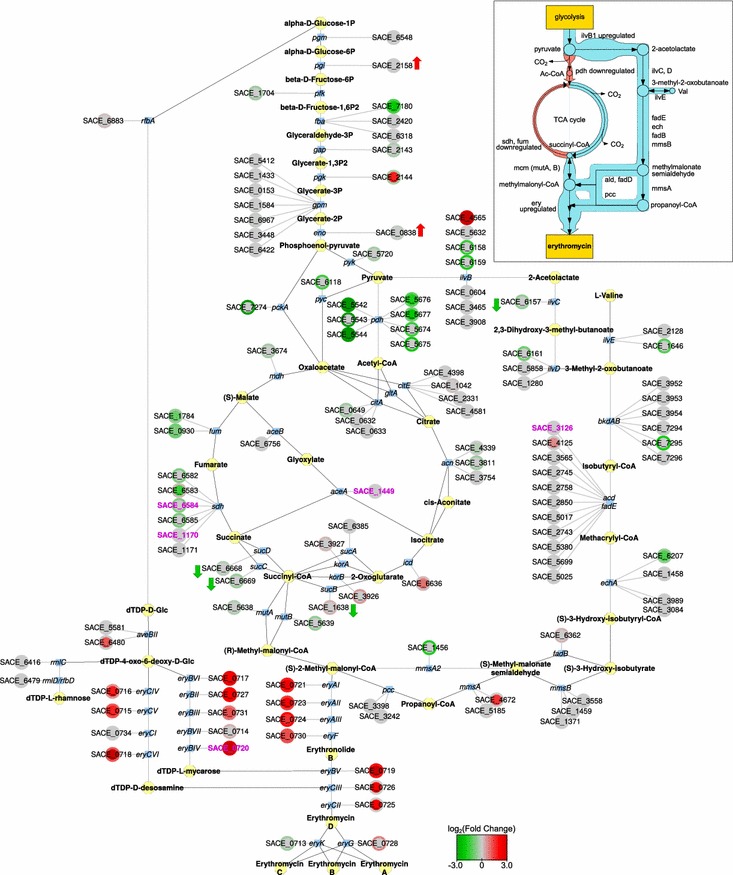
Visualisation of differences in glycolysis, TCA-cycle, valine metabolism and erythromycin biosynthesis at three omics levels. Presented data is from fermentation time point t2. Node (label) legend: circle: gene (locus tag), blue rectangle: protein (protein name), yellow octangle: metabolite (metabolite name). Circle centre and rim colours represent log2 (fold change between HP and WT) for gene and protein expression, respectively (see colour key). A red arrow next to a circle indicates upregulation in our 2D gel experiment, and a green arrow indicates downregulation. Gene mutations in the HP strain are indicated by magenta coloured gene labels. The inset shows a schematic representation of the proposed changes in metabolic flow between HP and WT strains. In the HP strain, key points of entry into the TCA cycle are likely restricted due to downregulation of pdh, sdh, and fdh (shown as thin red lines). Combined with the upregulation of ilvB, this could divert the flow of metabolites towards the biosynthesis/degradation pathway of branched-chain amino acids (shown as thick blue lines), resulting in increased supply of methylmalonyl-CoA and propionyl-CoA, key precursors for erythromycin synthesis
Re-engineering of erythromycin overproducing phenotype in the WT strain S. erythraea
Using a straightforward gene-overexpression approach, several genes putatively involved in increased erythromycin yield in HP strain were introduced in trans into the WT strain. We focused our work on three genes/operons that, based on the omics data, would most likely increase the metabolic flow towards erythromycin, when constitutively overexpressed in S. erythraea: (1) putative acetolactate synthase (ilvB1–SACE_4565) which catalyses the first dedicated step of branched-chain amino acid biosynthesis; (2) operon encoding putative branched-chain ketoacid dehydrogenase subunits (bkdOp–SACE_3952-54); and (3) operons encoding putative methylmalonate-semialdehyde dehydrogenase homologues, acyl-CoA dehydrogenase, enoyl-CoA hydratase and 3-hydroxybutyrate dehydrogenase (mmsOp1– SACE_1456-59 and mmsOp2– SACE_4672-73).
All constructs were expressed under the control of the strong constitutive promoter PermE* [27]. In addition, variants of selected genes with HA-tag on the C-terminus were also expressed, to allow evaluation of expression profiles by applying western blot analysis (Additional file 12), as described previously [28]. Average erythromycin yields of all groups of transformants are presented as a box plot diagram (Fig. 6). Notably, transformants constitutively overexpressing mmsOp1 construct showed highest erythromycin yield increase (approx. 2.5 fold higher yield compared to WT strain). In addition, transformants overexpressing the ilvB1 gene and bkd operon also showed significantly increased erythromycin yield, reaching 74 and 67 % erythromycin yield increase compared to the WT strain, respectively. In contrast, erythromycin yield of transformants overexpressing mmsOp2 construct did not significantly differ from the control strain. Surprisingly, when mmsOp1 operon and ilvB1 gene were constitutively expressed together, average erythromycin yield increase of 75 % was measured, which is comparable to the yield increase achieved by ilvB1 overexpression alone, and slightly lower compared to the transformants overexpressing only the mmsOp1 gene.
Fig. 6.
