

Abstract
Background
Our previous studies showed that the non-psychoactive cannabinoid, cannabidiol (CBD), ameliorates the clinical symptoms in mouse myelin oligodendrocyte glycoprotein (MOG)35-55-induced experimental autoimmune encephalomyelitis model of multiple sclerosis (MS) as well as decreases the memory MOG35-55-specific T cell (TMOG) proliferation and cytokine secretion including IL-17, a key autoimmune factor. The mechanisms of these activities are currently poorly understood.
Methods
Herein, using microarray-based gene expression profiling, we describe gene networks and intracellular pathways involved in CBD-induced suppression of these activated memory TMOG cells. Encephalitogenic TMOG cells were stimulated with MOG35-55 in the presence of spleen-derived antigen presenting cells (APC) with or without CBD. mRNA of purified TMOG was then subjected to Illumina microarray analysis followed by ingenuity pathway analysis (IPA), weighted gene co-expression network analysis (WGCNA) and gene ontology (GO) elucidation of gene interactions. Results were validated using qPCR and ELISA assays.
Results
Gene profiling showed that the CBD treatment suppresses the transcription of a large number of proinflammatory genes in activated TMOG. These include cytokines (Xcl1, Il3, Il12a, Il1b), cytokine receptors (Cxcr1, Ifngr1), transcription factors (Ier3, Atf3, Nr4a3, Crem), and TNF superfamily signaling molecules (Tnfsf11, Tnfsf14, Tnfrsf9, Tnfrsf18). “IL-17 differentiation” and “IL-6 and IL-10-signaling” were identified among the top processes affected by CBD. CBD increases a number of IFN-dependent transcripts (Rgs16, Mx2, Rsad2, Irf4, Ifit2, Ephx1, Ets2) known to execute anti-proliferative activities in T cells. Interestingly, certain MOG35-55 up-regulated transcripts were maintained at high levels in the presence of CBD, including transcription factors (Egr2, Egr1, Tbx21), cytokines (Csf2, Tnf, Ifng), and chemokines (Ccl3, Ccl4, Cxcl10) suggesting that CBD may promote exhaustion of memory TMOG cells. In addition, CBD enhanced the transcription of T cell co-inhibitory molecules (Btla, Lag3, Trat1, and CD69) known to interfere with T/APC interactions. Furthermore, CBD enhanced the transcription of oxidative stress modulators with potent anti-inflammatory activity that are controlled by Nfe2l2/Nrf2 (Mt1, Mt2a, Slc30a1, Hmox1).
Conclusions
Microarray-based gene expression profiling demonstrated that CBD exerts its immunoregulatory effects in activated memory TMOG cells via (a) suppressing proinflammatory Th17-related transcription, (b) by promoting T cell exhaustion/tolerance, (c) enhancing IFN-dependent anti-proliferative program, (d) hampering antigen presentation, and (d) inducing antioxidant milieu resolving inflammation. These findings put forward mechanism by which CBD exerts its anti-inflammatory effects as well as explain the beneficial role of CBD in pathological memory T cells and in autoimmune diseases.
Electronic supplementary material
The online version of this article (doi:10.1186/s12974-016-0603-x) contains supplementary material, which is available to authorized users.
Keywords: Autoimmune, Memory T cells, Gene expression, Cannabidiol
Background
Natural (Cannabis derived), synthetic, and endogenous cannabinoids were shown to exert potent anti-inflammatory effects in various models of inflammation (reviewed by [1, 2]), including T cell-mediated autoimmunity [3]. However, most of the experiments focused on the effects of THC, the main psychoactive Cannabis constituent, and on THC-like ligands that interact with either the CB1 cannabinoid receptors (mostly expressed on neurons) or the CB2 receptors (abundant on immune cells). Another phytocannabinoid, cannabidiol (CBD) has been recently gaining a major interest as a potent immunomodulatory compound [4]. CBD has a very weak affinity toward the CB1 and CB2 cannabinoid receptors and thus lacks CB1-mediated psychoactivity [5]. Moreover, CBD proved to have very low toxicity when examined in humans [6].
Indeed, CBD was observed to induce anti-inflammatory effects in animal models of T cell-mediated collagen-induced arthritis [7], autoimmune diabetes [8], and autoimmune hepatitis [9]. Recently, we have reported that CBD administered systemically ameliorated clinical symptoms in mouse myelin oligodendrocyte glycoprotein (MOG)35-55-induced experimental autoimmune encephalitis (EAE) model of multiple sclerosis (MS), a neurodegenerative autoimmune disease resulting in progressing paralysis and initiated by autoreactive T cells targeting myelin sheaths [10, 11]. We showed that CBD diminishes CNS immune infiltration, microglial activation, and axonal damage in these EAE mice [12]. Our observations were confirmed by other groups [13–15]. The mechanisms of these beneficial regulatory CBD activities are not yet understood.
Autoimmune pathologies, including MS/EAE, are driven by transformed subsets of T cells called memory T cells. These autoreactive memory T cells are falsely primed by antigen-presenting cells (APC) to target own cells leading to tissue degeneration and disease development including type I diabetes, rheumatoid arthritis, and MS. Memory T cells exhibit high proliferation potential in response to self-antigens along with high pathogenic effector functions controlled by specific signaling pathways [16]. Autoimmune memory T cells (including those that target myelin sheath and lead to MS development) secrete interleukin (IL)-17 cytokine in retinoic acid receptor-related orphan receptor γ-T (RORγt)/signal transducer and activator of transcription 3 (STAT3)-dependent manner and were defined as autoimmune Th17 phenotype [17–19]. Adoptive transfer of such encephalitogenic T cell clones to healthy animals results in rapid and severe MS-like symptoms [20, 21] and antigen re-activation of quiescent, circulating memory T cells may contribute to MS relapses in relapsing–remitting MS forms [22]. Therapeutic targeting of these memory T cells seems to be difficult although this strategy proved to be efficient [23].
The effects of cannabinoids, including CBD, on these antigen-specific memory T cells driving autoimmune pathologies are not well described and the mechanisms of these activities are not known. We have recently shown that CBD is able to decrease the function of encephalitogenic Th17 cells. Using a highly myelin-specific memory T cell line recognizing the MOG35-55 myelin epitope (TMOG) we showed that CBD decreases the production and release of IL-17 from encephalitogenic TMOG cells as well as of IL-6 [24], a cytokine controlling Th17 differentiation [25]. CBD also decreased the phosphorylation of STAT3 [26], a pathway known to control Th17-like function of memory TMOG cells [27]. In parallel, we observed that CBD boosted anti-inflammatory processes in these activated memory T cells including increased production of anti-inflammatory IL-10 cytokine and increased activity of several regulatory transcription factors including STAT5 and EGR2 [26].
To study the transcriptional mechanisms involved in the CBD immunoregulatory effects, we profiled gene expression in total mRNA isolated from activated TMOG cells treated with CBD using microarrays. Detailed bioinformatics analyses allowed us to identify gene networks, pathways and upstream regulators that mediate the CBD suppressory effects. We show that CBD downregulates the transcription of various proinflammatory genes controlling Th17 function of encephalitogenic T cells while enhancing IFN-dependent anti-proliferative genetic program and potentiating the expression of genes hampering APC/T communication and antigen presentation. Moreover, a number of anti-oxidant transcripts exerting anti-inflammatory effects were upregulated by CBD in activated memory T cells.
Methods
Reagents
Lyophilized MOG35-55 peptide [MEVGWYRSPFSRVVHLYRNGK] purchased from GenScript (Piscataway, NJ, USA) was reconstituted in sterile PBS and the stock solution stored in aliquots at −20 °C. CBD (kindly obtained from Prof. Raphael Mechoulam, the Hebrew University of Jerusalem, Israel) was dissolved in ethanol. The dose of 5 μM CBD used here was selected based on our previous studies in which we showed that CBD at this concentration significantly inhibits MOG35-55-induced TMOG cell proliferation and their Th17 activity, i.e., IL-17 expression and release [12, 24, 26]. Fetal calf serum (FCS) and other tissue culture reagents were obtained from Biological Industries (Kibbutz Beit HaEmek, Israel).
TMOG stimulation and CD4+ microbead purification from APC/TMOG co-cultures
The MOG35-55-specific T cell line (TMOG) was maintained as described before [12, 24, 26, 28]. APCs were freshly isolated from spleens of 8-week naïve male C57BL/6 mice just before the experiments. Dissociated spleen cells were plated (50 × 106 cells/10 cm plate) in RPMI-1640 medium containing 2.5 % FCS, 100 μg/ml streptomycin, 100 U/ml penicillin, 2 mM L-glutamine, and 50 μM β-mercaptoethanol. After 2 h at 37 °C in 5 % CO2 humidified air to allow APC adherence, the media with non-adherent cells was removed and the adherent APCs gently washed with Ca++/Mg++ containing PBS and covered with a new medium. Then, 2.5 × 106 of TMOG cells were added and APC/TMOG co-cultures were stimulated immediately with 5 μg/ml of MOG35-55 for 8 h in the presence or absence of CBD at 5 μM. The incubation time of 8 h was chosen based on previous time- and dose-response experiments [24, 26, 29]. CBD was added just prior to the addition of the MOG35-55. After 8 h of incubation, the media containing mostly TMOG cells (but not the adherent APC cells) were carefully collected and spun down for 10 min at 2,000 rpm. The cell pellet was washed in PBS containing 0.5 % BSA and 2 mM EDTA, centrifuged again, and resuspended in 90 μl of this buffer. To improve the purity of collected cells, CD4 (L3T4) magnetic beads (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) were added to the cell suspension for positive selection of CD4+ cells as described earlier [26, 29]. The mRNA isolated from purified TMOG cells was subjected to global mRNA microarray expression analysis followed by quantitative real time reverse transcription polymerase chain reaction (qPCR) for validation of selected gene products.
RNA extraction, microarray transcript analysis, and validation by qPCR
Purified TMOG cells were lysed and subjected to RNA extraction (5Prime, Darmstadt, Germany) as described earlier [26, 29, 30]. For comparative microarray analysis, 200 ng of total RNA were amplified, labeled and hybridized onto Illumina MouseRef-8 v 2.0 Expression Bead-Chip (Illumina Inc., San Diego, CA, USA) as described earlier [26, 29]. Raw data were log2 transformed and normalized using quantile normalization. The data is presented throughout the manuscript as fold change unless stated otherwise. Statistical microarray analysis and gene expression analysis of the raw data were performed at the Informatics Center for Neurogenetics and Neurogenomics core at UCLA using R scripts (www.r-project.org) and Bioconductor packages (http://www.bioconductor.org; [31]) as described [32].
Pathway and global functional analyses were performed using QIAGEN’s Ingenuity Pathway Analysis (IPA®, QIAGEN Redwood City CA, USA www.qiagen.com/ingenuity). Genes that met the p value cutoff of 0.005 for differential expression were used to build the gene networks using IPA tools.
