

Abstract
Objective
OPA1 mutations cause protein haploinsufficiency leading to dominant optic atrophy (DOA), an incurable retinopathy with variable severity. Up to 20% of patients also develop extraocular neurological complications. The mechanisms that cause this optic atrophy or its syndromic forms are still unknown. After identifying oxidative stress in a mouse model of the pathology, we sought to determine the consequences of OPA1 dysfunction on redox homeostasis.
Methods
Mitochondrial respiration, reactive oxygen species levels, antioxidant defenses, and cell death were characterized by biochemical and in situ approaches in both in vitro and in vivo models of OPA1 haploinsufficiency.
Results
A decrease in aconitase activity suggesting an increase in reactive oxygene species and an induction of antioxidant defenses was observed in cortices of a murine model as well as in OPA1 downregulated cortical neurons. This increase is associated with a decline in mitochondrial respiration in vitro. Upon exogenous oxidative stress, OPA1‐depleted neurons did not further exhibit upregulated antioxidant defenses but were more sensitive to cell death. Finally, low levels of antioxidant enzymes were found in fibroblasts from patients supporting their role as modifier factors.
Interpretation
Our study suggests that the pro‐oxidative state induced by OPA1 loss may contribute to DOA pathogenesis and that differences in antioxidant defenses can explain the variability in expressivity. Furthermore, antioxidants may be used as therapy as they could prevent or delay DOA symptoms in patients.
Introduction
Dominant optic atrophy (DOA) is characterized by moderate to severe loss of visual acuity with insidious onset in early childhood. DOA penetrance may be as low as 40% and prevalence is 1:50,000 worldwide.1 Up to now, there is no effective treatment for this complex pathology. The majority of DOA patients (~75%) harbor mutation in the OPA1 gene coding for a mitochondrial GTPase.2 There are 280 different OPA1 mutations (http:/mitodyn.org), the majority of which result in premature termination ensuing OPA1 haploinsufficiency.1 There is marked inter‐ and intrafamilial variability in the rate of disease progression, and recent studies show a severe multisystemic disorder associated with some OPA1 mutations (DOA+ syndrome).3, 4, 5, 6 These patients present additional neurological complications, such as ataxia, sensorineural deafness, sensory motor neuropathy as well as chronic progressive external ophtalmoplegia and myopathy. Moreover, a recent case described two DOA families which also presented parkinsonism.7 Altogether these recent findings are of major pathophysiological importance, and highlight the widespread deleterious consequences of OPA1 mutations, not only for retinal ganglionic cells (RGCs) but also for other neuronal populations.6, 8, 9
The OPA1 gene encodes a mitochondrial inner membrane protein facing the intermembrane space10 with various functions including inner membrane fusion, cristae structuration, mitochondrial DNA maintenance, mitochondrial energetic modulation, and protection from apoptosis.10 Patients' skin fibroblasts, muscles, or lymphoblasts revealed impairments in mitochondrial morphology,11, 12, 13 respiration, and energetics, and also displayed a loss of mitochondrial DNA integrity and an increase in sensitivity to apoptosis.8, 14, 15 However, it is still not clear if DOA patients do or do not develop energetic defects (see for review refs. 10, 16). Mitochondria from DOA or DOA+ mice models present an increase in mitochondrial fragmentation, cristae disorganization, mitophagy, and cytochrome c oxidase deficiency. In Caenorhabditis elegans, mutations of the OPA1 gene ortholog, eat‐3, cause mitochondrial fragmentation and hypersensitivity to oxidative stress, but do not lead to cell death.17 Heterozygous mutations of OPA1 in Drosophila melanogaster result in a shortened life span, increased reactive oxygen species (ROS) production, sensitivity to oxidative stress, and defects in the activity of respiratory chain complexes.18, 19 These two DOA invertebrate models link the critical generation of ROS with OPA1 dysfunction. We sought to investigate if ROS production promotes DOA pathogenesis in mammals using various complementary models such as (1) a DOA mouse model,20 (2) a canonical ex vivo neuronal model of OPA1 haploinsufficiency,21 and (3) fibroblasts from DOA and DOA+ patients.22
Our results show that OPA1 haploinsufficiency lead to a decrease in mitochondrial respiration, accompanied by an increase in mitochondrial ROS production. This associated increase is linked to a decrease in aconitase activity and an increase in antioxidant defenses, in both primary cultured neurons and in mice. ROS production is countered by the activation of the nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) transcription factor and its downstream targets. Loss of functional OPA1 imbalanced the redox state, because additional exogenous oxidative stress challenged both the antioxidant response and the viability of OPA1‐depleted neurons. In addition, we found altered expression of antioxidant genes at the protein level in fibroblasts from patients with DOA. On the basis of these new findings, we propose a model in which reduced functional OPA1 leads to an imbalance in cellular redox state, which sensitizes cells to exogenous pro‐oxidative stresses. We believe this phenomenon is a molecular mechanism underlying DOA pathogenesis.