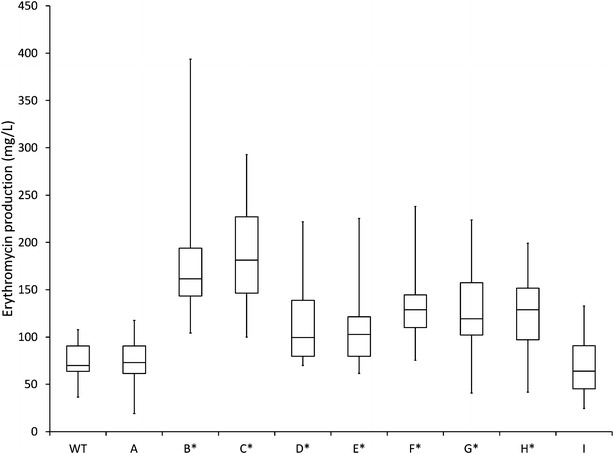
Effect of overexpressed genes on erythromycin production, compared to the WT strain. Box plot diagram of erythromycin production by the WT strain and transformants, determined by the microbiological assay; WT: control 1, NRRL2338, A: control 2, WT + pSet152, B: WT + mmsOp1 (pABE60), C: WT + mmsOp1-HA (pABE87), D: WT + ilvB1 (pABE61), E: WT + ilvB1-HA (pABE62), F: WT + mmsOp1-HA + ilvB1-HA (pABE89), G: WT + ilvB1-HA + mmsOp1-HA (pABE88), H: WT + bkdOp (pABE56), I: WT + mmsOp2 (pABE95). Boundaries of the boxes indicate the 1st and the 3rd quartile of the sample populations. Horizontal lines represent the median values and whiskers indicate the highest and lowest values of the results. Asterisks denote statistically significant (p < 0.05) differences between transformants compared to control samples 1 and 2. The data were analysed using SAS/STAT program as described in the “Methods” section
Discussion
By applying comparative genomic, transcriptomic and proteomic analyses, we have gained a comprehensive view of the differences between WT and HP strains of S. erythraea. On the one hand we identified entire functional groups, e.g. metabolic pathways or biological processes (Table 1) that are expressed at different levels in the two strains. On the other hand, we used co-expression analysis to identify several possible mechanisms that could connect the genomic mutations with altered expression levels. In our analyses we assumed that mutations act directly on the affected gene, rather than through potential polar effects on downstream genes, which allowed some interpretation in terms of differences in strain physiology (Fig. 3). In general, we observed that genes/proteins related to several key physiological processes are differentially expressed in the HP strain. For example, genes related to the translation machinery (e.g. ribosomes), biosynthesis of secondary metabolites and proteolysis are significantly upregulated in HP whereas on the other hand, several key central metabolic pathways, such as the TCA-cycle and fatty acid metabolism as well as DNA repair, nucleotide metabolism and oxidative stress response are significantly downregulated. While some of these physiological features are clearly related to increased erythromycin biosynthesis, rational explanations or even just putative roles of some of the other differences observed between WT and HP strains are still to be uncovered.
In addition to the observed increased expression of dedicated erythromycin biosynthetic genes, which clearly contributes to higher yield, the supply of the main biosynthetic precursor methylmalonyl-CoA could very well be a key factor. Some genes predicted to be involved in a methylmalonyl-CoA feeder pathway are overexpressed in the HP strain, while other pathways that could contribute to a higher supply of this precursor are downregulated. It was previously demonstrated that methylmalonyl-CoA pool can either be filled or drained by the methylmalonyl-CoA mutase-catalysed reaction converting the TCA cycle intermediate succinyl-CoA and methylmalonyl-CoA. When S. erythraea is cultivated on carbohydrate based media this reaction proceeds in direction from methylmalonyl-CoA to succinyl-CoA, thereby consuming methylmalonyl-CoA into the TCA cycle rather than supplying precursors for erythromycin biosynthesis [29]. The observed downregulation of the TCA cycle in the HP strain may reduce this drain on the methylmalonyl-CoA pool and lead to higher erythromycin yield. In addition, our genomic and transcriptomic data show that in the HP strain the isocitrate lyase gene (SACE_1449) carries a mutation in the 5′-UTR and is significantly downregulated at the gene expression level, suggesting a possible role of conversion of methylmalonyl-CoA to succinyl-CoA as an anaplerotic step in the HP strain. Furthermore, novel findings from our transcriptomic and proteomic analyses suggest that, rather than through the TCA cycle glucose can be converted to methylmalonyl-CoA via a bypass involving genes related to branched-chain amino acid metabolism (Fig. 5). This hypothesis is also supported by the observed increased erythromycin yield in engineered strains overexpressing different genes/operons of the branched chain amino acid metabolism (Fig. 6). While branched-chain amino acids have been previously suggested as a potential source of methylmalonyl-CoA [18, 21] our results indicate that parts of biosynthesis and degradation pathways of branched-chain amino acids could operate together to provide metabolic flux to methylmalonyl-CoA (Fig. 5). Biosynthesis and degradation of e.g. valine are believed to intersect at the point of 3-methyl-2-oxobutanoate, so the metabolic flux to methylmalonyl-CoA could actually avoid valine as an intermediate (Fig. 5 inset).