Weighted Gene Co-expression Network Analysis (WGCNA; [33]) (http://labs.genetics.ucla.edu/horvath/htdocs/CoexpressionNetwork/) was applied to complete the functional characterization of gene expression data. WGCNA is an analysis method that recognizes co-expression networks based on topological overlap between genes and considers the correlation of two genes with each other and the degree of their shared correlations within the network. Briefly, genes consistently present on arrays with high coefficient of covariation and high connectivity were selected for network construction and were hierarchically clustered. Clusters of highly interconnected genes (modules) were determined based on their topological overlap using a dynamic tree-cutting algorithm. Such modules were visualized using VisANT (http://visant.bu.edu). The gene expression pattern was condensed within a module to a “module eigengene” (ME), which is a weighted summary of gene expression in the module and can be correlated to traits. The relationships between genes within modules can be identified, and follow up analyses can focus on hub genes (genes with highest connectivity in the module) and corresponding pathways driving gene expression changes without any a priori assumptions about gene function. At the final stage, the modules of interest were annotated using gene ontology (GO) functional and biological categories using online tool (http://geneontology.org). The WGCNA method has been used in a large number of recent transcriptional studies to reveal functional gene networks [34, 35].
qPCR analysis
Selected gene products found by microarray analysis to be affected by CBD were validated by qPCR as described [26, 30]. The cDNA of each chosen gene was amplified using a pair of specific primers presented in Additional file 1: Table S1. β2-microglobulin (β2mg) gene product was used for normalization [30]. The qPCR runs were repeated 3–4 times using mRNA preparations from independent experiments.
ELISA assay
The enzyme-linked immunosorbent assay (ELISA) was performed as described earlier [24]. Shortly, the TMOG cells were cultured in assay medium in 24-well plates (0.25 × 106 cells per well) together with splenic APC (5 × 106 cells/well). CBD at 5 μM was added to the cells just before the addition of MOG35-55 at 5 μg/ml. After 24 h of incubation, the cell-conditioned media were collected, spun down, and analyzed for IL-1β and IL-3 concentrations by ELISA (R&D Systems, Minneapolis, MN, USA). The incubation time of 24 h was chosen based on our previous observations [24].
Statistical analysis of qPCR and ELISA data
qPCR and ELISA data are expressed as the mean ± SEM of 3–4 independent experiments and analyzed for statistical significance using one way analysis of variance (ANOVA), followed by Newman-Keul’s post-hoc test. p < 0.05 was considered significant. Graph Pad Prism program (La Jolla, CA, USA) was used for statistical analysis of the data.
Results
Gene expression profile of activated TMOG cells treated with CBD
We recently described the detailed gene expression profile of TMOG activated by MOG35-55 [29] and demonstrated a powerful proinflammatory effect of MOG35-55 activation at the transcriptional level confirming the Th17 function of activated TMOG cells [24, 26].
Here, we examined the effects of CBD co-incubation on MOG35-55-induced transcriptional profile of TMOG. Samples of mRNA were prepared from purified TMOG cells (CD4+ positive selection), co-cultured earlier with pre-attached APC, and stimulated with MOG35-55 at 5 μg/ml for 8 h in the presence or absence of CBD at 5 μM.
Microarray analysis based on the threshold of p < 0.005 revealed that 2755 transcripts (within ~25,600 targets present on this Illumina set) were differentially regulated across the treatments. Of these, stimulation with MOG35-55 upregulated the expression of 842 gene transcripts and downregulated 1094 gene probes (Fig. 1a). Addition of CBD to MOG35-55-stimulated TMOG cells resulted in a total of 968 upregulated gene transcripts and in 1330 downregulated transcripts as compared to control (without CBD and MOG35-55) samples. Out of these, 81 gene products were significantly (p < 0.005) upregulated by the addition of CBD and 82 gene products were downregulated by CBD as compared to the level observed in MOG35-55-stimulated cells without CBD (Fig. 1b). TMOG cells stimulated with MOG35-55 alone or co-incubated with CBD shared the same 627 upregulated and 852 downregulated gene products. Twenty eight transcripts were upregulated and 30 downregulated in TMOG cells incubated with CBD given alone or by CBD+MOG35-55 combination. Only one gene product was downregulated by MOG35-55-only stimulation and by CBD-only treatment. Interestingly, 13 individual transcripts were always upregulated and 37 were always downregulated by all experimental conditions.
Fig. 1.
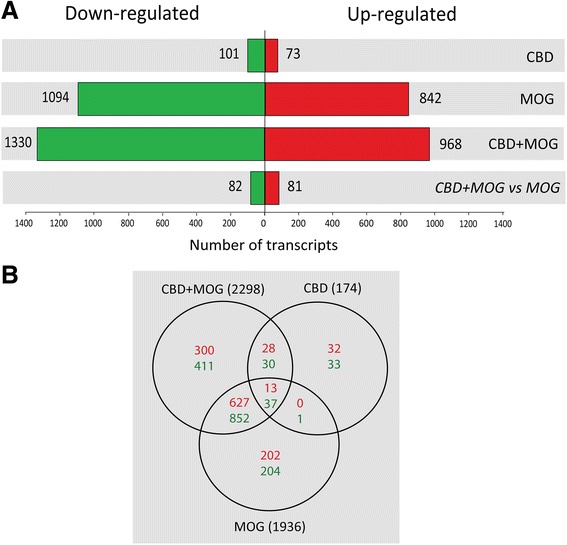
Number of gene transcripts affected in MOG35-55-stimulated TMOG cells in the presence or absence of CBD. mRNA prepared from TMOG cells co-cultured with APC and stimulated with MOG35-55 in the presence or absence of CBD was subjected to microarray analysis as described in Methods. a Number of differentially expressed transcripts that were either significantly upregulated (red) or downregulated (green) across the different treatment conditions vs control untreated cells (p < 0.005). b Venn’s diagram illustrating the number of transcripts affected (up- and downregulation) by CBD, MOG35-55, or CBD+MOG35-55 treatments (p < 0.005) and their overlap
Gene by gene analysis
Only genes reaching the criteria of p < 0.005 and fold change of 0.2 (i.e., 20 % change) were included in further analysis of CBD effects on transcriptional activity of stimulated TMOG. Several approaches were employed to characterize the CBD modulation of self-antigen activation of TMOG. These included (1) identification of the most CBD-suppressed transcripts within those activated by MOG35-55, (2) identification of MOG35-55-upregulated transcripts that were not affected by CBD, (3) identification of the most CBD-upregulated transcripts, and (4) identification of the effect of CBD on the MOG35-55 downregulated genes.
Table 1 displays the transcripts upregulated by stimulation with MOG35-55 and significantly reduced in the presence of CBD. These transcripts include: cytokines such as Il3 mRNA (downregulated by CBD by 72 %), Il1b mRNA (reduced by 70 %), Xcl1 (by 49 %), and Il12a (by 24 %); cytokine and chemokine receptors’ mRNAs (e.g., Ifngr1 by 60 %, Cxcr1 by 50 %); transcription factors and regulators (Crem by 50 %, Ier3 by 31 %, Atf3 by 28 %); TNF-family signaling members (Tnfrsf18 by 40 %, Tnfsf11 and Tnfsf14 by 30 %, Tnfrsf9 by 21 %); signaling elements including those affecting adhesion (Amica1 by 21 %), growth (Igfbp4 by 50 %), cell structure and trafficking (Vps37b by 60 %, Tubb2d by 50 %) as well as lipid/cholesterol synthesis and metabolism (Dgat1 by 60 %, Fdps by 50 %). These changes indicate that CBD has a profound suppressing effect on various MOG35-55-activated genes in TMOG cells. Several of the gene transcripts that were reduced by the combined CBD+MOG35-55 treatment (vs MOG35-55 only) were also reduced by incubation with CBD alone (vs control) including Il1b (by 70 %), Cxcr1 (by 30 %), Ifngr1 (by 60 %), Crem (by 50 %), Tnfrs18 (by 50 %), Igfbp4 (by 50 %), Vps37b (by 60 %), Tubb2b (by 50 %), Fdps (by 50 %), and Dgat1 (by 60 %).
Table 1.
List of transcripts which expression is upregulated by MOG35-55 stimulation and this upregulation is suppressed by CBD (p < 0.005)
| Symbol | Gene name | Accession number | MOG alone [fold]$ | MOG+CBD [fold]$ | Change [%] | CBD alone [fold]$ |
|---|---|---|---|---|---|---|
| Cytokines, chemokines, and receptors | ||||||
| Xcl1 | Chemokine (C motif) ligand 1 | NM_008510.1 | 30.0 | 15.3 | −49 % | 1.0 |
| Il3 | Interleukin 3 | NM_010556.2 | 25.4 | 7.2 | −72 % | 1.0 |
| Cxcl9 | Chemokine (C-X-C motif) ligand 9 | NM_008599 | 7.4 | 5.4 | −27 % | 0.9 |
| Gpr83 | G protein-coupled receptor 83 | NM_010287 | 3.9 | 1.8 | −53 % | 0.9 |
| Il12a | Interleukin 12A | NM_008351 | 2.3 | 1.5 | −24 % | 0.9 |
| Cxcr1 | Chemokine (C-X-C motif) receptor 1 (Il8ra, CD181) | NM_178241.1 | 1.7 | 0.9 | −50 % | 0.7 |
| Il1b | Interleukin 1, beta | NM_008361 | 1.5 | 0.5 | −70 % | 0.3 |
| Ifngr1 | Interferon gamma receptor 1 | NM_10511.1 | 0.8 | 0.3 | −60 % | 0.4 |
| Transcription factors and regulators | ||||||
| Ier3 | Immediate early response 3 | NM_133662.1 | 5.5 | 3.8 | −31 % | 1.3 |
| Atf3 | Activating transcription factor 3 | NM_007498.2 | 4.7 | 3.4 | −28 % | 1.1 |
| Nr4a3 | Nuclear receptor subfamily 4, group A, member 3 | NM_015743.1 | 4.1 | 3.2 | −22 % | 1.0 |
| Crem | cAMP responsive element modulator | NM_013498 | 1.9 | 0.9 | −50 % | 0.5 |
| TNF-family signaling elements | ||||||
| Tnfsf11 | Tumor necrosis factor ligand superfamily member 11 (Receptor activator of nuclear factor kappa-B ligand (RANKL)) | NM_011613.2 | 11.8 | 8.2 | −31 % | 0.9 |
| Tnfsf14 | Tumor necrosis factor ligand superfamily member 14 | NM_019418.1 | 5.0 | 3.5 | −30 % | 1.0 |
| Tnfrsf9 | Tumor necrosis factor receptor superfamily, member 9 | NM_011612.2 | 4.2 | 3.3 | −21 % | 0.8 |
| Tnfrsf18 | Tumor necrosis factor receptor superfamily, member 18 | NM_009400.1 | 1.3 | 0.7 | −40 % | 0.5 |
| Signaling elements | ||||||
| Sphk1 | Sphingosine kinase | NM_011451 | 12.3 | 8.2 | −34 % | 1.0 |
| Amica1 | Adhesion molecule interacting with CXADR antigen | NM_001005421.2 | 5.0 | 3.9 | −21 % | 1.2 |
| Axud1 | Cysteine/serine-rich nuclear protein 1/TGF-beta induced apoptosis protein 3 | NM_153287.2 | 4.4 | 3.5 | −20 % | 0.9 |
| Cdk5r1 | Cyclin-dependent kinase 5 activator 1 | NM_009871.2 | 3.9 | 3.1 | −20 % | 0.9 |
| Igfbp4 | Insulin-like growth factor binding protein 4 | NM_010517.2 | 2.3 | 1.1 | −50 % | 0.5 |
| Vps37b | Vacuolar protein sorting 37 homologue B | NM_177876.2 | 1.5 | 0.6 | −60 % | 0.4 |
| Dgat1 | Diacylglycerol O-acyltransferase 1 | NM_010046.2 | 1.0 | 0.4 | −60 % | 0.4 |
| Tubb2d | Tubulin, beta 2b | NM_023716.1 | 0.8 | 0.5 | −50 % | 0.5 |
| Fdps | Farnesyl diphosphate synthase | NM_134469 | 0.8 | 0.4 | −50 % | 0.5 |
$The results are presented as fold change vs control (without MOG35-55 or CBD). Numbers in italics indicate a significant effect of CBD alone (p < 0.005) in non-stimulated TMOG
It is important to note (see Table 2) that several of the MOG35-55-upregulated proinflammatory transcripts were not affected by CBD (given in the presence of MOG35-55). These include transcripts of several cytokines (e.g., Csf2, Tnf, Ifng), MOG35-55-upregulated chemokines (e.g., Ccl3, Ccl4, Cxcl10), regulatory glycoproteins (e.g., Sema7a), transcription factors mediating tolerogenic mechanisms (Egr2, Egr1), and several IFN-related transcriptional and signaling regulators (e.g., Tbx21, Ifit3).