Materials and Methods
Cell culture
Wistar rats
All animals (n = 45 pregnant rats, 350 embryos) in this study were ethically maintained and used as ethical laws of CNRS (Centre National de la Recherche Scientifique) and FRBT (Fédération de Recherche en Biologie de Toulouse). The animal facility called Genotoul Anexpo is maintained in accordance to F.E.L.A.S.A organism (Federation of European Laboratory Animal Science Associations). Animals were housed in groups of six and kept in a specific pathogen‐free and temperature‐controlled facility on a 12‐h light/dark cycle. Pregnant Wistar rats are delivered by Janvier company 48 h before being sacrificed and were handled in a calm room. Day 17 embryos were removed from pregnant Wistar rats (Janvier, France) under intraperitoneal pentobarbitol (Sigma‐Aldrich, UK) anesthesia. The rats were then immediately sacrificed. Cortices were dissected, enzymatically dissociated with papain (10 U/mL, Sigma‐Aldrich, UK), then exposed for 5 min in a solution that inactivated papain: DNAse I (Invitrogen, Thermo Fisher Scientific, Waltham, USA) and B27 (Gibco, Thermo Fisher Scientific, Waltham, USA), diluted in Phosphate Buffer Saline (PBS) 1X with d‐glucose (33 mmol/L, Sigma‐Aldrich, UK). Cells were dissociated by trituration and filtered through a membrane (70 μm, BD Falcon). Cells were then purified through a BSA solution (8%, Sigma‐Aldrich, UK) diluted in Neurobasal A‐25 (Invitrogen, Thermo Fisher Scientific, Waltham, USA). Dishes, with or without glass coverslips, were coated with poly‐d‐lysine (0.1 mg/mL, Sigma‐Aldrich, UK) 24 h prior to culturing. For each experiment, cortices from 8 to 12 embryos per rat are mixed. Experiments were reproduced three to eight times. Cultures were grown in Neurobasal® (Eurobio) supplemented with B27 (Invitrogen, Thermo Fisher Scientific, Waltham, USA), 2 mmol/L glutamine, 0.1% penicillin and streptomycin (Gibco, Thermo Fisher Scientific, Waltham, USA), 250 U/mL amphotericin (Invitrogen, Thermo Fisher Scientific, Waltham, USA), and 1 mmol/L lactic acid (Sigma‐Aldrich, UK) at a density of 6 × 105 cells per cm2.
Cortical neurons (1 × 106) were electroporated using the Rat Neuron Nucleofector Kit (Amaxa, Lonza) according to the manufacturers' optimized protocol with 3 μg of control luciferase‐targeting (siCtrl, D‐001210‐02, Dharmacon GE, UK) or OPA1‐targeting (siOPA1, target sequence GAUUGUGCCUGACUUUAUA, Dharmacon GE, UK) small interfering RNA (Dharmacon GE, UK).
Patients' fibroblasts
Fibroblasts obtained by skin biopsy from consenting DOA patients and healthy volunteers were cultured in Dulbecco's modified Eagle's medium 4.5 g/L glucose (DMEM, Invitrogen, Thermo Fisher Scientific, Waltham, USA), supplemented with 10% Fetal Bovine Serum (FBS). penicillin (100 U/mL) and streptomycin (100 mg/mL), and maintained for up to 20 passages. The use of patient fibroblasts for research purpose in neurogenetics was approved by the “Comité de Protection des Personnes Ouest II – Angers” (No. CB 2014/02).
Opa1 +/− mice
Mice were kept in a 12‐h light/12‐h dark cycle with food and water available ad libitum in full‐barrier facilities free of specific pathogens. Mouse breeding and all experimental procedures were performed according to ethical laws of CNRS (Centre National de la Recherche Scientifique) and Fédération de Recherche en Biologie de Toulouse (FRBT). The animal facility called Genotoul Anexpo is maintained in accordance to F.E.L.A.S.A organism (Federation of European Laboratory Animal Science Associations). Two sanitary controls are effectuated per year following the set point CO66. The B6;C3‐Opa1 329‐355del mouse strain (abbreviated Opa1 +/− mouse) has been previously described by Alavi and colleagues.13, 20, 23 Wild‐type (Opa1 +/+) littermates were systematically used as control. All examined mice were sacrificed either at 4 or 10 months old. After sacrifice, adult brains cortices were isolated and separated from hippocampus and cerebellum area. Each of those areas has been snap frozen in liquid nitrogen and stored at −80°C. Cortices were thawed on ice and 20 mg of tissue as been isolated and processed for immunoblot experiments.
Measurement of oxygen consumption and ATP/ADP levels
Oxygen consumption rates (OCR) were performed using the XF24 Extracellular Flux Analyser (Seahorse Bioscience, North Billerica, MA). Neurons (3 × 105) transfected with siCtrl or siOPA1 were plated on XF24 microplates 6 days before OCR measurements. Dual‐analyte sensor cartridges were soaked in XF Calibrant Solution (Seahorse Biosciences) in 24‐wells cell culture microplates overnight at 37°C to hydrate the probes. One hour prior to experimentation, injection ports on the sensor cartridge were filled with oligomycin (0.6 μmol/L), carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP) (6 μmol/L), and rotenone (50 nmol/L) plus antimycin A (0.182 μmol/L). Plates were then loaded into the XF24 instrument for calibration. For oxygen consumption measurement, growth media of neurons were replaced with incubation media (DMEM supplemented with NaCl (143 mmol/L), phenol red (3 mg/mL), glucose (10 mmol/L), glutamine (2 mmol/L), and pyruvate (2 mmol/L) at pH 7.4, and kept at 37°C, 1 h prior experimentation. The XF24 microplate was then loaded into the Seahorse XF24 analyzer following the manufacturer's instructions.