In addition to the metabolic pathways involved in methylmalonyl-CoA supply, the results of our transcriptomic and proteomic studies also suggest a number of other possible mechanisms underlying increased erythromycin yield, most of which could not be validated experimentally in the scope of this study but represent a rich source of directions for future work. For example, the expression correlation analysis revealed a coordinated regulation of the ery cluster and several other PKS clusters (pks2, pks6 and pks8) and a terpenoid biosynthesis cluster (tpc4). However, to achieve higher erythromycin titres, the HP strain has to avoid wasting resources on producing “undesired” secondary metabolites. For pks2 and pks8 this could be achieved by a separate mechanism of downregulation. The sigma factor SACE_5521, which appears to be connected to the expression of these two PKS clusters (Fig. 3c), would be an interesting target for further experimental characterization. The other two clusters, pks6 and tpc4, are overexpressed alongside ery, but appear to be affected by mutations in SACE_4563 and tauD (SACE_4651), respectively. This could actually be advantageous to erythromycin production, as pks6 also encodes feeder pathway proteins such as acetolactate synthase (ilvB-SACE_4565), which, when overexpressed, help divert substrate supply (precursors) from the primary carbon metabolism into the pool of common PKS substrate metabolites. Notably, overexpression of pks6 cluster was also observed in other high-producing S. erythraea strains [18, 21]. This suggests that feeder enzymes of the SACE_4561-76 cluster continue supplying precursors, but its PKS is disabled by mutations and unable to consume the substrates, leaving more available for erythromycin biosynthesis. Thus, subverting rather than completely inactivating alternative PKS or NRPS clusters may represent one of the mechanisms for increasing the production of erythromycin or other secondary metabolites.
All experimental data generated in this study have been integrated into a spreadsheet (Additional file 2), which represents a useful tool for the identification of new targets for metabolic engineering of S. erythraea in the future. Notably, as the same WT strain (NRRL2338) was used as a reference in our work and in the two previous studies on independently derived erythromycin high-producing strains Px [21] and E3 [18], the available data from these studies are also included in Additional file 2 columns AN-BD. The comparative genome analysis of the strains Px [21], E3 [18] and ABE1441 (this work) shows that 60 mutations are present in all three strains (see Additional file 2), disregarding the initially excluded WT sequencing errors [19]. Out of 147 genes affected by mutations in ABE1441, 27 genes are only mutated in this high producing strain and have WT sequence in the Px and E3 strains. Each of these 27 genes harbours one variation (24 SNVs, two MNVs and one deletion; see Additional file 2). Interestingly, the uniquely mutated genes in ABE1441 include a significantly reduced expression levels of the isocitrate lyase (SACE_1449) gene, which further confirms a reduced role of the TCA cycle intermediates (e.g. succinyl-CoA) as sources for the supply of methylmalonyl-CoA extender unit in our HP strain. The high number of common mutations in the ABE1441, Px and E3 strains suggests there was a common ancestor in the strain improvement history, from which three separate development lines were subsequently initiated. In addition, several groups of genes, such as erythromycin biosynthetic genes and pks6 (SACE_4561-76) were found to be upregulated in all three industrial strains while in contrast, the TCA cycle-related genes were downregulated.
It is important to stress that the high relevance of our results is based on a particular effort that we made to carry out omics analyses of S. erythraea cultures in fermentation conditions that most closely resemble conditions in the real industrial setting. Cultivation of WT and HP strains was carried out on the bioreactor scale where key process parameters such as pH and pO2 were controlled. Our work demonstrates that if relevant (industrial) cultivation conditions are applied and different bioinformatics and statistics approaches are used for data analysis, meaningful hypotheses can be generated and rapidly validated. Erythromycin is produced in more than 8000 tons per year [30]. To our knowledge, final yields of erythromycin at the industrial scale do not exceed 10 g/L, which is significantly less than the yields of most other mature high-volume active pharmaceutical ingredients. Considering relatively limited success in erythromycin yield improvement over decades, we believe that omics and metabolic engineering approaches based on the data presented in this work and in previously published studies [18, 19, 21] will be of particular importance for future strain and process development efforts.
Conclusions
In conclusion, the present study contributes a significant step forward in our understanding of how erythromycin biosynthesis is coordinated with the primary carbon metabolism and with the activities of other secondary metabolic pathways of S. erythraea such as pks2, pks6, and pks8. Based on the data from omics analyses and subsequent metabolic engineering experiments we identified valine biosynthesis/degradation pathway to be of importance in providing the precursors for erythromycin biosynthesis in the HP strain. It is particularly interesting to note that in this strain an increased expression of a “competing” PKS cluster, pks6 might further contribute to the efficiency of substrate supply for erythromycin biosynthesis. Thus, the omics data reported here suggest several future strategies for erythromycin strain improvement by metabolic engineering. Beyond that, this study demonstrates that our integrated omics approach, based on the bioprocess-guided experimental design and targeted and non-targeted data analysis, in combination with valuable literature information [18, 19, 21], generated tangible results which can be rapidly transferred to the industrial setting.
Methods
Genome sequencing and analysis
Since the reference WT S. erythraea genome was sequenced several years ago [15], we first aimed to investigate whether this reference genome assembly contains sequencing errors. We therefore analysed the recently published Illumina RNAseq reads of the same WT strain [19] [SRA accession SRX1277529], 15.7 G bases in total, and mapped them to the reference genome. Consistent variations between reference and mapped sequence reads (SNPs, insertions and deletions) were regarded as putative errors in the reference genome for all our further comparative genomic analyses. Variant analysis was performed in CLC Genomics Workbench v 6.5 (QIAGEN). Mapping to reference genome was performed using the following parameters: no masking, mismatch cost 2, insertion cost 3, deletion cost 3, length fraction 0.8, similarity fraction 0.95, global alignment ‘No’, auto-detect paired distances ‘Yes’, non-specific match handling ‘Map randomly’. CLC’s Probabilistic Variant Detection tool was used to determine potential sequencing errors of the reference genome. To ensure high confidence of variance detection, non-specific matches and broken pairs were ignored, variant presence was required in both forward and reverse reads, minimum coverage at variation sites was set to 10, variant probability to 90 % and maximum expected variants to 2.