Table 2.
List of transcripts which expression is upregulated by MOG35-55 stimulation and not affected by CBD
| Symbol | Gene name | Accession number | MOG alone [fold]$ | MOG+CBD [fold]$ | CBD alone [fold]$ |
|---|---|---|---|---|---|
| Cytokines | |||||
| Csf2 | Colony stimulating factor 2 | NM_009969.2 | 77.0 | 67.1 | 1.1 |
| Tnf | Tumor necrosis factor alpha | NM_013693.1 | 15.3 | 13.8 | 1.1 |
| Ifng | Interferon gamma | NM_008337.1 | 10.8 | 9.5 | 0.8 |
| Chemokines | |||||
| Ccl3 | Chemokine (C-C motif) ligand 3 | NM_011337.1 | 54.1 | 48.6 | 0.9 |
| Ccl4 | Chemokine (C-C motif) ligand 4 | NM_013652.1 | 40.2 | 37.3 | 0.9 |
| Cxcl10 | Chemokine (C-X-C motif) ligand 10 | NM_021274.1 | 9.0 | 9.8 | 0.9 |
| Glycoproteins | |||||
| Sema7a | Semaphorin 7A (GPI membrane anchor) | NM_011352.2 | 4.6 | 4.3 | 0.8 |
| Transcription factors | |||||
| Egr2 | Early growth response 2 | NM_010118.1 | 20.1 | 18.9 | 1.3 |
| Egr1 | Early growth response 1 | NM_007913.2 | 10.7 | 11.7 | 1.5 |
| Tbx21 | T-box 21 | NM_019507.1 | 2.2 | 2.5 | 0.9 |
| Signaling elements | |||||
| Ndrg1 | N-myc downstream regulated 1 | NM_010884.1 | 6.2 | 6.7 | 2.5 |
| Ifit3 | Interferon-induced protein with tetratricopeptide repeats 3 | NM_010501.1 | 5.6 | 5.9 | 0.9 |
$The results are presented as fold change vs control (without MOG35-55 or CBD). Numbers in italics indicate a significant effect of CBD alone (p < 0.005)
Gene transcripts that were significantly upregulated in the presence of CBD in MOG35-55-stimulated TMOG cells are listed in Table 3. Among these transcripts we find mainly membrane and transmembrane molecules known to negatively regulate T cell activation, e.g., via disruption of APC/T interactions as well as of antigen processing and presentation. These include transcripts of Lag3 (increase of 210 %), Btla (80 %), and CD69 (39 %). Moreover, CBD given alone or in combination with MOG35-55 enhanced the transcription of Trat1 (an intracellular element trafficking CTLA-4 inhibitory T cell receptor into the membrane) by 60 and 200 %, respectively. CBD had a remarkable boosting effect on the transcriptional activity of numerous IFN-dependent genes known to execute IFN-driven anti-proliferative responses including Mx2, Irf4, Ifit2, Ephx1, Rsad2 (Viperin), Ets2, Trim30, and Tyki mRNAs. In addition, CBD had an enhancing effect on a number of transcripts associated with GTP activation and signaling, protein transport, and turnover in T cells such as Tagap, Gfod1, Tnfsf8 (CD30), Nfatc1, Gbp11, Ppic, Dusp6, and Slc3a2 (CD98) mRNAs.
Table 3.
List of genes which transcription is enhanced by CBD in TMOG cells (p < 0.005)
| Symbol | Gene name | Accession number | MOG alone [fold]$ | MOG+CBD [fold]$ | Change [%] | CBD alone [fold]$ |
|---|---|---|---|---|---|---|
| MOG35-55-upregulated and CBD-enhanced | ||||||
| Surface negative regulators | ||||||
| Lag3& | Lymphocyte-activation gene 3 | NM_008479.1 | 1.7 | 5.2 | +210 % | 1.7 |
| Btla | B and T lymphocyte associated | NM_177584.3 | 1.8 | 3.2 | +80 % | 2 |
| Cd69 | Cluster of Differentiation 69 | XM_132882.1 | 3.9 | 5.4 | +39 % | 1.4 |
| IFN-dependent transcripts | ||||||
| Rsad2 | Radical S-adenosyl methionine domain containing 2 | NM_021384.2 | 4.9 | 9.5 | +94 % | 1.6 |
| Trim30 | Tripartite motif-containing 30 | NM_009099 | 2.2 | 3.1 | +40 % | 1.1 |
| Mx2 | Myxovirus (influenza virus) resistance 2 | NM_013606 | 6.0 | 8.2 | +37 % | 1.0 |
| Irf4 | Interferon regulatory factor 4 | NM_013674.1 | 4.5 | 5.3 | +18 % | 1.4 |
| Signaling elements | ||||||
| Gbp11 | Guanylate binding protein 11 | NM_01039647.1 | 3.2 | 7 | +120 % | 1.1 |
| Ppic | Peptidylprolyl isomerase C | NM_008908.1 | 1.8 | 3.2 | +80 % | 1.8 |
| Tyki | Thymidylate kinase family LPS-inducible member | NM_020557.3 | 4 | 5.7 | +42 % | 1 |
| Tagap | T-cell activation RhoGTPase activating protein | NM_145968.1 | 4.7 | 6.4 | +35 % | 1.4 |
| Tnfsf8 | Tumor necrosis factor (ligand) superfamily, member 8 | NM_009403.2 | 2.2 | 2.9 | +31 % | 1.1 |
| Dusp6 | Dual specificity phosphatase 6 | NM_026268.1 | 3.4 | 4.3 | +26 % | 1.4 |
| Gfod1 | Glucose-fructose oxidoreductase domain containing 1 | NM_001033399.1 | 6.2 | 7.8 | +26 % | 1.0 |
| Nfatc1& | Nuclear factor of activated T-cells, cytoplasmic 1 | NM_16791 | 3.3 | 4 | +21 % | 1.3 |
| Only CBD upregulated | ||||||
| Anti-oxidant and anti-inflammatory | ||||||
| Mt1 | Metallothionein 1 | NM_013602.2 | 0.5 | 2.3 | +330 % | 3 |
| Slc30a1 | Solute carrier family 30 (zinc transporter), member 1 | NM_009579 | 0.9 | 1.8 | +100 % | 1.8 |
| Mt2 | Metallothionein 2 | NM_008630.1 | 1 | 1.9 | +80 % | 2.3 |
| Negative regulators of T cell activity | ||||||
| Trat1 | T cell receptor associated transmembrane adaptor 1 | NM_198297 | 0.7 | 2.1 | +200 % | 1.6 |
| IFN-dependent transcripts | ||||||
| Ephx1 | Epoxide hydrolase 1 | NM_010145.2 | 0.7 | 1.8 | +170 % | 2.4 |
| Ets2 | V-ets avian erythroblastosis virus | NM_011809.2 | 1.3 | 2.4 | +80 % | 1.3 |
| Signaling elements | ||||||
| Ddr1 | Discoidin domain receptor tyrosine kinase 1 | NM_007584.1 | 1.1 | 3 | +170 % | 2.4 |
| Pitpnm2 | Phosphatidylinositol transfer protein, membrane-associated 2 | NM_011256.1 | 1.2 | 2.9 | +150 % | 2.6 |
| Gpnmb | Glycoprotein (transmembrane) nmb | NM_053110.2 | 0.4 | 0.9 | +90 % | 2.2 |
| Asb2 | Ankyrin repeat and SOCS box-containing protein 2 | NM_023049.1 | 0.29 | 0.52 | +79 % | 1.38 |
| Slc3a2 | Solute carrier family 3 member 2 | NM_008577.2 | 1.3 | 2.3 | +70 % | 1.5 |
$The results are presented as fold change vs control (without MOG35-55 or CBD). Numbers in italics indicate a significant effect of CBD (p < 0.005) in non-stimulated TMOG. &result reported previously [25]
Several transcripts including anti-oxidant and anti-inflammatory as well as negative regulators of T cell activity were increased by CBD even that they were not affected by MOG35-55. These included the zinc transporter Slc30a1, metallothioneins (Mt1 and Mt2), heme oxygenase (Hmox1), and mRNA of a protein preserving endoplasmic reticulum homeostasis (Herpud1) (Table 3, Figs. 2 and 3).
Fig. 2.
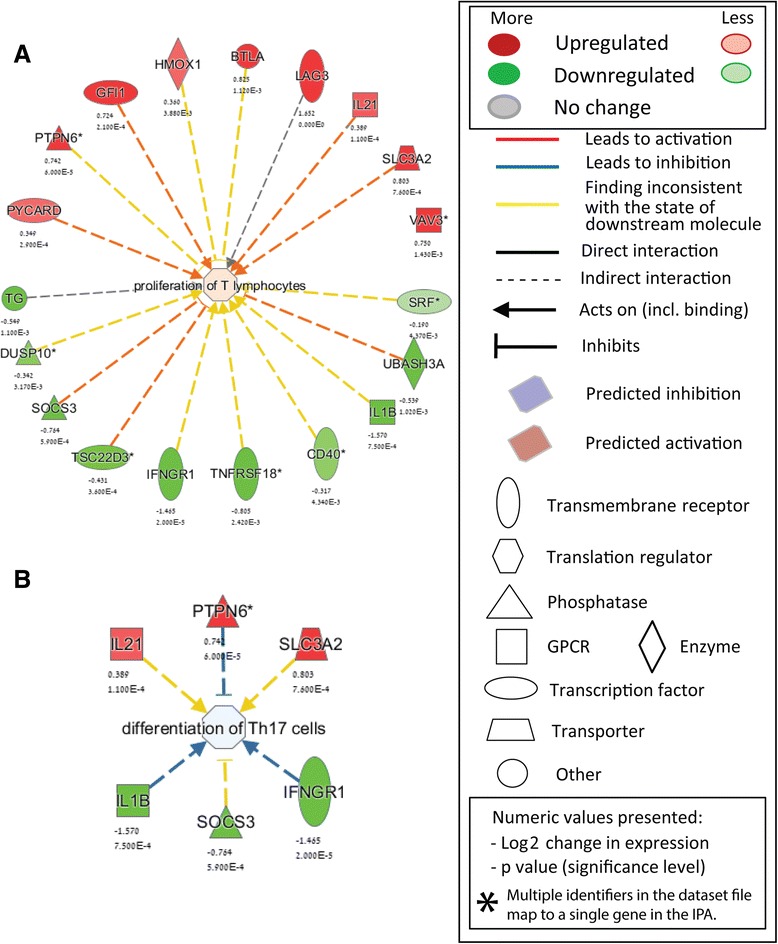
IPA-identified CBD-enriched functional terms in activated TMOG cells. a Genes forming “Proliferation of T lymphocytes” annotation found to be affected by CBD (b). Genes forming “Differentiation of Th17 cells” annotation found to be affected by CBD. The direction of change is color-coded with green implying downregulation and red implying upregulation. Log2 values expressing CBD-induced change vs MOG35-55-only-stimulated cells and respective p values are provided below each transcript name. Explanation of symbols is provided in the Legend
Fig. 3.