ATP and ADP measurements in siOPA1‐ or siCtrl‐transfected neurons were determined using a bioluminescence technique using an ATP monitoring kit as described previously.24
Immunoblot analysis
Neurons, human fibroblasts, and 20 mg of DOA mice models and control littermate mice cortices were lysed for 30 min in a buffer containing 50 mmol/L Tris‐HCL pH 7.5, 250 mmol/L NaCl, 5 mmol/L Ethylene Diamine Tetraacetic acid (EDTA), 5 mmol/L Ethylene Glycol Tetraacetic Acid (EGTA), 1 mmol/L dithiothreitol, 0.1% Triton X‐100, 0.1% SDS, 1% deoxycholate, 1% tergitol‐type NP‐40, and protease inhibitors (“Complete” protease inhibitor mixture, Roche Applied Science, UK). After sonication, cell lysates were centrifuged at 20,000 g at 4°C for 10 min. Lysate from frozen mice cortices were obtained using the same protocol, but adding a step of dounce homogenization.
Total protein concentration was determined in the supernatant using the Bradford protein assay (Bio‐Rad). Proteins (100–200 μg) were separated by SDS‐PAGE (8–15%) and transferred onto nitrocellulose membranes (Whatman, Protran, Sigma‐Aldrich, UK). Free binding sites were blocked with 5% nonfat dry milk, 0.2% Tween 20 in Tris buffer saline, pH 7.6 (blocking buffer). The membranes were probed with various primary antibodies: anti‐OPA1 (1/300, BD Bioscience, USA), anti‐actin (1/25000, Chemicon, Merck Millipore, UK), anti‐HSP60 (1/8000, Sigma‐Aldrich, UK), anti‐citrate synthase (1/3000, Abcam, Cambridge, USA), anti‐VDAC (1/1000, Abcam, Cambridge, USA), anti‐TOM20 (1/2000, Abcam, Cambridge, USA), anti‐aconitase (1/500, Abcam, Cambridge, USA), anti‐SOD1 and anti‐SOD2 (1/2000, Epitomics, Abcam, Cambridge, USA), anti‐catalase (1/3000, Abcam, Cambridge, USA), anti‐NQO1 (1/3000, Abcam, Cambridge, USA), anti‐mitofilin (1/1000 Abcam, Cambridge, USA), anti‐MFN1 (1/1000 , Abnova, Taipei City, Taiwan), and anti‐GSTP1 (1/8000, Oxford Biochemical Research, Euromedex, France), and incubated overnight at 4°C in blocking buffer. After chemiluminescent detection of horseradish peroxidase‐conjugated secondary antibody (1/10000, Abcam, Cambridge, USA), scanned photographic films were quantitatively analyzed using Image J software.
Immunocytochemistry
Neurons were fixed for 15 min at 37°C with PBS containing 3.7% formaldehyde and permeabilized for 15 min in PBS, 1% bovine serum albumin, 0.3% Triton™ X‐100, and for 10 min at −20°C in methanol. Nonspecific sites were blocked for 1 h in PBS containing 5% goat serum, 3% bovine serum albumin, and 0.5% Tween 20 (blocking solution). Polyclonal antibodies against NRF2 (1/50, Santa Cruz Biotechnology, Dallas, TX, USA) were incubated overnight at 4°C in blocking solution. Neurons were then incubated with Alexa fluor 488‐conjugated secondary antibodies (1/300, Molecular Probes), labeled with 0.25 μg/mL Hoechst in PBS over 5 min and mounted in Mowiol. Immunolabeling was visualized under a fluorescence microscope (Nikon Eclipse 80i or Zeiss 710 Big) and images were acquired using NIS‐Element (Nikon Digital Sight DUS2 camera) or ZEN 2011 software. Nucleus raw integrated densities (sum of pixel values) of NRF2 by μm2 in neurons were measured using Image J software and confocal images.
Aconitase and catalase activities and GSH/GSSG measurements
Aconitase activities measurements were performed using a protocol described previously.25 The photochrome was measured at 340 nm using the UVIKON Spectrophotometer 922. Catalase activity was determined by measuring decomposition of H2O2 at 240 nm as described previously.26 Reduced (GSH) and oxidized (GSSG) measurements were performed by reverse‐phase high‐performance liquid chromatography as described previously.26 For both aconitase and catalase activities measurement, 20 mg of DOA mice and control mice cortices were used per replicate of experiment and 1 × 106 cortical neurons ex vivo transfected with either siCtrl or siOPA1 has been harvested for the assay.