The erythromycin high-producing ABE1441 industrial strain (HP) has been developed by Acies Bio Ltd based on the acquired intermediate producing strain. The genome of ABE1441 was sequenced using 454 sequencing technology (Roche) at Macrogen Inc., South Korea. Genomic reads were aligned to the reference genome in CLC Genomics Workbench (detailed read mapping metrics are available in Additional file 13) and CLC’s Probabilistic Variant Detection was used to determine variations. Variations that were identical to those found by aligning RNAseq reads described above [19] were flagged as WT genome sequencing errors. Some of the variations flagged as sequencing errors as well as several true HP variations were confirmed by PCR cloning and Sanger sequencing of fragments in WT and HP strains.
Cultivation of strains and sample collection
Propagation of the S. erythraea WT strain and HP strain as well as genetically engineered strains was done on ABSM4 agar plates (1 % corn starch, 1.1 % corn steep liquor, 0.3 % (NH4)2SO4, 0.3 % NaCl, 0.3 % CaCO3, 2 % agar) for 2 weeks at 30 °C. Laboratory scale fermentation in liquid culture for estimation of erythromycin productivity of transformant strains was done in 50-ml scale. Seed cultures of all strains were prepared in the ABVM1 medium (3 % corn steep liquor, 3 % sucrose, 0.4 % (NH4)2SO4, 0.6 % CaCO3) at 30 °C and 220 rpm for 48 h.
Cultivation experiments for comparative transcriptomic and proteomic analyses were carried out in 5 L bioreactors (Sartorius Biostat B) with 3.5 L of ABPM8 production medium, operated at 30 °C, 1 vvm airflow and 350–900 rpm agitation. Bioreactors were inoculated with 10 vol. % seed culture. Dissolved oxygen was maintained above 20 % with increasing agitation and aeration rate during the bioprocess. Foaming was controlled by automatic addition of antifoam SAG5693 (Momentive). Samples from bioreactor were taken out regularly and the following parameters were determined (packed mycelium volume (PMV-%), pH, glucose concentration, erythromycin concentration). Samples for qPCR, microarray and proteomic analysis were taken during the bioprocess and stored at −80 °C until analysis as described below.
To evaluate erythromycin yield of genetically engineered strains production phase was carried out in the ABPM8 medium (3.6 % soybean flour, 3.6 % corn starch, 0.24 % (NH4)2SO4, 0.72 % CaCO3, 0.5 % soybean oil) inoculated with 10 % (v/v) of the seed culture. We have now updated the Methods section with the following sentence. “Cultivations were performed in 50 mL Falcon tubes (sealed with foam plugs) at 30 °C and 220 rpm for 7 days. The working volume was 5 mL. 2 % glucose and 0.67 % n-propanol were added at the time of inoculation. and 1 % glucose and 0.34 % n-propanol were added after 24 h of cultivation. Apramycin (50 µg/ml) and thiostrepton (25 µg/ml solid and 5 µg/ml liquid media) were added to the solid and liquid media as required.
Sampling points for transcriptomic and proteomic analysis were selected according to bioprocess parameters and marker gene expression, taking into account that different fermentations have different dynamics, as described in more detail in the “Results” section.
Transcriptomics
Nucleotide sequences of the coding regions of S. erythraea were obtained from the Saccharopolyspora erythraea Genome Project Web Site (http://jblseqdat.bioc.cam.ac.uk/gnmweb/files.html). Agilent custom gene expression microarrays of 8×15 k format were designed using Agilent e-array (https://earray.chem.agilent.com/earray/). Positions of the predicted mutations were excluded from probe design. 60-mer probes were designed using Tm matching methodology (Tm = 80 °C, trimming allowed). Single antisense probe per gene (altogether 7216 probes) was designed and printed on microarrays in two replicates.
Cultures were fixed and RNA was isolated as described previously [28]. RNA was purified using Rneasy MinElute (Qiagen) and the quality and quantity were controlled by 2100 Bioanalyzer and RNA 6000 Nano LabChip Kit (Agilent Technologies). Sample labelling and hybridization were performed at IMGM (Germany). 300 ng of total RNA was spiked using One-Color RNA Spike-In Mix (Agilent Technologies) and subjected to reverse transcription, subsequent in vitro transcription, Cy3 labelling using Full Spectrum™ MultiStart Primers for T7 IVT (System Biosciences) and One-Color Quick-Amp Labelling Kit (Agilent Technologies) following manufacturer’s protocols. cRNA yield and integrity were determined using NanoDrop ND-1000 (Thermo Scientific) and 2100 Bioanalyzer (Agilent Technologies). cRNA was cleaned and hybridized to microarrays using Gene Expression Hybridization Kit (Agilent Technologies) following manufacturer’s protocols. Hybridized microarrays were washed using Gene Expression Wash Buffers (Agilent Technologies) followed by drying with acetonitrile. Fluorescent signal intensities were detected with Scan Control A.8.4.1 software (Agilent Technologies) on the Agilent DNA Microarray Scanner. Feature Extraction 10.7.1.1 software (Agilent Technologies) was used for feature extraction and quality control.
Raw microarray data was quality checked by inspecting signal and noise density plots, box plots, position images and QC spot intensities. Sample clustering was performed and two outlier WT samples were removed from analysis (one from time point t1 and another from time point t3).
Preliminary qPCR analysis and qPCR for validation of microarray results were carried out for selected marker genes on three independent fermentations of WT in HP strains. Primers and probes for 7 genes involved in primary metabolism and erythromycin production were designed as Custom TaqMan Gene Expression Assays (Life Technologies) and are shown in Additional file 12. Reverse transcription, qPCR setup and analysis were performed as described previously [28]. Preliminary qPCR analysis was performed on dense time point series (15–20 time points), where two genes, related to erythromycin biosynthesis were analysed (Additional file 3) while microarray validation analysis was performed at selected time points for all marker genes (Additional file 4).