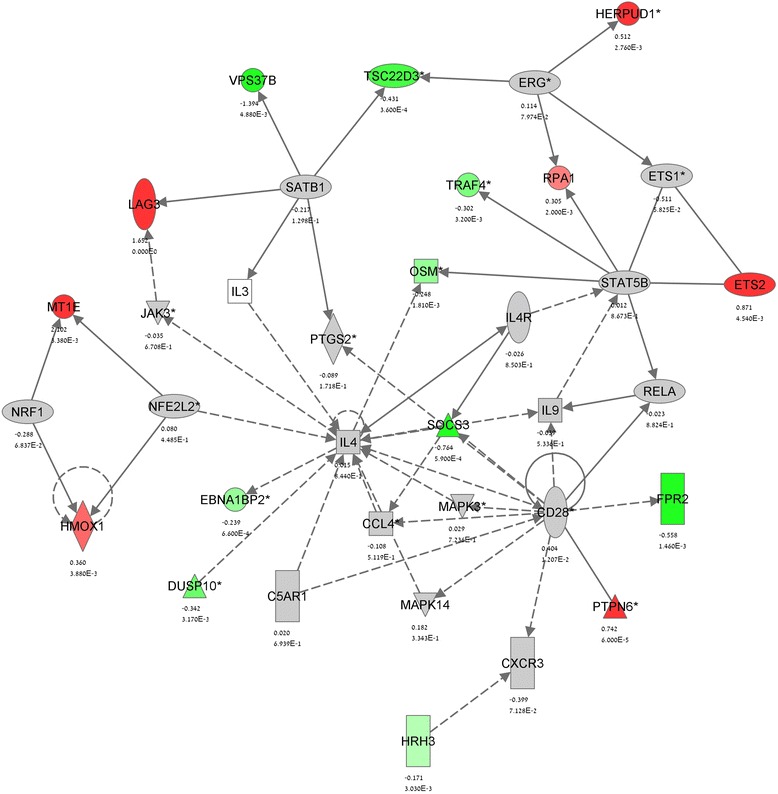
IPA gene network analysis of CBD effect in stimulated TMOG cells—Network 1. Network analysis of the genes which expression is significantly (p < 0.005) affected by CBD in MOG35-55-stimulated TMOG cells was performed using the IPA software as described in Methods. The network display, symbols, and biological relationships between the genes are explained in Fig. 2. Genes in grey were shown in the literature to interact with the colored gene products that appear in the scheme
At the last step, we focused on MOG35-55-downregulated transcripts and studied the effect of CBD on their levels. Table 4 shows a list of several highly MOG35-55-suppressed transcripts that were not affected by CBD. The list comprises of representatives of various gene families and functions. Interestingly, MOG35-55-activation led to a dramatic decrease in transcription of several IL-17 receptors (Il17re, transcript variant 2, Il17re variant 1, and Il17rc; see also [29]) and this downregulation was not affected by the presence of CBD.
Table 4.
MOG35-55-most-downregulated genes (p < 0.005) that are not affected by CBD
| Symbol | Definition | Accession | MOG alone [fold]$ | MOG+CBD [fold]$ | CBD alone [fold]$ |
|---|---|---|---|---|---|
| Il17re | Interleukin 17 receptor E, variant 2 | NM_001034029.1 | 0.23 | 0.23 | 1.04 |
| Sytl2 | Synaptotagmin-like 2, variant 5 | NM_001040088.1 | 0.25 | 0.27 | 1.03 |
| Il17re | Interleukin 17 receptor E, variant 1 | NM_145826.2 | 0.26 | 0.27 | 1.11 |
| Tmem176a | Transmembrane protein 176A | NM_025326.2 | 0.28 | 0.30 | 1.07 |
| Ian3 | Immune associated nucleotide 3 | NM_146167.2 | 0.29 | 0.23 | 0.94 |
| Klrd1 | Killer cell lectin-like receptor, subfamily D, member1 | NM_010654.1 | 0.30 | 0.29 | 0.95 |
| Klri2 | Killer cell lectin-like receptor family I member 2 | NM_177155.2 | 0.31 | 0.35 | 1.01 |
| Sit1 | Suppression inducing transmembrane adaptor 1 | NM_019436.1 | 0.32 | 0.29 | 0.84 |
| Il17rc | Interleukin 17 receptor C | NM_134159.2 | 0.33 | 0.36 | 0.95 |
| Golph2 | Type II Golgi transmembrane protein | NM_027307 | 0.35 | 0.35 | 0.94 |
$The results are presented as fold change vs control (without MOG35-55 or CBD)
Identification of functional networks affected by CBD in activated TMOG
As described above, CBD treatment of the MOG35-55 treated TMOG cells resulted in upregulation of 81 transcripts and in downregulation of 82 transcripts. These genes were uploaded onto the IPA analysis program to identify the functional subsets, pathways, gene networks, and upstream regulators targeted by CBD. Figure 2 illustrates two IPA-identified CBD-enriched GO-functional terms in activated TMOG cells, “Proliferation of T lymphocytes” (Fig. 2a) and “Differentiation of Th17 cells” (Fig. 2b). The “Proliferation of T lymphocytes” functional annotation was recognized by IPA to be upregulated in activated TMOG as a result of CBD co-incubation due to upregulated transcription of Pycard, Ptpn6, Gfi1, Hmox1, Btla, Lag3, Il21, Slc3a2 and Vav3 mRNAs and due to parallel suppression of Tg, Dusp10, Socs3, Tsc22d3, Ifngr1, Tnfsf18, Cd40, Il1b, Ubash3a and Srf mRNAs (Fig. 2a). In addition, within the “Differentiation of Th17 cells" annotation, IPA classified the upregulated Il21, Ptpn6, Slc3a2 and the downregulated Il1b, Socs3, and Ifngr1 transcripts and recognized it to be suppressed by CBD in stimulated TMOG (Fig. 2b).
IPA analysis identified several other canonical pathways that are affected by CBD in MOG35-55-stimulated TMOG (Table 5). These included “IL-10 signaling” and “IL-6 signaling” (p < 0.005), followed by “cAMP-mediated signaling”, “IGF-1 signaling”, “Fcg receptor mediated phagocytosis in macrophages and monocytes”, “Superpathway of cholesterol biosynthesis”, and “T helper cell differentiation” pathways (p < 0.05).
Table 5.
IPA-identified “top canonical pathways” enriched by CBD treatment in stimulated TMOG cells
| Canonical pathways | Annotated genes | p value |
|---|---|---|
| IL-10 signaling | Hmox1, Il1b, Jun, Socs3 | 0.0039 |
| IL-6 signaling | Il1b, Jun, Socs3, Srf, Vegfa | 0.0042 |
| cAMP-mediated signaling | Crem, Fpr2, Gnal, Grm6, Hrh3 | 0.024 |
| IGF-1 signaling | Igfbp4, Jun, Socs3, Srf | 0.015 |
| Fcγ receptor mediated phagocytosis in macrophages and monocytes | Appc3, Hck, Hmox1, Vav3 | 0.012 |
| Superpathway of cholesterol biosynthesis | Fdps, Sqle | 0.016 |
| T helper cell differentiation | CD40, Ifbgr1, Il21 | 0.042 |
IPA identified the gene networks affected by CBD in MOG35-55 stimulated TMOG cells (Figs. 3 and 4). Within these gene networks IPA denoted the main upstream regulators which function is modulated by CBD. Accordingly, Network 1 (Fig. 3) links the effects of CBD to the modulation of IL-4, STAT5B, SATB1, CD28, NRF2/NFE2L2, and NRF1 while Network 2 (Fig. 4) links the effects of CBD to IL-2 and IL-1β as well as to NFKB1 and EGR2.
Fig. 4.
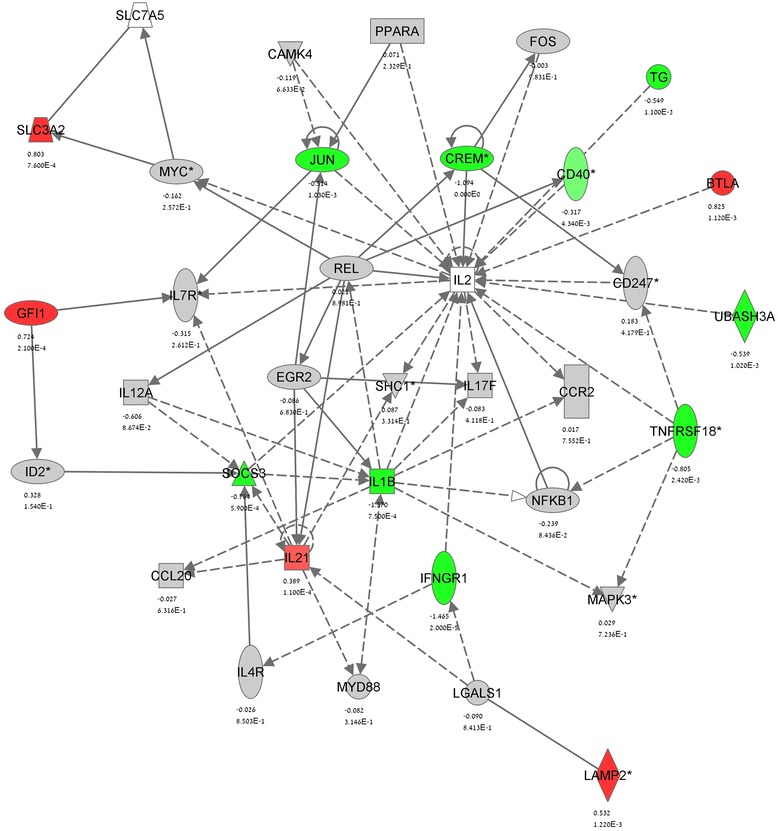
IPA gene network analysis of the CBD modulation of stimulated TMOG cells—Network 2. See Figs. 2 and 3 for details
Altogether, we show here that more cytokines, chemokines, receptors, and signaling pathways were down-regulated by CBD (Table 1, Figs. 1 and 2) than up-regulated (Table 3) which is in agreement with the reduction of TMOG cell proliferation and their Th17 function shown by us before [12, 24]. However, among up-regulated genes we found mainly regulatory T cell molecules known to inhibit T cell proliferation and function.