Rotenone treatment and cellular viability
Acute oxidative stress was induced in DIV6 (6 days in vitro) cultured neurons by 1 h incubation in neurobasal medium containing 500 nmol/L rotenone (Sigma‐Aldrich, UK). Then, rotenone was removed and cells were incubated for 3 h in neurobasal medium.
Neuronal viability was estimated by trypan blue exclusion and counting picnotic nuclei after DAPI staining as described in Bertholet et al.21
Statistical analysis
Most of the experiments were statistically treated with Student's paired t‐test because of the systematic comparison between siCtrl and siOPA1 neurons. OCRs between siCtrl and siOPA1 neurons were investigated using a two‐way analysis of variance (ANOVA) with a Sidak's multiple comparison test. Nucleus NRF2 raw integrated densities in siCtrl and siOPA1 neurons were carried out using a nonparametric test (Mann–Whitney test). The results presented in Figures 1 and 3 were assessed by a two‐way ANOVA with a Sidak's multiple comparison test. P values: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 were considered statistically significant.
Figure 1.
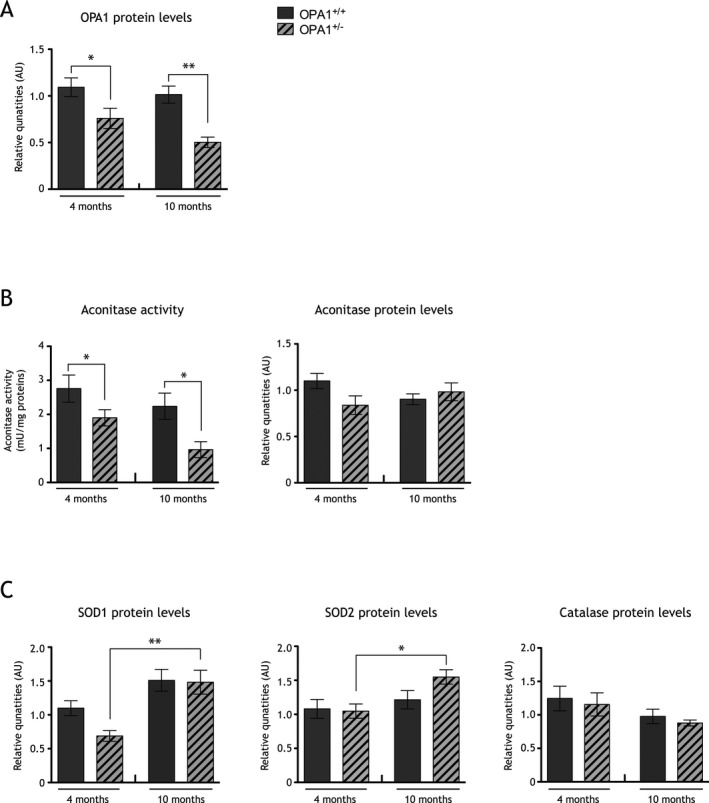
Redox state is imbalanced in a dominant optic atrophy (DOA) mice model. (A) Histograms representative of OPA1 protein quantities, assessed by immunoblot, in cortices of 4 and 10 months old control mice (gray bars) and Opa1 +/− mice (striped bars). OPA1 quantities are significantly decreased in both Opa1 +/− 4 (0.70 ± 0.10 arbitrary units [AU]) and 10 (0.50 ± 0.05 AU) months old mice when compare to 4 (1.09 ± 0.10 AU) and 10 (1.01 ± 0.09 AU) months old Opa1 +/+ mice. Results are expressed as mean ± standard error of the mean (SEM) for 4 and 10 months old mice (n = 6–9). Statistical significance was determined by a two‐way analysis of variance (ANOVA), *P < 0.05, **P < 0.01. (B) Histogram representative of aconitase activities in cortices of 4 and 10 months old Opa1 +/− (striped bars) and Opa1 +/+ control mice (gray bars) cortices. Aconitase activity is significantly lower in 4 (1.56 ± 0.11 mU/mg) and 10 (0.97 ± 0.23 mU/mg) months old Opa1 +/− mice when compared to 4 (2.95 ± 0.40 mU/mg) and 10 (2.24 ± 0.38 mU/mg) months old control Opa1 +/+ mice, respectively. Aconitase proteins quantities are unchanged in DOA mice model's cortices. Results are expressed as mean ± SEM (n = 9 for 4 months old mice and n = 6 for 10 months old mice). Statistical significance was determined by a two‐way ANOVA, *P < 0.05. (C) Histogram representative of SOD1, SOD2, and catalase proteins quantities relative to actin, assessed by immunoblot, in 4 and 10 months old Opa1 +/− (striped bars) and Opa1 +/+ (gray bars) mice cortices. SOD1 proteins quantities are significantly increased in 10 months old (1.48 ± 0.17 AU) Opa1 +/− mice compared to 4 months old (0.76 ± 0.08 AU) Opa1 +/− mice. Likewise, SOD2 proteins quantities are significantly increased in 10 months old (1.55 ± 0.11 AU) Opa1 +/− mice compared to 4 months old (0.98 ± 0.09 AU) Opa1 +/− mice. Catalase quantity is unchanged in Opa1 +/+ and Opa1 +/− 4 and 10 months old mice. Results are expressed as mean ± SEM (n = 9 for 4 months old mice and n = 6 for 10 months old mice). Statistical significance was determined using a two‐way ANOVA, *P < 0.05, **P < 0.01.