Proteomics
At selected time points 4 ml samples of fermentation broth were taken and cells were sonicated in the presence of 7 M urea, 2 M thiourea, 4 % (w/v) CHAPS, 65 mM DTT, and a protease inhibitor cocktail. Analysis of proteins in the cell extract was performed by 2-D electrophoresis [31] and LC–MS spectral count.
For LC–MS spectral count, the cell extract was separated on a 12 % SDS-PAGE gel and whole protein lanes were cut into eight bands. Following gel destaining, disulphide reduction and trypsin digestion, the peptides were extracted from the gel and loaded onto an LC–MS/MS system composed of a trapping column, an analytical column, and an Orbitrap LTQ Velos mass spectrometer (Thermo Scientific). MS/MS spectra were obtained by fragmentation of the nine most intense precursor ions from the full MS scan. The database search and quantification by spectral counting were performed using the MaxQuant proteomics software [32, 33] using the S. erythraea protein database obtained from the Saccharopolyspora erythraea Genome Project Web Site.
2-D electrophoresis was run on IPG strips (pH 4–7) for the first dimension and a 12 % SDS-PAGE gel for the second dimension, then gels were stained with SYPRO Ruby (Invitrogen). Triplicate gels for each sample were matched to provide an average gel sample. Full experimental details of proteomic analysis are provided in Additional file 13.
Data analysis
Differential gene expression analysis was performed in the R statistical environment using limma package [34]. Features with signal intensity lower than ‘the average background signal increased for 2-times standard deviation’ in less than three samples were excluded from further analysis. Raw data were log-transformed, quantile-normalized and averaged between the two replicated probes. Log fold changes (logFC) for comparison between HP and WT strain in each time point were calculated. Statistics for the comparisons were calculated using the eBayes function. Genes having p value <0.01 and |logFC| > 1.0 were considered to be significantly differentially expressed. Additionally, gene expression profiles of the two strains were analysed using regression based approach implemented in the maSigPro Bioconductor package [35].
Enrichment analysis was performed using normalized averaged signals for each time point separately in GSEA desktop application v2.2.2 using “gene_set” permutation type, “Signal2Noise” method for ranking genes and S. erythraea KEGG BRITE or GO ontology (generated by Blast2Go software, BioBam) to build gene sets. KEGG gene sets with less than 8 or more than 400 and GO gene sets with less than 15 and more than 500 genes were excluded from analysis. Gene sets with enrichment nominal p value <0.01 for KEGG and p < 0.05 for GO were considered to be statistically significant.
For LC–MS spectral counts protein abundance ratio between HP and WT strain (logFC) of the same samples as used for transcriptomics was calculated for time points t1 and t2. LogFC was calculated for proteins with at least one spectral count above 10. Proteins with |logFC| > 1.5 were considered to be differentially abundant. LogFC values for time points t3 and t4 were not calculated because of the extensive proteolytic degradation of the samples from the HP strain in these time points. Spots on 2-D electrophoresis gels were visualized and quantified using the 2-D Dymension software, version 2.02 (Syngene), on the basis of their normalized volumes, defined as the spot volume divided by the total volume over the whole set of gel spots. Expression changes (HP/WT) for particular time point were considered as significant when the intensity of the corresponding spots reproducibly differed by more than 1.5-fold in a normalized volume (p value <0.05). The differentially expressed proteins were cut from the gel and identified by mass spectrometry as describe in detail in Additional file 13.
Analysis of correlation coefficients was carried out using a custom-written R script. Three separate matrices of Pearson’s correlation coefficients were calculated, one including all experimental data simultaneously, a second using only data from the WT strain, and a third using only data from the HP strain. To calculate p-values for correlation coefficients in each matrix, we randomly reshuffled the order of all measurements corresponding to each gene and recalculated the correlation matrix using the reshuffled data, which yielded random correlation coefficients without changing the distribution of expression levels for each gene. The distribution of these randomized correlation coefficients was then used to calculate the probability of a given value of a correlation coefficient appearing at random (p value). We also calculated the FDR q values. The calculation of correlation coefficients and q values, as well as the random reshuffling of experimental data was accomplished using built-in functions in R.
For each gene, we calculated the sum of squared correlation coefficients to all members of the ery cluster and the maximum squared correlation coefficient to any gene harbouring a mutation in the HP strain. These values, calculated using overall as well as WT- and HP-only correlations, were used to filter genes. Typically only the top 5 % of genes according to each criterion were considered. The correlations between selected genes were visualized in Cytoscape and filtered to produce visually interpretable figures. This typically required very stringent criteria with p values for individual correlations <0.005.
Genomic variation, microarray, qPCR, 2-D PAGE and shotgun proteomics results generated in this study were additionally integrated with gene annotations from several sources and gene expression/variation results from three recently published transcriptomic studies [19–21]. Comprehensive presentation of all data from our genomic, transcriptomic and proteomic experiments as well as data from relevant previous studies is presented in Additional file 2.
Overexpression of selected genes in S. erythraea
To confirm that the observed differences between WT and HP strains indeed result in increased erythromycin yield, we overexpressed several genes, related to branched-chain amino acid metabolism in the WT strain. Selected genes/putative operons were amplified using S. erythraea genomic DNA as template. Sequences of primers used for amplification of target DNA segments are presented in Additional file 12. In all cases NdeI restriction site was introduced at the 5′-end and XbaI site was inserted at the 3′-end, after the stop codon. The PCR amplified fragments were cloned into the pSet152-derived plasmid into which the constitutive PermE* promoter [27] had been previously cloned, thus creating a series of pABE vectors (Additional file 14). Correct assembly of the PCR products was confirmed by sequencing. In the cases of ilvB gene and mmsOp constructs additional variants with a HA-tag on the C-terminus of the proteins were prepared in order to enable confirmation of functional expression of overexpressed proteins by western blotting.