Validation by ELISA and qPCR
Several gene products that were identified by microarray analysis as differentially regulated were subjected to validation by either qPCR using β2-microglobulin as a reference gene) or by ELISA (for the quantification of protein secretion). Figure 5a shows that TMOG stimulation with MOG35-55 resulted in increased IL-1β protein secretion and that co-incubation with CBD lowers it back to control level. Moreover, TMOG stimulation with MOG35-55 increased IL-3 secretion by circa tenfold and this level was suppressed by CBD by 40 % (Fig. 5b).
Fig. 5.
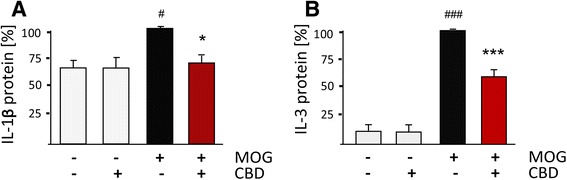
ELISA of IL-1β and IL-3 cytokines released from MOG35-55-stimulated TMOG cells in the presence or absence of CBD. APC/TMOG were stimulated with MOG35-55 at 5 μg/ml for 24 h in the presence or absence of CBD at 5 μM and the amounts of (a) IL-1β and (b) IL-3 in the culture media determined. 100 % is equivalent to maximal MOG35-55 effect which refers to 15 ± 6 pg/ml for IL-1β and to 377 ± 100 pg/ml for IL-3. Statistics (one way ANOVA) and symbols: a F(3,6) = 6.7, p < 0.05. b F(3,15) = 80.25, p < 0.001; Symbols: # p < 0.05, ### p < 0.01 vs Control, *p < 0.05, ***p < 0.001 vs MOG-35-55-stimulated cells
Table 6 shows examples of qPCR analysis of selected gene products in stimulated TMOG cells. TMOG stimulation with MOG35-55 resulted in a high upregulation of the mRNA level of Xcl1 chemokine (62.7-fold) and this level was suppressed by CBD by circa fourfold. The expression of Il12a was also increased by MOG35-55 (4.3-fold) and reduced to 2.6-fold when CBD was added to MOG35-55-stimulated TMOG cells. Stimulation of TMOG cells with MOG35-55 increased also the expression of Dusp6 by 2.4-fold that was further increased by co-incubation with CBD (by around 40 %). The table also shows several other genes which transcription was significantly enhanced by CBD alone including Btla, Lag3, and Irf4. The transcription of Btla, Lag3, and of Irf4 was further increased when MOG35-55 and CBD were added together. Interestingly, Il10 mRNA was increased only when MOG35-55 and CBD were applied together but not following MOG35-55 or CBD given alone. These observations are in agreement with the mRNA expression observed using the gene profiling method.
Table 6.
qPCR validation of various transcripts in mRNA prepared from MOG35-55-stimulated TMOG cells and the effect of CBD
| Symbol | Gene name | MOG alone [fold] $ | CBD+MOG [fold] $ | CBD alone [fold] $ | One-way ANOVA |
|---|---|---|---|---|---|
| Xcl1 | Chemokine (C Motif) Ligand 1 | 62.7 ± 0.7## | 17.9 ± 11.4** | 1.6 ± 0.1 | F(3,8) = 12.6, p < 0.01 |
| IL12a | Interleukin 12 | 4.3 ± 0.1# | 2.6 ± 0.3* | 0.8 ± 0.1 | F(3,8) = 6.0, p < 0.05 |
| Btla | B- and T-lymphocyte attenuator | 2.8 ± 0.4## | 7.1 ± 0.4*** | 6.0 ± 0.5### | F(3,8) = 60.9, p < 0.001 |
| Dusp6 | Dual specificity phosphatase 6 | 2.4 ± 0.4 | 3.3 ± 0.7# | 1.9 ± 0.3 | F(3,8) = 3.3, p = 0.07 |
| Irf4 | Interferon regulatory factor 4 | 3.4 ± 0.6## | 7.9 ± 0.5*** | 3.7 ± 0.3## | F(3,8) = 43.3, p < 0.001 |
| Lag3 & | Lymphocyte-activation gene 3 | 11.5 ± 0.3### | 25.9 ± 2.3*** | 5.8 ± 1.5* | F(3,8) = 114, p < 0.001 |
| IL10 & | Interleukin 10 | 1.5 ± 0.3 | 3.4 ± 0.8# | 1.4 ± 0.3 | F(3,11) = 3.6, p = 0.05 |
$Data are expressed as fold change vs control, not stimulated cells (equal to 1). # p < 0.05, ## p < 0.01, ### p < 0.01 vs control TMOG cells. *p < 0.05, **p < 0.01, ***p < 0.001 vs MOG-35-55-stimulated TMOG cells. &results reported previously [25]
Weighted gene co-expression network analysis (WGCNA)
We applied WGCNA analysis and clustering to complement the functional characterization of the changes in gene expression induced by CBD in activated TMOG. As mentioned in Methods, WGCNA clusters functionally related transcripts into modules in an unsupervised manner based on the expression pattern across the samples. The result of this clustering can be viewed as a dendrogram (Fig. 6a) in which each branch corresponds to a group of co-expressed genes (a module) that is designated a color and will be referred to by its color in the rest of the manuscript.
Fig. 6.
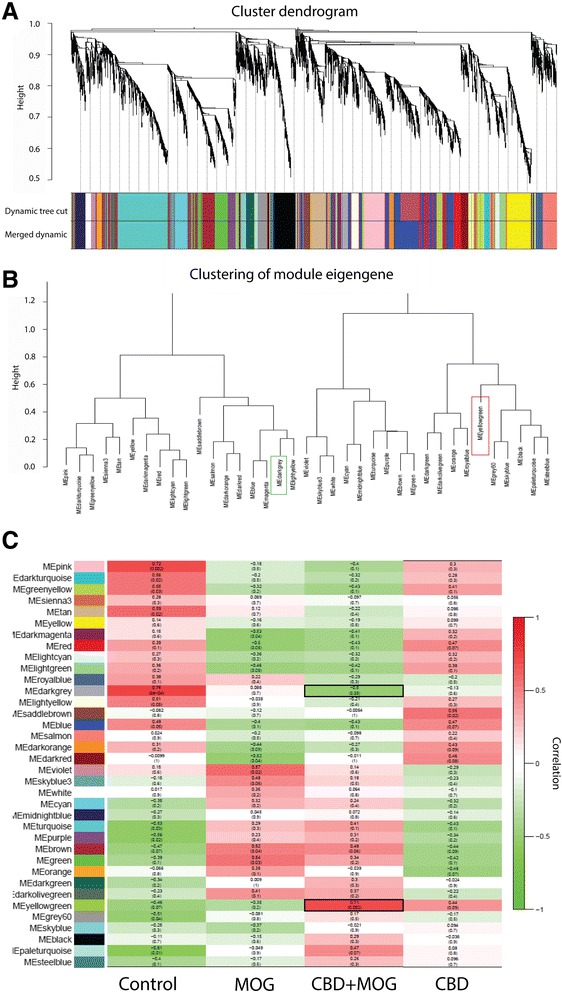
WGCNA-clustered gene modules and their correlation to CBD effect in MOG35-55-stimulated TMOG cells. WGCNA analysis identifies distinct modules of coexpressed genes. a Dendrogram showing topological overlap of genes and their relation to modules which are color-coded below. b clustering of resulting modules. Color frames indicate the main modules that are positively (red frame) and negatively (green frame) correlated to CBD effect in MOG35-55-stimulated TMOG. c module-trait relationship plots showing Spearman’s correlation between module principal components (a.k.a. module eigengenes [ME]; in color-coded rows) and the treatments. The top value in each square represents the correlation coefficient between the ME and the treatment. The bottom value in parentheses is the relevant p value. Modules highly correlated with CBD+MOG35-55 treatment are emphasized using black frames
WGCNA analysis grouped the affected transcripts into 36 discrete modules (Fig. 6b). Pearson correlation of these modules revealed modules that were the most significantly correlated to each of the treatments (Fig. 6c). The darkgrey module was shown to be the most negatively correlated (r = −0.5, p = 0.05) and the yellowgreen as the most positively correlated (r = 0.71, p = 0.002) to the combined CBD+MOG35-55 treatment. This stays in contrast to MOG35-55 effect where the darkgrey gene coexpression module was recognized as positively correlated (r = 0.088, p = 0.7) and the yellowgreen module negatively correlated (r = 0.35, p = 0.2). This further indicates that CBD reverses gene interactions’ patterns induced by MOG35-55 stimulation of TMOG. In search for functional interpretation of these associations, we annotated the CBD+MOG35-55-correlated modules with GO categories to highlight the main biological functions linked to the CBD effect in stimulated TMOG cells. The GO categories identified within the darkgrey module (containing 94 genes Additional file 1: Table S2) seemed to be associated predominantly with “antigen processing and presentation of exogenous peptide antigen via MHC class II” (GO:0019882, GO:0002504 GO:0019886 GO:0002495 GO:0002478 GO:0019884). This is probably due to the high representation within this module of genes regulating antigen processing, e.g., H2-Ab1, CD9, Tmem66, CD74, Tlr2, and Fcrla. Other GO categories identified within this the darkgrey module include “immune system processes” (GO:0002376), “defense response” (GO:0006952), “inflammatory response” (GO:0006954), “reactive nitrogen species metabolic process” (GO:2001057), “regulation of chemokine production” (GO:0032642, all within Biological Process terms), and “binding” (GO:0005488; within Molecular Function terms).
The yellowgreen module (containing 48 genes Additional file 1: Table S2) includes genes such as Ptpnm2, Slc30a1 and Btla. GO analysis qualified the yellowgreen assigned terms into “regulation of immune system processes” (GO:0002682), “response to stimulus” (GO:0048583), “signaling” (GO:0023051), “cell communication” (GO:0010646), “cell adhesion” (GO:0030155), “signal transduction” (GO:0009966; within Biological Process terms), “guanyl-nucleotide exchange factor activity” (GO:0005085; within Molecular Function terms), and “external side of plasma membrane” (GO:0009897; Cellular Component terms). In summary, the WGCNA analysis demonstrated that the main effect of CBD is to restrain the MOG35-55 activation of TMOG predominantly by targeting processes regulating antigen processing and presentation.
Discussion
In the present study, we characterized the CBD effects on the transcriptional activity of mouse MOG35-55-activated encephalitogenic TMOG cells, the latter known to induce autoimmune pathologies. We previously showed that CBD decreases IL-17 and IL-6 mRNA production and protein secretion in MOG35-55-stimulated TMOG co-cultured with APC while increasing anti-inflammatory IL-10 mRNA synthesis [24, 26]. In parallel, we showed that CBD negatively regulates STAT3 phosphorylation while enhancing STAT5 activity, main positive and negative regulators of Th17 differentiation, respectively [36], further confirming the regulatory effect of CBD on Th17 function of memory T cells [26]. The present gene profiling reveals that CBD targets a relatively small number of genes (163) within a large group of those significantly affected by MOG35-55 stimulation (reaching 1936 transcripts). Bioinformatics analysis performed on these CBD affected transcripts assembled them into various pathways and gene networks mediating the effects of CBD on the MOG35-55-activated memory T cells. In line with our previous studies, this gene profiling confirms that CBD affects various processes controlling “Th17 differentiation”, including “IL-6 signaling” (a pathway promoting memory Th17 generation) and anti-inflammatory “IL10 signaling” (known to restrain Th17 function and Th17-mediated autoimmunity [25, 37]).