Figure 3.
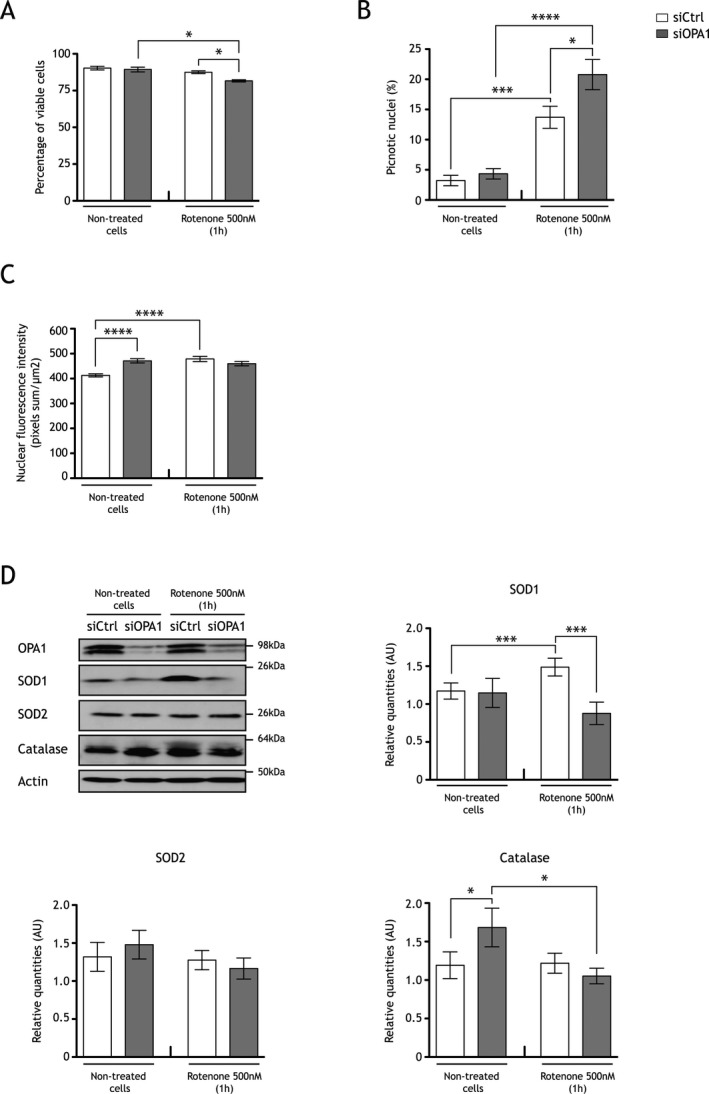
Oxidative stress challenges viability of OPA1 downregulated neurons, which do not induce the NRF2 pathway. (A) Trypan blue exclusion assay were performed after acute rotenone treatment (1 h, 500 nmol/L rotenone treatment) in neurons transfected with (gray) or without siOPA1 (white). Viable cells, which exclude the coloring agent, are lower in siOPA1 neurons treated with rotenone (75.37 ± 2.13%) than in control cells treated with rotenone (83.9 ± 1.82%). Results are expressed as mean ± SEM (n = 5). Statistical significance was determined by a two‐way analysis of variance (ANOVA), *P < 0.05. (B) Percentage of picnotic nuclei determined after Hoechst staining is higher in siOPA1 (gray bars, 20.78 ± 2.50%) than in siCtrl neurons (white bars, 13.71 ± 1.82%) upon rotenone treatment. Results are expressed as mean ± SEM (n = 4, 100 nuclei per conditions). Statistical significance was determined by a two‐way ANOVA, *P < 0.05, ***P < 0.001, ****P < 0.0001. (C) Histogram representing NRF2 nuclear fluorescence intensity (pixels sum/μm2, Image J software), which is higher in siOPA1 neurons (gray bars, 471.4 ± 8.5 pixels sum/μm2) than in controls (white bars, 412.8 ± 6.1 pixels sum/μm2) in basal condition. After rotenone treatment, NRF2 nuclear fluorescence intensity is increased in controls (478.7 ± 10.4 pixels sum/μm2), but not in siOPA1 neurons (459.9 ± 8.8 pixels sum/μm2). Results are expressed as mean ± SEM (≈800 cells per condition, n = 5). Statistical significance was determined by a two‐way ANOVA, ****P < 0.0001. (D) Representative immunoblots and histograms of SOD1, SOD2, and catalase proteins quantities relative to actin in siOPA1 (gray bars) and siCtrl neurons (white bars) with or without rotenone treatment. Upon stress, SOD1 quantity is increased in controls (without rotenone: 1.10 ± 0.10 AU; with rotenone: 1.49 ± 0.11 AU), but not in siOPA1‐transfected neurons (without rotenone: 1.15 ± 0.19 AU; with rotenone: 0.88 ± 0.15 AU). SOD2 quantities are the same in siCtrl‐ and siOPA1‐transfected neurons with or without rotenone. Upon rotenone treatment catalase protein level is unchanged in controls (without rotenone: 1.19 ± 0.17 AU; with rotenone: 1.22 ± 0.13 AU), while it is decreased in siOPA1 neurons (without rotenone: 1.67 ± 0.25 AU; with rotenone: 1.05 ± 0.1 AU). Results are expressed as mean ± SEM (n = 7). Statistical significance was determined by a two‐way ANOVA, *P < 0.05, ***P < 0.001.