Simultaneous overexpression of ilvB1 gene and mms operon genes was also carried out. For this purpose two pSet152 based vectors were constructed (pABE88 and pABE89) in which the ilvB1 gene and mms operon were positioned in different orders. In pABE89 ilvB1 gene was cloned upstream of the mms operon genes, whereas in pABE88 construct ilvB1 was positioned downstream of the mms operon. In both cases, expression of the upstream gene was driven directly by the PermE* promoter, while for expression of the downstream gene a RBS was introduced upstream of the gene’s start codon. The obtained plasmid constructs were introduced into S. erythraea WT strain using standard conjugation procedure [36]. The obtained independent colonies were cultivated in shake flasks and erythromycin yields were estimated by microbiological assay and HPLC analyses according to previously described procedures [28].
For testing the effect of different mutations on erythromycin production, compared to the WT strain, at least 20 independent colonies (transformants) were tested for each engineered strain in two consecutive independent experiments. Each independent colony (transformant) was tested in duplicates. Yields of erythromycin were calculated with SAS/STAT software using means and the univariante procedure to test the normality of distribution. Using the GLM model, data were calculated as least mean square and are presented as an average change observed from all experiments when comparing least mean square values to the wild-type control least mean square value of each experiment.
Availability of supporting data
Genome sequence reads of the S. erythraea ABE1441 strain were deposited at NCBI SRA under accession SRX1277529.
The microarray data were deposited in NCBI’s Gene Expression Omnibus [37] and are accessible through GEO Series Accession number GSE65007 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=65007).
The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium [38] via the PRIDE partner repository with the dataset identifiers PXD002472 for spectral counts and PXD002677 for 2D PAGE analyses.
Authors’ contributions
KK and VM carried out cultivation of S. erythraea in shaker and fermenter level with sampling and analysed the morphology of transformant strains as well as carried out molecular cloning, conjugation of S. erythraea, genetic analysis of ex-conjugants and western blot analyses. KK and PJ participated in preparation of cell extracts for comparative proteomic analysis and analysed proteomic data obtained by 2-D PAGE. JH set up and carried out analytical methods for erythromycin determination. MP performed NGS data and variation analysis, ŠB, MP and AR analysed microarray and qPCR data. ID performed co-expression network analysis. RV, MF and BT performed mass spectrometry analysis and proteomic data processing and evaluation. ŠF was involved in study design, data interpretation and statistical analysis. ID, MP, ŠB, KG, HP and GK were involved in the conceptualization and design of the study, data interpretation and manuscript preparation. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank the European Regional Development Fund and the Government of Slovenia, Ministry of Education, Science and Sport, for the award of the “KC Brin competence centre” grant which provided most of the funding for this study (VM, MT, MH, MP, ŠB, ŠF, JH, HP, GK, RV, MF, BT). We are grateful to the European Social Fund and the Ministry of Economic Development and Technology for the “KROP” Grant (Contract No. C2130-14-090124) and employment of ID. KK is Grateful for the award of the “young researcher” funding by Slovenian Technology Agency—TIA/SPIRIT (contract No. P-MR-09/103). This work was also supported by the Slovenian Research Agency (ARRS), Grants (J4-2195 to PJ, P1-0140 and J1-4121 to BT and J4-4149 to ŠF). We would like to thank Neža Turnšek and Katja Stare for excellent technical assistance.
Competing interests
JH, ŠF and GK are shareholders of Acies Bio Ltd.
Abbreviations
- 2-D
two dimensional
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1- propanesulfonate
- CoA
coenzyme A
- DE
differentially expressed
- DTT
dithiothreitol GSEA: gene set enrichment analysis
- HA
human influenza hemagglutinin
- HP
S. erythraea high-producing strain ABBE1441
- LC–MS
liquid chromatography–mass spectrometry
- MNV
multiple nucleotide variation
- NRPS
non-ribosomal peptide synthetase
- ORF
open reading frame
- TCA
tricarboxylic acid
- PAGE
polyacrylamide gel electrophoresis
- PCR
polymerase chain reaction
- PMV
packed mycelium volume
- PKS
polyketide synthase
- qPCR
quantitative real-time PCR
- RBS
ribosomal binding site
- SDS
sodium dodecyl sulphate
- rpm
revolutions per minute
- SNV
single nucleotide variation
- UTR
untranslated region
- vvm
volume of air under standard conditions per volume of liquid per minute
- WT
S. erythraea wild type strain NRRL2338
Additional files
10.1186/s12934-016-0496-5 40 putative differences between the WT DNA sequence and published RNA-seq data [SRA Accession SRP014619] that were considered to be sequencing errors of the original genome sequence.
10.1186/s12934-016-0496-5 An integrated table of all obtained genomic, transcriptomic and proteomic data as well as data obtained and published in previous studies (Peano et al., 2012; Marcellin et al., 2013, Li et al., 2011).
10.1186/s12934-016-0496-5 A graphic representation of erythromycin biosynthesis genes eryK and eryAI in 6 individual fermentations of wild type and high producer strain of S. erythraea, analysed by qPCR.
10.1186/s12934-016-0496-5 A table for validation of microarray data by qPCR.