CBD upregulated the transcription of 81 genes and downregulated 82 genes in MOG35-55-stimulated TMOG. For example, CBD was found to decrease the MOG35-55-upregulated mRNA levels of various proinflammatory mediators including interleukins (Il3, Il1b, and Il12a), chemokines (Xcl1 and Cxcl9) and receptors (Cxcr1, Ifngr1, Gpr83), molecules known to be involved in Th17 generation and function [38–40]. In addition, CBD downregulated elements of various signaling cascades, including members of the TNF superfamily (e.g., Tnfsf11, Tnfrsf18), molecules mediating cell adhesion (e.g., Amica1) growth, structure, and metabolism (e.g., Cdk5r1, Tubb2d, Dgat1). The effect of CBD on the TNF superfamily members was found to be very diverse. This is not surprising since the TNF superfamily consists of a large number of molecules involved in multiple cellular processes including inflammation, cell growth, cell cycle, differentiation, metabolism, and cytokine production [41]. The mRNA of the cAMP-responsive element modulator (CREM) transcription factor was also significantly suppressed by CBD in activated encephalitogenic TMOG cells. CREM activity was shown to control IL-2 and IL-17 expression during CD4 lineage commitment and distribution [42]. Indeed, human autoimmune T cells were shown to display increased expression of CREM which accounted for their increased IL-17 release and autoimmunity (e.g., in systemic lupus erythematosus and MS-like pathologies [43, 44]). Thus, the decrease in Crem mRNA found here may serve as an important contribution to the CBD-induced suppression of the Th17-like activity of the TMOG cells. This downregulation of Crem mRNA by CBD does not seem to be mediated via the cannabinoid receptors CB1 and CB2, both known to affect adenylyl cyclase activity and cAMP formation [45, 46]. This is due to the fact that CBD was shown to have negligible affinity toward these receptors [47]. Moreover, we showed previously that CBD effects on TMOG do not involve these receptors [12, 24].
Several of the MOG35-55-induced transcripts were preserved at high levels or even further upregulated in the presence of CBD. Egr2 transcription factor was one of the highest upregulated representative of this group along with a number of cytokines (Csf2, Ifng, Tnf), chemokines (Ccl3, Ccl4, Cxcl10) and regulatory glycoproteins (Sema7a). This gene expression pattern may indicate a promotion by CBD of tolerogenic conditions driving exhaustion of T cells (anergy) and thus leading to the functional deactivation of T cells [48]. Indeed, EGR2 is an acknowledged T cell anergy marker which induction turns on a genetic program driving cell cycle arrest, eventually dampening immune responses [49, 50] including IL-17 production [51, 52]. Indeed, we have recently shown that CBD induces Egr2-dependent anergy in encephalitogenic T cells and that CBD enhances the expression of several other anergy signature genes including Il10 and Lag3 [26].
MOG35-55-upregulated Ifng mRNA production and secretion were not affected by CBD in stimulated TMOG cells [24]. This result was reproduced by the current microarray analysis showing no effect of CBD on the IFNγ upstream transcription factor Tbx21 mRNA. On the other hand, the present results show that CBD has a potent enhancing effect on several IFN-dependent transcripts including Irf4, Ifit3, Rsad2/Viperin, and Mx2. Type I (IFN-α and -β) and type II (IFN-γ) interferons are known to mediate innate immune responses and inhibit the proliferation of virus-infected cells [53]. IFNs exert antiproliferative and immunomodulatory effects and IFNγ expressing cells were shown to counteract Th17 activity [54]. Alterations in IFN-stimulated gene patterns (including in Ifit, Mx2, Cxcl10) and/or in Jak/STAT activity, a main IFN-dependent pathway, cause a failure in immunological tolerance resulting in T cell-mediated autoimmune pathologies (e.g., type I diabetes) [55]. Thus, we assume that CBD-enhanced IFN-controlled pathways may contribute to CBD anti-proliferative effects in activated memory T cells, as already observed by us previously [12].
Interferon regulatory factor 4 (IRF4, a lymphocyte-specific nuclear factor) was shown to be a core promoter of other IFN-dependent genes such as Rsad2 and Ifit3 [56]. IRF4 was reported to mediate the differentiation of naive CD4+ T cells into various T helper lineages [57]. IRF4 potently synergizes with NFAT signaling to enhance the promoter activity of anti-inflammatory IL-4 cytokine [58]. Indeed, in our hands Nfatc1 mRNA, an element of the NFAT signaling axis, was upregulated by CBD.
The transcription of several genes was downregulated by MOG35-55 stimulation. This group of genes included IL-17 receptors (Il17re variant 1 and 2, Il17rc). Interestingly, CBD did not reverse this Il17r mRNA downregulation suggesting that CBD does not interfere with intrinsic negative feedback mechanisms restraining the activity of memory T cells.
Antigen-induced T cell activation occurs following peptide presentation by major histocompatibility complex class II (MHCII) molecules on APC cells. A number of molecules negatively regulate these APC/T interactions [59]. Our study shows that CBD is a powerful inducer of negative regulators of APC/T interactions as CBD increased the mRNA levels of: B and T lymphocyte attenuator (Btla/Cd272); lymphocyte-activation gene 3 (Lag3/Cd223); cluster of differentiation 69 (Cd69) and of T cell receptor associated transmembrane adaptor 1/T-cell receptor interacting molecule (Trat1/TRIM). These molecules are members of a broad, structurally related group of immunoglobulin receptors that were found to serve as markers of various inducible T cell regulatory phenotypes involved in Th17 suppression and amelioration of MS-like pathologies [59–62]. For example, LAG3 is a CD4 receptor homologue that by interfering with MHCII on APC upon antigen exposure inhibits the function and expansion of memory T cells [63] decreasing IL-17 and increasing IL-10 secretions, this way preventing autoimmunity in mice [64, 65]. Mice lacking the CD69 transmembrane receptor develop aggravated forms of autoimmune pathologies including arthritis, contact dermatitis, allergic asthma, and autoimmune myocarditis [66]. Indeed, CD69-deficient T cells demonstrate increased differentiation into Th17 cells, higher IL-17 release as well as Jak3/Stat5 and RORγt pathways’ increased activities [67] (Martin 2010). Similarly, BTLA ligation with its ligand, herpes virus entry mediator (HVEM/TNFRSF14), was shown to attenuate T cell proliferation [68, 69] and to promote anergy [70]. Functional BTLA/HVEM pathway maintains immune tolerance and prevents autoimmunity (for review see [71] and BTLA-deficiency in mice leads to T cell-mediated rheumatoid arthritis, lymphocytic infiltration, autoimmune hepatitis-like diseases, and EAE [72–74]. CTLA-4 (CD152) is an inhibitory receptor which in its inactive state is located intracellularly and upon demand is trafficked by TRAT1/TRIM protein (that is upregulated by CBD) to the membrane [75]. CTLA-4 negatively competes on T cells with CD28, a co-stimulatory T cell molecule, in binding to the co-activators CD80 and CD86 on APC cells [76, 77]. TRAT1/TRIM-dependent increase in CTLA4 expression led to suppression of encephalitogenic T cells expansion and of EAE development [78]. In agreement with this, impaired CTLA-4 expression on T cells was found to correlate with increased IL-17 release and more severe EAE [79]. Moreover, CTLA-4-deficient mice show an autoimmune phenotype with T cell-mediated organ destruction [80–82]. Thus, CBD upregulation of these powerful inhibitors of APC/T communication should exert robust immunoregulatory effect in MOG35-55 activated TMOG resulting in their suppressed activity. Indeed, complement WGCNA analysis confirmed the inhibitory role of CBD in processes related to antigen presentation and processing and APC/TMOG interactions. Moreover, these observations are in agreement with our previous report showing that CBD decreases the activation of B cells (the main APC in the periphery) incubated with TMOG as well as MHCII expression on these cells [26].
Reactive oxygen species generated primarily by macrophages and damaged neurons, increase immune response and T cell activation, propagate demyelination and neurodegeneration [83]. The present gene profiling links the CBD effects to nuclear factor-erythroid 2-related factor 2 (Nfe2l2/Nrf2)-driven antioxidant genetic program, known to exert immunoregulatory effects [84, 85]. Our results show that CBD enhances the expression of Nrf2 target genes including Heme oxygenase-1 (Hmox-1 encoding HO-1 protein) and metallothioneins (Mt1 and Mt2). Indeed, Mt(−/−) mice show increased macrophage and T-lymphocyte infiltration into the CNS as well as increased cytokine release, oxidative stress and axon damage as compared to their wildtype EAE littermates [86]. HO-1 was shown to diminish inflammatory reactions, e.g., via induction of the anti-inflammatory IL10 cytokine and modulation of Jak/STAT pathway [87]. Indeed, pharmacological or genetic induction of Nrf2-dependent anti-oxidant genes (i.e., Hmox1, Mt1, and Mt2) in EAE mice led to reduction in IL-17 levels and decreased EAE severity [88]. Moreover, Hmox1−/− C57BL/6 mice develop enhanced EAE-like paralysis and CNS demyelination while HO-1 restoration diminished EAE progression and relapses [89]. Interestingly, this protective effect of HO-1 has been associated with inhibition of MHCII expression by APCs via mechanisms involving carbon monoxide, an antioxidant product of HO-1 activity. Thus, CBD induction of Nfe2l2/Nrf2-dependent antioxidant genes may be one of the key mechanisms driving the anti-inflammatory effects of this cannabinoid in TMOG cells and EAE. Similar CBD regulation of anti-oxidant genes was reported by us in quiescent and LPS-stimulated microglial cells [30, 90]. These findings suggest that Nrf2-dependent immunoregulation may be an important feature of this cannabinoid contributing to its anti-inflammatory effects. However, we cannot rule out that CBD is also affecting several other pathways, e.g., receptors for A2A as well as dimers of CB2/5HT1A [91]. In addition, CBD was shown to increase the endocannabinoid levels, known to modulate immune functions [92], thus it may indirectly exert immunoregulatory actions on T cells through CB2 receptors, previously suggested to modulate T cell-mediated autoimmunity [93].
Conclusions
In summary, gene expression profiling followed by bioinformatics analyses demonstrate that CBD exerts its immunoregulatory effects in activated memory TMOG cells via several mechanisms: (a) by suppressing proinflammatory transcription and inhibiting pathways that favor Th17 differentiation and function, (b) by promoting a transcriptional set up leading to T cell exhaustion/tolerance, (c) by enhancing IFN-dependent anti-proliferative program, (d) by potentiating intrinsic T cell co-inhibitory mechanisms hampering APC/T communication and efficient antigen presentation, and (e) by inducing antioxidant milieu resolving inflammation.
Abbreviations
APC antigen-presenting cells, CBD cannabidiol, EAE experimental autoimmune encephalomyelitis, GO gene ontology, IFN interferon, IL interleukin, IPA ingenuity pathway analysis, MOG35-55 myelin oligodendrocyte glycoprotein 35-55, TMOG MOG35-55-specific T cells, WGCNA weighted gene co-expression network analysis
Acknowledgements
The TMOG cell line was kindly provided by the laboratory of Professor Avraham Ben-Nun from the Immunology Department at the Weizmann Institute of Science, Rehovot, Israel.