Results
Opa1 +/− transgenic mice cortices show imbalanced redox state
To address the question of the implication of ROS as a determinant of DOA pathogenesis in mammals, we investigated whether a redox imbalance could be evidenced in vivo in a mouse model of DOA.20
As expected, a decrease in OPA1 levels was evidenced in Opa1 +/− transgenic mice cortices compared to litter mate Opa1 +/+ mice (Fig. 1A). Cortices from 4 and 10 months old DOA mice were analyzed for aconitase activity. Aconitase was shown to be highly sensitive to oxidation due to damaged FeS core and inhibition of its activity is routinely used as a signature of increased mitochondrial ROS production.27, 28, 29 Downregulation of OPA1 correlated with a 47% and 57% inhibition of aconitase activity in 4 and 10 months old Opa1 +/− transgenic mice compared to litter mate Opa1 +/+ mice, respectively (Fig. 1B). Moreover, aconitase activity was slightly decreased (38%) from 4 to 10 months in Opa1 +/− transgenic mice (Fig. 1B). This drop could not be attributed to a change in protein quantity since aconitase protein levels were unchanged (Fig. 1B).
Decrease in aconitase activity strongly supports higher mitochondrial ROS production in Opa1 +/− mice. However, to reinforce our data we monitored antioxidant defenses induction as a consequence of increased ROS levels. The protein levels of superoxide dismutases 1 and 2 (SOD1 and SOD2), that catalyze the dismutation of superoxide anion into hydrogen peroxide, and the levels of catalase, detoxifying hydrogen peroxide into water and oxygen, were estimated by immunoblotting in 4 and 10 months old Opa1 +/− mice cortices and control (Fig. 1C). The data reveal an increase in SOD1 and SOD2, while catalase is unchanged in 10 months Opa1 +/− mice cortices when compared to 4 months Opa1 +/− mice. No difference was observed in control mice (Fig. 1C).
Altogether these findings suggest that during the 6 months interval (between 4 and 10 months) there is an excess of mitochondrial ROS in Opa1 +/− mice that may induce an antioxidant response.
OPA1 downregulation imbalances neuronal redox state in vitro and activates NRF2 pathway
To decipher the molecular mechanisms underlying the excess of ROS in DOA mice, we used a previously characterized in vitro neuronal model of OPA1 haploinsufficiency.21
Primary cortical neurons transfected with siRNA directed against OPA1 mRNA (siOPA1) showed 70% decrease in OPA1 protein levels at day 6 post‐transfection when compared to neurons treated with control siRNA (siCtrl) (Fig. 2A). Interestingly, citrate synthase, HSP60, VDAC, and TOM20 levels were unchanged (Fig. 2A), showing that mitochondria quantity was not affected by OPA1 downregulation.
Figure 2.
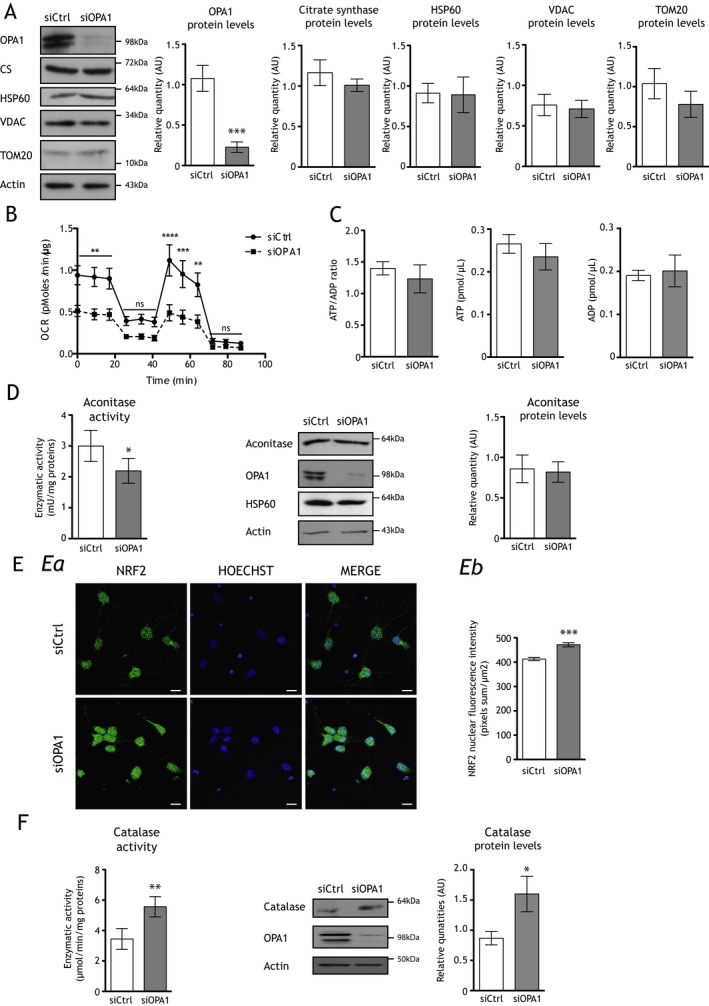
OPA1 downregulation decreases mitochondrial respiration, induces the nuclear translocation of NRF2, and increases both catalase quantity and activity in cortical neurons ex vivo. (A) Representative immunoblots and histograms showing protein levels of OPA1 (inner membrane), citrate synthase (matrix), HSP60 (matrix), VDAC (outer membrane), and TOM20 (outer membrane) relative to actin in siOPA1‐ (gray bars) and siCtrl‐transfected (white bars) neurons. Only OPA1 protein quantity is drastically decreased (0.23 ± 0.06 AU) in siOPA1 neurons when compared to controls (1.08 ± 0.16 AU). Results are expressed as mean ± SEM (n = 5–8). Statistical significance was determined by Student's paired t‐test, ***P < 0.001. (B) Oxygen consumption rates were measured at 6 days in vitro (DIV6) in neurons transfected with control small interfering RNA (siCtrl, black