10.1186/s12934-016-0496-5 A table of overrepresentation of GO Molecular function gene sets (KEGG pathways or GO Biological processes) in S. erythraea HP compared to WT strain in individual time points (t1-t4).
10.1186/s12934-016-0496-5 A graphical representation of clusters of genes with differential time-course profiles between the HP and WT strains.
10.1186/s12934-016-0496-5 A graphic representation of regulatory genes whose expression profiles exhibit strong correlations to ery genes.
10.1186/s12934-016-0496-5 Results of the 2-D PAGE experiments: representation of 2-D gels of WT and HP strain in different time points (t1-t4), comparison of 2-D images between HP and WT in t1 and t2.
10.1186/s12934-016-0496-5 A heatmap representation of LC–MS proteomics results on individual proteins belonging to selected metabolic pathways.
10.1186/s12934-016-0496-5 A table of differentially expressed proteins according to 2-D PAGE: KEGG Pathway classification of differentially expressed proteins in time points 1 and 2.
10.1186/s12934-016-0496-5 A table of genes and proteins with apparently opposite regulation patterns in transcriptomic and high-throughput proteomic profiling.
10.1186/s12934-016-0496-5 Figures representing western blot analyses of constitutively over-expressed ilvB1 gene and mms operon (mmsOp).
10.1186/s12934-016-0496-5 A detailed description of selected methods: preliminary qPCR analysis of key erythromycin related genes, ABE1441 strain sequencing and read mapping metrics, proteomic analysis (2-D electrophoresis, spectral counting), oligonucleotide primers used in genetic engineering of S. erythraea.
10.1186/s12934-016-0496-5 A table with description of strains and plasmids generated and used in this study and graphical representation of over-expressed operons/genes.
Contributor Information
Katarina Karničar, Email: katarina.karnicar@aciesbio.com.
Igor Drobnak, Email: igor.drobnak@ki.si.
Marko Petek, Email: marko.petek@nib.si.
Vasilka Magdevska, Email: vasilka.magdevska@aciesbio.com.
Jaka Horvat, Email: jaka.horvat@aciesbio.com.
Robert Vidmar, Email: robert.vidmar@ijs.si.
Špela Baebler, Email: spela.baebler@nib.si.
Ana Rotter, Email: ana.rotter@nib.si.
Polona Jamnik, Email: polona.jamnik@bf.uni-lj.si.
Štefan Fujs, Email: stefan.fujs@aciesbio.com.
Boris Turk, Email: boris.turk@ijs.si.
Marko Fonovič, Email: marko.fonovic@ijs.si.
Kristina Gruden, Email: kristina.gruden@nib.si.
Gregor Kosec, Email: gregor.kosec@aciesbio.com.
Hrvoje Petković, Email: hrvoje.petkovic@aciesbio.com.
References
- 1.Zhou Z, Gu J, Du YL, Li YQ, Wang Y. The-omics era-toward a systems-level understanding of Streptomyces. Curr Genomics. 2011;12(6):404–416. doi: 10.2174/138920211797248556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hibi M, Yukitomo H, Ito M, Mori H. Improvement of NADPH-dependent bioconversion by transcriptome-based molecular breeding. Appl Environ Microbiol. 2007;73(23):7657–7663. doi: 10.1128/AEM.01754-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee KH, Park JH, Kim TY, Kim HU, Lee SY. Systems metabolic engineering of Escherichia coli for l-threonine production. Mol Syst Biol. 2007;3:149. doi: 10.1038/msb4100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park JH, Lee SY, Kim TY, Kim HU. Application of systems biology for bioprocess development. Trends Biotechnol. 2008;26(8):404–412. doi: 10.1016/j.tibtech.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Sindelar G, Wendisch VF. Improving lysine production by Corynebacterium glutamicum through DNA microarray-based identification of novel target genes. Appl Microbiol Biotechnol. 2007;76(3):677–689. doi: 10.1007/s00253-007-0916-x. [DOI] [PubMed] [Google Scholar]
- 6.Lee JW, Na D, Park JM, Lee J, Choi S, Lee SY. Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol. 2012;8(6):536–546. doi: 10.1038/nchembio.970. [DOI] [PubMed] [Google Scholar]
- 7.Berdy J. Bioactive microbial metabolites. J Antibiot (Tokyo) 2005;58(1):1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- 8.Adrio JL, Demain AL. Genetic improvement of processes yielding microbial products. FEMS Microbiol Rev. 2006;30(2):187–214. doi: 10.1111/j.1574-6976.2005.00009.x. [DOI] [PubMed] [Google Scholar]
- 9.Parekh S, Vinci VA, Strobel RJ. Improvement of microbial strains and fermentation processes. Appl Microbiol Biotechnol. 2000;54(3):287–301. doi: 10.1007/s002530000403. [DOI] [PubMed] [Google Scholar]
- 10.Lum AM, Huang J, Hutchinson CR, Kao CM. Reverse engineering of industrial pharmaceutical-producing actinomycete strains using DNA microarrays. Metab Eng. 2004;6(3):186–196. doi: 10.1016/j.ymben.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Baltz RH. Strain improvement in actinomycetes in the post genomic era. J Ind Microbiol Biotechnol. 2011;38(6):657–666. doi: 10.1007/s10295-010-0934-z. [DOI] [PubMed] [Google Scholar]
- 12.Martin JF, Liras P. Cascades and networks of regulatory genes that control antibiotic biosynthesis. Subcell Biochem. 2012;64:115–138. doi: 10.1007/978-94-007-5055-5_6. [DOI] [PubMed] [Google Scholar]
- 13.Gravius B, Bezmalinovic T, Hranueli D, Cullum J. Genetic instability and strain degeneration in Streptomyces rimosus. Appl Environ Microbiol. 1993;59(7):2220–2228. doi: 10.1128/aem.59.7.2220-2228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minas W. Production of erythromycin with Saccharopolyspora erythraea. In: Barredo JL, editor. Microbial processes and products. New York city: Humana Press; 2005. pp. 65–90. [Google Scholar]