Funding
This work was supported by the Dr Miriam and Sheldon G. Adelson Medical Research Foundation. AJ was supported by the Israeli Ministry for Absorption in Science. We acknowledge the support of the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691).
Availability of data and materials
There is no data, software, databases, application/tool available apart from the reported in the present study. Data supporting the conclusions are presented in the manuscript.
Authors’ contributions
EK designed and performed the experiments, carried out the IPA analysis, and wrote the article. FG and GC carried out microarray experiments and analyzed the data. AJ performed the mRNA extraction and the qPCR analysis of gene expression. NK and EK provided the know-how in TMOG cell lines establishing and culturing. EK, AJ, FG, GC, NK, and ZV contributed to discussions of article content, and to reviewing and editing the manuscript before submission.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All mice used in these studies were maintained according to the guidelines of the Institutional Animal Care and Use Committee (IACUC). Animal experiments were approved and performed by the Weizmann Institute of Science and the Tel Aviv University IACUCs.
Additional files
List of primers used for qPCR validation of mRNA levels of selected genes in purified TMOG cells. Table S2. WGCNA-assigned gene content of yellowgreen and darkgrey modules, classified as being the most correlated with CBD treatment in MOG35-55-stimulated TMOG. (DOC 233 kb)
References
- 1.Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–11. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- 2.Nagarkatti P, Pandey R, Rieder SA, Hegde VL, Nagarkatti M. Cannabinoids as novel anti-inflammatory drugs. Future Med Chem. 2009;1:1333–1349. doi: 10.4155/fmc.09.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eisenstein TK, Meissler JJ. Effects of Cannabinoids on T-cell Function and Resistance to Infection. J Neuroimmune Pharmacol. 2015;10:204–16. doi: 10.1007/s11481-015-9603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernández-Ruiz J, Sagredo O, Pazos MR, García C, Pertwee R, Mechoulam R, Martinez-Orgado J. Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid? Br J Clin Pharmacol. 2013;75(2):323–33. doi: 10.1111/j.1365-2125.2012.04341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas BF, Gilliam AF, Burch DF, Roche MJ, Seltzman HH. Comparative receptor binding analyses of cannabinoid agonists and antagonists. J Pharmacol Exp Ther. 1998;285:285–92. [PubMed] [Google Scholar]
- 6.Zuardi AW. Cannabidiol: from an inactive cannabinoid to a drug with wide spectrum of action. Rev Bras Psiquiatr. 2008;30:271–80. doi: 10.1590/S1516-44462008000300015. [DOI] [PubMed] [Google Scholar]
- 7.Malfait AM, Gallily R, Sumariwalla PF, Malik AS, Andreakos E, Mechoulam R, Feldmann M. Proc Natl Acad Sci U S A. 2009;97:9561–9566. doi: 10.1073/pnas.160105897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss L, Zeira M, Reich S, Har-Noy M, Mechoulam R, Slavin S, Gallily R. Cannabidiol lowers incidence of diabetes in non-obese diabetic mice. Autoimmunity. 2006;39:143–151. doi: 10.1080/08916930500356674. [DOI] [PubMed] [Google Scholar]
- 9.Hegde VL, Nagarkatti PS, Nagarkatti M. Role of myeloid-derived suppressor cells in amelioration of experimental autoimmune hepatitis following activation of TRPV1 receptors by cannabidiol. PLoS One. 2011;6:e18281. doi: 10.1371/journal.pone.0018281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Zwam M, Huizinga R, Melief MJ, Wierenga-Wolf AF, van Meurs M, Voerman JS, Biber KP, Boddeke HW, Höpken UE, Meisel C, Meisel A, Bechmann I, Hintzen RQ, 't Hart BA, Amor S, Laman JD, Boven LA. Brain antigens in functionally distinct antigen-presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med (Berl) 2009;87:273–286. doi: 10.1007/s00109-008-0421-4. [DOI] [PubMed] [Google Scholar]
- 11.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 12.Kozela E, Lev N, Kaushansky N, Eilam R, Rimmerman N, Levy R, Vogel Z. Cannabidiol inhibits pathogenic T cells, decreases spinal microglial activation and ameliorates multiple sclerosis-like disease in C57BL/6 mice. Br J Pharmacol. 2011;163:1507–19. doi: 10.1111/j.1476-5381.2011.01379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mecha M, Feliú A, Iñigo PM, Mestre L, Carrillo-Salinas FJ, Guaza C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: a role for A2A receptors. Neurobiol Dis. 2013;59:141–150. doi: 10.1016/j.nbd.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 14.Duchi S, Ovadia H, Touitou E. Nasal administration of drugs as a new non-invasive strategy for efficient treatment of multiple sclerosis. J Neuroimmunol. 2013;258:32–40. doi: 10.1016/j.jneuroim.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Rahimi A, Faizi M, Talebi F, Noorbakhsh F, Kahrizi F, Naderi N. Interaction between the protective effects of cannabidiol and palmitoylethanolamide in experimental model of multiple sclerosis in C57BL/6 mice. Neuroscience. 2015;290:279–87. doi: 10.1016/j.neuroscience.2015.01.030. [DOI] [PubMed] [Google Scholar]
- 16.Podojil JR. Miller SD (2009) Molecular mechanisms of T-cell receptor and costimulatory molecule ligation/blockade in autoimmune disease therapy. Immunol Rev. 2009;229:337–55. doi: 10.1111/j.1600-065X.2009.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 18.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’Schea JJ. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–81. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Yura M, Takahashi I, Serada M, Koshio T, Nakagami K, Yuki Y, Kiyono H. Role of MOG-stimulated Th1 type “light up” (GFP+) CD4+ T cells for the development of experimental autoimmune encephalomyelitis (EAE) J Autoimmun. 2001;17:17–25. doi: 10.1006/jaut.2001.0520. [DOI] [PubMed] [Google Scholar]
- 21.Shevach EM. Animal models for autoimmune and inflammatory diseases. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current protocols in immunology. New York: Wiley; 2011. pp. 15.0.1–15.0.5. [Google Scholar]
- 22.Okuda Y, Okuda M, Apatoff BR, Posnett DN. The activation of memory CD4(+) T cells and CD8(+) T cells in patients with multiple sclerosis. J Neurol Sci. 2005;235:11–17. doi: 10.1016/j.jns.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Aharoni R. Immunomodulation neuroprotection and remyelination—the fundamental therapeutic effects of glatiramer acetate: a critical review. J Autoimmun. 2014;54:81–92. doi: 10.1016/j.jaut.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Kozela E, Juknat A, Kaushansky N, Rimmerman N, Ben-Nun A, Vogel Z. Cannabinoids decrease the th17 inflammatory autoimmune phenotype. J Neuroimmune Pharmacol. 2013;8:1265–76. doi: 10.1007/s11481-013-9493-1. [DOI] [PubMed] [Google Scholar]
- 25.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 26.Kozela E, Juknat A, Kaushansky N, Ben-Nun A, Coppola G, Vogel Z. Cannabidiol, a non-psychoactive cannabinoid, leads to EGR2-dependent anergy in activated encephalitogenic T cells. J Neuroinflammation. 2015;12:52. doi: 10.1186/s12974-015-0273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardoll DM, Drake CG. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 28.Kaushansky N, Zhong MC, Kerlero de Rosbo N, Hoeftberger R, Lassmann H, Ben-Nun A. Epitope specificity of autoreactive T and B cells associated with experimental autoimmune encephalomyelitis and optic neuritis induced by oligodendrocyte-specific protein in SJL/J mice. J Immunol. 2006;177:7364–76. doi: 10.4049/jimmunol.177.10.7364. [DOI] [PubMed] [Google Scholar]
- 29.Kozela E, Juknat A, Gao F, Coppola G, Kaushansky N, Vogel Z. Insights into gene expression of activated pathogenic autoimmune T cells—studies in experimental multiple sclerosis-like model. Immunome Res. 2016;12:108. doi:10.4172/1745-7580.10000108.