line) or small interfering RNA against OPA1 (siOPA1, dotted line). Spontaneous mitochondrial respiration is significantly lower in siOPA1‐transfected neurons (0.48 ± 0.07 pmol/min per μg) than in controls (0.92 ± 0.11 pmol/min per μg). After 0.6 μmol/L oligomycin injection, cell respiration is also significantly lower in siOPA1 neurons (0.20 ± 0.03 pmol/min per μg) than in controls (0.40 ± 0.06 pmol/min per μg). After 6 μmol/L FCCP injection, maximal respiration is significantly lower siOPA1 neurons (0.44 ± 0.09 pmol/min per μg) than in controls (0.59 ± 0.16 pmol/min per μg). Finally, 50 nmol/L rotenone and 0.18 μmol/L antimycin A injections inhibit mitochondrial respiration. Results are expressed as mean ± SEM (n = 3). Statistical significance was determined by a two‐way analysis of variance (ANOVA), **P < 0.01 and ***P < 0.001. (C) ATP over ADP ratio is unchanged in siOPA1 neurons (gray bar) when compared to siCtrl‐treated (white bar) cells. Results are expressed as mean ± SEM (n = 3). (D) Aconitase activity is lower in siOPA1‐treated neurons (2.2 ± 0.4 mU/mg) (gray bars) when compared to control cells (3.0 ± 0.5 mU/mg) (white bars). Results are expressed as mean ± SEM (n = 5). Statistical analysis was determined by Student's paired t‐test, *P < 0.05. Representative immunoblots and histogram showing that OPA1 downregulation in neurons has no effect on the aconitase quantity relative to actin. Results are expressed as mean ± SEM (n = 6). Statistical significance was determined by Student's paired t‐test, *P < 0.05. (E) a: Representative micrographs of NRF2 immunolabeling (green) and Hoechst DNA staining (blue) in siOPA1 and siCtrl neurons. b: Histogram represents fluorescence intensity (pixels sum/μm2) of nuclear NRF2 determined by Image J software, which is higher in siOPA1 neurons (471.4 ± 8.5 pixels sum/μm2) (gray bars) than in controls (412.8 ± 6.2 pixels sum/μm2) (white bars). Results are expressed as mean ± SEM (≈800 cells per condition, n = 5). Statistical significance was determined by a two‐way ANOVA, ***P < 0.001. Scale bar: 5 μm. (F) Catalase activity is increased in siOPA1 neurons (5.56 ± 0.66 μmol/min per mg) when compared to controls (3.44 ± 0.69 μmol/min per mg). Results are expressed as mean ± SEM (n = 5). Representative immunoblots and protein quantities of catalase in siOPA1 (gray bars) and siCtrl (white bars) neurons relative to actin. Catalase quantity is increased in siOPA1 (1.6 ± 0.33 AU) when compared to controls (0.85 ± 0.14 AU). Results are expressed as mean ± SEM (n = 7). Statistical significance was determined by Student's paired t‐test, *P < 0.05, **P < 0.01.
While OPA1 downregulation is known to negatively impact respiration in various cell lines,30, 31, 32 we evaluated here for the first time the effect of OPA1 downregulation on respiration in neurons using the Seahorse XF24 analyzer (Seahorse Bioscience) (Fig. 2B). In siOPA1‐ and siCtrl‐transfected neurons, rotenone and antimycin treatment considerably inhibited the OCR, showing that more than 95% of respiration was due to mitochondria. In siCtrl‐transfected neurons, oligomycin inhibited ATP‐linked respiration, while addition of the protonophore FCCP, which uncouples oxidation and phosphorylation, resulted in maximal OCR. In siOPA1‐transfected neurons, spontaneous respiration was reduced by 32%, when compared to siCtrl‐treated neurons. Furthermore, the ATP‐linked respiration and the maximal OCR were reduced by 34% and 46%, respectively. Contrary to siCtrl‐transfected cells, the maximal OCR in siOPA1‐treated cells was not significantly different than spontaneous OCR.