- 15.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol. 2007;25(4):447–453. doi: 10.1038/nbt1297. [DOI] [PubMed] [Google Scholar]
- 16.Carata E, Peano C, Tredici SM, Ferrari F, Tala A, Corti G, Bicciato S, De Bellis G, Alifano P. Phenotypes and gene expression profiles of Saccharopolyspora erythraea rifampicin-resistant (rif) mutants affected in erythromycin production. Microb Cell Fact. 2009;8:18. doi: 10.1186/1475-2859-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang X, Liu S, Yu YT, Li YX, Li YY. Identifying modules of coexpressed transcript units and their organization of Saccharopolyspora erythraea from time series gene expression profiles. PLoS One. 2010;5(8):e12126. doi: 10.1371/journal.pone.0012126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li YY, Chang X, Yu WB, Li H, Ye ZQ, Yu H, Liu BH, Zhang Y, Zhang SL, Ye BC, et al. Systems perspectives on erythromycin biosynthesis by comparative genomic and transcriptomic analyses of S. erythraea E3 and NRRL23338 strains. BMC Genom. 2013;14:523. doi: 10.1186/1471-2164-14-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcellin E, Mercer TR, Licona-Cassani C, Palfreyman RW, Dinger ME, Steen JA, Mattick JS, Nielsen LK. Saccharopolyspora erythraea’s genome is organised in high-order transcriptional regions mediated by targeted degradation at the metabolic switch. BMC Genom. 2013;14:15. doi: 10.1186/1471-2164-14-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peano C, Bicciato S, Corti G, Ferrari F, Rizzi E, Bonnal RJ, Bordoni R, Albertini A, Bernardi LR, Donadio S, et al. Complete gene expression profiling of Saccharopolyspora erythraea using GeneChip DNA microarrays. Microb Cell Fact. 2007;6:37. doi: 10.1186/1475-2859-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peano C, Tala A, Corti G, Pasanisi D, Durante M, Mita G, Bicciato S, De Bellis G, Alifano P. Comparative genomics and transcriptional profiles of Saccharopolyspora erythraea NRRL 2338 and a classically improved erythromycin over-producing strain. Microb Cell Fact. 2012;11:32. doi: 10.1186/1475-2859-11-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Wang Z, Chu J, Xi B, Zhuang Y. The glucose RQ-feedback control leading to improved erythromycin production by a recombinant strain Saccharopolyspora erythraea ZL1004 and its scale-up to 372-m(3) fermenter. Bioprocess Biosyst Eng. 2015;38(1):105–112. doi: 10.1007/s00449-014-1248-8. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Fuente A. From ‘differential expression’ to ‘differential networking’—identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010;26(7):326–333. doi: 10.1016/j.tig.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS Comput Biol. 2008;4(8):e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13(4):227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magdevska V, Gaber R, Goranovic D, Kuscer E, Boakes S, Duran Alonso MB, Santamaria RI, Raspor P, Leadlay PF, Fujs S, et al. Robust reporter system based on chalcone synthase rppA gene from Saccharopolyspora erythraea. J Microbiol Methods. 2010;83(2):111–119. doi: 10.1016/j.mimet.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Kirm B, Magdevska V, Tome M, Horvat M, Karnicar K, Petek M, Vidmar R, Baebler S, Jamnik P, Fujs S, et al. SACE_5599, a putative regulatory protein, is involved in morphological differentiation and erythromycin production in Saccharopolyspora erythraea. Microb Cell Fact. 2013;12:126. doi: 10.1186/1475-2859-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reeves AR, Brikun IA, Cernota WH, Leach BI, Gonzalez MC, Weber JM. Effects of methylmalonyl-CoA mutase gene knockouts on erythromycin production in carbohydrate-based and oil-based fermentations of Saccharopolyspora erythraea. J Ind Microbiol Biotechnol. 2006;33(7):600–609. doi: 10.1007/s10295-006-0094-3. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Wang Z, Chu J, Xi B, Zhuang Y. The glucose RQ-feedback control leading to improved erythromycin production by a recombinant strain Saccharopolyspora erythraea ZL1004 and its scale-up to 372-m(3) fermenter. Bioprocess Biosystems Engineering. 2015;38(1):105–112. doi: 10.1007/s00449-014-1248-8. [DOI] [PubMed] [Google Scholar]
- 31.Gorg A. Two-dimensional electrophoresis. Nature. 1991;349(6309):545–546. doi: 10.1038/349545a0. [DOI] [Google Scholar]
- 32.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 33.Cox J, Michalski A, Mann M. Software lock mass by two-dimensional minimization of peptide mass errors. J Am Soc Mass Spectrom. 2011;22(8):1373–1380. doi: 10.1007/s13361-011-0142-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smyth GK. limma: Linear Models for Microarray Data. In: Gentleman R, Carey V, Huber W, Irizarry R, editors. Bioinformatics and computational biology solutions using r and bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 35.Conesa A, Nueda MJ, Ferrer A, Talon M. maSigPro: a method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics. 2006;22(9):1096–1102. doi: 10.1093/bioinformatics/btl056. [DOI] [PubMed] [Google Scholar]
- 36.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces genetics. Norwich: John Innes Foundation; 2000. [Google Scholar]
- 37.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32(3):223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]