- 30.Juknat A, Pietr M, Kozela E, Rimmerman N, Levy R, Gao F, Coppola G, Geschwind D, Vogel Z. Microarray and pathway analysis reveal distinct mechanisms underlying cannabinoid-mediated modulation of LPS-induced activation of BV-2 microglial cells. PLoS One. 2013;8:e61462. doi: 10.1371/journal.pone.0061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coppola G. Designing, performing, and interpreting a microarray-based gene expression study. Methods Mol Biol. 2011;793:417–39. doi: 10.1007/978-1-61779-328-8_28. [DOI] [PubMed] [Google Scholar]
- 33.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 34.Park CC, Gale GD, de Jong S, Ghazalpour A, Bennett BJ, Farber CR, Langfelder P, Lin A, Khan AH, Eskin E, Horvath S, Lusis AJ, Ophoff RA, Smith DJ. Gene networks associated with conditional fear in mice identified using a systems genetics approach. BMC Syst Biol. 2011;5:43. doi: 10.1186/1752-0509-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fogel BL, Cho E, Wahnich A, Gao F, Becherel OJ, Wang X, Fike F, Chen L, Criscuolo C, De Michele G, Filla A, Collins A, Hahn AF, Gatti RA, Konopka G, Perlman S, Lavin MF, Geschwind DH, Coppola G. Mutation of senataxin alters disease-specific transcriptional networks in patients with ataxia with oculomotor apraxia type 2. Hum Mol Genet. 2014;23(18):4758–69. doi: 10.1093/hmg/ddu190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, O’Schea JJ, Laurence A. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–54. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujio K, Okamura T, Yamamoto K. The Family of IL-10-secreting CD4+ T cells. Adv Immunol. 2010;105:99–130. doi: 10.1016/S0065-2776(10)05004-2. [DOI] [PubMed] [Google Scholar]
- 38.Chavany C, Vicario-Abejón C, Miller G, Jendoubi M. Transgenic mice for interleukin 3 develop motor neuron degeneration associated with autoimmune reaction against spinal cord motor neurons. Proc Natl Acad Sci U S A. 1998;95(19):11354–9. doi: 10.1073/pnas.95.19.11354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–41. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 40.Mony JT, Khorooshi R1, Owens T. Chemokine receptor expression by inflammatory T cells in EAE. Front Cell Neurosci. 2014;8:187. doi: 10.3389/fncel.2014.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Croft M, Duan W, Choi H, Eun SY, Madireddi S, Mehta A. TNF superfamily in inflammatory disease: translating basic insights. Trends Immunol. 2012;33:144–52. doi: 10.1016/j.it.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hedrich CM, Crispin JC, Rauen T, Ioannidis C, Apostolidis SA, Lo MS, Kyttaris VC, Tsokos GC. cAMP response element modulator α controls IL2 and IL17A expression during CD4 lineage commitment and subset distribution in lupus. Proc Natl Acad Sci U S A. 2012;109:16606–11. doi: 10.1073/pnas.1210129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rauen T, Hedrich CM, Juang YT, Tenbrock K, Tsokos GC. cAMP-responsive element modulator (CREM)α protein induces interleukin 17A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J Biol Chem. 2011;286:43437–46. doi: 10.1074/jbc.M111.299313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, Rauen T, Crispín JC, Tsokos GC. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124:2234–45. doi: 10.1172/JCI73411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogel Z, Barg J, Levy R, Saya D, Heldman E, Mechoulam R. Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J Neurochem. 1993;61:352–355. doi: 10.1111/j.1471-4159.1993.tb03576.x. [DOI] [PubMed] [Google Scholar]
- 46.Rhee MH, Bayewitch M, Avidor-Reiss T, Levy R, Vogel Z. Cannabinoid receptor activation differentially regulates the various adenylyl cyclase isozymes. J Neurochem. 1998;71:1525–1534. doi: 10.1046/j.1471-4159.1998.71041525.x. [DOI] [PubMed] [Google Scholar]
- 47.Showalter VM, Compton DR, Martin BR, Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther. 1996;278:989–999. [PubMed] [Google Scholar]
- 48.Anderson PO, Manzo BA, Sundstedt A, Minaee S, Symonds A, Khalid S, Rodriguez-Cabezas ME, Nicolson K, Li S, Wraith DC, Wang P. Persistent antigenic stimulation alters the transcription program in T cells, resulting in antigen-specific tolerance. Eur J Immunol. 2006;36:1374–85. doi: 10.1002/eji.200635883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harris JE, Bishop KD, Phillips NE, Mordes JP, Greiner DL, Rossini AA, Czech MP. Early growth response gene-2, a zinc-finger transcription factor, is required for full induction of clonal anergy in CD4+ T cells. J Immunol. 2004;173:7331–7338. doi: 10.4049/jimmunol.173.12.7331. [DOI] [PubMed] [Google Scholar]
- 50.Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, Blackford A, Horton MR, Drake C, Schwartz RH, Powell JD. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol. 2005;6:472–480. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- 51.Zhu B, Symonds AL, Martin JE, Kioussis D, Wraith DC, Li S, Wang P. Early growth response gene 2 (Egr-2) controls the self-tolerance of T cells and prevents the development of lupuslike autoimmune disease. J Exp Med. 2008;205:2295–307. doi: 10.1084/jem.20080187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miao T, Raymond M, Bhullar P, Ghaffari E, Symonds AL, Meier UC, Giovannoni G, Li S, Wang P. Early growth response gene-2 controls IL-17 expression and Th17 differentiation by negatively regulating Batf. J Immunol. 2013;190:58–65. doi: 10.4049/jimmunol.1200868. [DOI] [PubMed] [Google Scholar]
- 53.Zahoor MA, Xue G, Sato H, Murakami T, Takeshima SN, Aida Y. HIV-1 Vpr induces interferon-stimulated genes in human monocyte-derived macrophages. PLoS One. 2014;9:e106418. doi: 10.1371/journal.pone.0106418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 55.Panarina M, Kisand K, Alnek K, Heilman K, Peet A, Uibo R. Interferon and interferon-inducible gene activation in patients with type 1 diabetes. Scand J Immunol. 2014;80:283–92. doi: 10.1111/sji.12204. [DOI] [PubMed] [Google Scholar]
- 56.Forero A, Moore PS, Sarkar SN. Role of IRF4 in IFN-stimulated gene induction and maintenance of Kaposi sarcoma-associated herpesvirus latency in primary effusion lymphoma cells. J Immunol. 2013;191:1476–1485. doi: 10.4049/jimmunol.1202514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huber M, Lohoff M. IRF4 at the crossroads of effector T-cell fate decision. Eur J Immunol. 2014;44:1886–1895. doi: 10.1002/eji.201344279. [DOI] [PubMed] [Google Scholar]
- 58.Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med. 2002;195:1003–1012. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–42. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan B, Zhao L, Fu F, Liu Y, Lin C, Wu X, Shen H, Yang Z. A novel nanoparticle containing MOG peptide with BTLA induces T cell tolerance and prevents multiple sclerosis. Mol Immunol. 2014;57:93–99. doi: 10.1016/j.molimm.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 61.Baitsch L, Legat A, Barba L, Fuertes Marraco SA, Rivals JP, Baumgaertner P, Christiansen-Jucht C, Bouzourene H, Rimoldi D, Pircher H, Rufer N, Matter M, Michielin O, Speiser DE. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS One. 2012;7:e30852. doi: 10.1371/journal.pone.0030852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okazaki T, Okazaki IM, Wang J, Sugiura D, Nakaki F, Yoshida T, Kato Y, Fagarasan S, Muramatsu M, Eto T, Hioki K, Honjo T. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med. 2011;208:395–407. doi: 10.1084/jem.20100466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol. 2003;33:970–979. doi: 10.1002/eji.200323382. [DOI] [PubMed] [Google Scholar]
- 64.Bettini M, Szymczak-Workman AL, Forbes K, Castellaw AH, Selby M, Pan X, Drake CG, Korman AJ, Vignali DA. Cutting edge: accelerated autoimmune diabetes in the absence of LAG-3. J Immunol. 2011;187(7):3493–98. doi: 10.4049/jimmunol.1100714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jha V, Workman CJ, McGaha TL, Li L, Vas J, Vignali DA, Monestier M. Lymphocyte activation gene-3 (LAG-3) negatively regulates environmentally-induced autoimmunity. PLoS One. 2014;9:e104484. doi: 10.1371/journal.pone.0104484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martín P, Sánchez-Madrid F. CD69: an unexpected regulator of TH17 cell-driven inflammatory responses. Sci Signal. 2011;4:pe14. doi: 10.1126/scisignal.2001825. [DOI] [PubMed] [Google Scholar]
- 67.Martín P, Gómez M, Lamana A, Cruz-Adalia A, Ramírez-Huesca M, Ursa MA, Yanez-Mo M, Sanchez-Madrid F. CD69 association with Jak3/Stat5 proteins regulates Th17 cell differentiation. Mol Cell Biol. 2010;30:4877–4889. doi: 10.1128/MCB.00456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, Allison JP, Murphy KM. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–9. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 69.Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 70.Hurchla MA, Sedy JR, Gavrieli M, Drake CG, Murphy TL, Murphy KM. B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly Induced in anergic CD4+ T cells. J Immunol. 2005;174:3377–3385. doi: 10.4049/jimmunol.174.6.3377. [DOI] [PubMed] [Google Scholar]
- 71.Wu YL, Liang J, Zhang W, Tanaka Y, Sugiyama H. Immunotherapies: the blockade of inhibitory signals. Int J Biol Sci. 2012;8:1420–1430. doi: 10.7150/ijbs.5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin SC, Kuo CC, Chan CH. Association of a BTLA gene polymorphism with the risk of rheumatoid arthritis. J Biomed Sci. 2006;13:853–860. doi: 10.1007/s11373-006-9113-7. [DOI] [PubMed] [Google Scholar]
- 73.Oya Y, Watanabe N, Owada T, Oki M, Hirose K, Suto A, Kagami S, Nakajima H, Kishimoto T, Iwamoto I, Murphy TL, Murphy KM, Saito Y. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheum. 2008;58:2498–510. doi: 10.1002/art.23674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Subudhi SK, Anders RA, Lo J, Sun Y, Blink S, Wang Y, Wang J, Liu X, Mink K, Degrandi D, Pfeffer K, Fu YX. The role of herpesvirus entry mediator as a negative regulator of T cell-mediated responses. J Clin Invest. 2005;115:711–717. doi: 10.1172/JCI200522982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Valk E, Leung R, Kang H, Kaneko K, Rudd CE, Schneider H. T cell receptor-interacting molecule acts as a chaperone to modulate surface expression of the CTLA-4 coreceptor. Immunity. 2006;25:807–821. doi: 10.1016/j.immuni.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 76.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-X. [DOI] [PubMed] [Google Scholar]
- 78.Hurwitz AA, Sullivan TJ, Sobel RA, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) limits the expansion of encephalitogenic T cells in experimental autoimmune encephalomyelitis (EAE)-resistant BALB/c mice. Proc Natl Acad Sci U S A. 2002;99:3013–3017. doi: 10.1073/pnas.042684699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vogel I, Kasran A, Cremer J, Kim YJ, Boon L, Van Gool SW, Ceuppens JL. CD28/CTLA-4/B7 costimulatory pathway blockade affects regulatory T-cell function in autoimmunity. Eur J Immunol. 2015;45:1832–1841. doi: 10.1002/eji.201445190. [DOI] [PubMed] [Google Scholar]
- 80.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 81.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 82.Romo-Tena J, Gómez-Martín D, Alcocer-Varela J. CTLA-4 and autoimmunity: new insights into the dual regulator of tolerance. Autoimmun Rev. 2013;12:1171–1176. doi: 10.1016/j.autrev.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 83.Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. 2004;251:261–8. doi: 10.1007/s00415-004-0348-9. [DOI] [PubMed] [Google Scholar]
- 84.Chen XL, Kunsch C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: a new therapeutic approach for the treatment of inflammatory diseases. Curr Pharm Des. 2004;10(8):879–91. doi: 10.2174/1381612043452901. [DOI] [PubMed] [Google Scholar]
- 85.Sandberg M, Patil J, D'Angelo B, Weber SG, Mallard C. NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology. 2014;79:298–306. doi: 10.1016/j.neuropharm.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Penkowa M, Espejo C, Martínez-Cáceres EM, Montalban X, Hidalgo J. Increased demyelination and axonal damage in metallothionein I+II-deficient mice during experimental autoimmune encephalomyelitis. Cell Mol Life Sci. 2003;60:185–197. doi: 10.1007/s000180300013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys. 2014;564:83–88. doi: 10.1016/j.abb.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 88.Pareek TK, Belkadi A, Kesavapany S, Zaremba A, Loh SL, Bai L, Cohen ML, Meyer C, Liby KT, Miller RH, Sporn MB, Letterio JJ. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci Rep. 2011;1:201. doi: 10.1038/srep00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chora AA, Fontoura P, Cunha A, Pais TF, Cardoso S, Ho PP, Lee LY, Sobel RA, Steinman L, Soares MP. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest. 2007;117:438–47. doi: 10.1172/JCI28844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Juknat A, Pietr M, Kozela E, Rimmerman N, Levy R, Coppola G, Geschwind D, Vogel Z. Differential transcriptional profiles mediated by exposure to the cannabinoids cannabidiol and Δ9-tetrahydrocannabinol in BV-2 microglial cells. Br J Pharmacol. 2012;165:2512–2528. doi: 10.1111/j.1476-5381.2011.01461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Burstein S. Cannabidiol (CBD) and its analogs: a review of their effects on inflammation. Bioorg Med Chem. 2015;23:1377–1385. doi: 10.1016/j.bmc.2015.01.059. [DOI] [PubMed] [Google Scholar]
- 92.Cabral GA, Rogers TJ, Lichtman AH. Turning over a new leaf: cannabinoid and endocannabinoid modulation of immune function. J Neuroimmune Pharmacol. 2015;10(2):193–203. doi: 10.1007/s11481-015-9615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL, Shriver LP, Ledent C, Cheng X, Carrier EJ, Mann MK, Giovannoni G, Pertwee RG, Yamamura T, Buckley NE, Hillard CJ, Lutz B, Baker D, Dittel BN. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat Med. 2007;13:492–7. doi: 10.1038/nm1561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
There is no data, software, databases, application/tool available apart from the reported in the present study. Data supporting the conclusions are presented in the manuscript.