Therefore, depletion of OPA1 in neurons induced a decrease in spontaneous, ATP‐linked respiration and maximal mitochondrial respiration without affecting the mitochondrial biomass. A decrease in mitochondrial respiration could lead to energetic failure. However, we showed that neither ATP nor ADP levels, and thus the ratio of ATP over ADP, changed upon OPA1 downregulation in cortical neurons (Fig. 2C). The ratio of ATP/ADP (and the amounts of these molecules) in the steady state is the result of synthesis and consumption. So if it remains stable while cellular respiration in phosphorylation conditions decreases, it could be the consequence of a consumption decrease to the same extent as synthesis. In the context of neurons it can be reflected by a decrease in the functionality of these cells. This was shown in Bertholet et al.21 by the reduction of synaptogenesis and dendritogenesis in OPA1 downregulated neurons.
Impaired MRC functioning could lead to an imbalance in the redox state because of increased electron leaks. In agreement, we showed that aconitase activity was reduced by 33% in siOPA1‐treated neurons (Fig. 2D), with no effect in aconitase protein quantity, suggesting that OPA1 downregulation would induce an increase in mitochondrial ROS levels in cultured neurons.21
Again, to reinforce these data we analyzed the NRF2 transcription factor pathway which accounts in large parts for the oxidative stress responses, and monitored some of its antioxidant target genes by analyzing SOD1, SOD2, and catalase protein levels.33 Since a hallmark of NRF2 activation is its nuclear translocation, we studied its intracellular localization. In neurons ex vivo, the decreased quantity of OPA1 induced a 1.3‐fold increase in nuclear NRF2 fluorescence compared to siCtrl cells (Fig. 2E). In siOPA1‐transfected neurons, no statistical differences in SOD1 and 2 (data not shown) were detected, while we uncovered an increase in both catalase quantity (88%) and activity (61%) (Fig. 2F).
We thus concluded that downregulation of OPA1 induced NRF2 activation leading to upregulation of, at least, one of its target genes, catalase.
Supplemental oxidative stress challenges viability of OPA1‐downregulated neurons
Since OPA1 downregulation leads to increased mitochondrial ROS levels that induce antioxidant response, we addressed the impact of an additional acute oxidative stress in OPA1 downregulated cells. A supplemental stress would therefore be deleterious for cell viability. Neurons were thus incubated with rotenone, a potent inhibitor of respiratory complex I that induces a major oxidative stress.34, 35 We checked this hypothesis by estimating the neuronal viability by trypan blue exclusion assay (Fig. 3A) as well as the number of picnotic nuclei by DAPI staining (Fig. 3B) of siCtrl‐ and siOPA1‐transfected neurons upon acute treatment of rotenone (1h, and 500 nmol/L). siOPA1‐transfected cells showed 8% decreased viability when compared to siCtrl‐treated neurons (Fig. 3A). Accordingly, the number of picnotic nuclei is increased by 34% in siOPA1 versus siCtrl rotenone‐treated neurons (Fig. 3B). These results suggest that antioxidant defenses may be overwhelmed in OPA1 downregulated conditions. We checked this hypothesis by analyzing the NRF2 nuclear translocation (Fig. 3C), as well as SOD1, SOD2, and catalase protein quantities (Fig. 3D). Upon acute rotenone treatment, NRF2 translocation was increased by 1.2‐fold in siCtrl neurons, while it was unchanged in siOPA1 neurons (Fig. 3C). Accordingly, a 1.5‐fold increase in SOD1 protein quantity was observed in control cells, while no change occurred in siOPA1 neurons (Fig. 3D). No difference was observed for SOD2 protein quantities in siOPA1 and siCtrl neurons treated or not with rotenone (Fig. 3D). Moreover, catalase protein quantity, which is increased by 32% in basal condition in siOPA1 neurons when compared to control neurons, did not increase and even diminished by 32%, suggesting that downregulation of OPA1 results in maximal catalase induction that cannot be further increased upon rotenone‐induced oxidative stress.
Cellular antioxidant defenses are impaired in certain fibroblasts from DOA patients
We next addressed the question of antioxidant defenses in DOA patients' fibroblasts (Table S1). SOD1, SOD2, and catalase were studied by immunoblot analysis (Fig. 4A). Fibroblasts from five healthy volunteers (C1–C5) and eight DOA patients (P1–P8) were analyzed. Even though heterogeneity exists between healthy volunteers and DOA patients in antioxidant defenses, some patients showed altered expression of antioxidant genes at the protein level (Fig. 4A and Fig. S2). DOA patients P1, P3, P5, and P7 expressed low levels of SOD1 and SOD2 proteins, while catalase was not different (Fig. 4A). To note, the levels of these antioxidant proteins are not correlated with the age of patients (Table S1 and Fig. S3).
Figure 4.
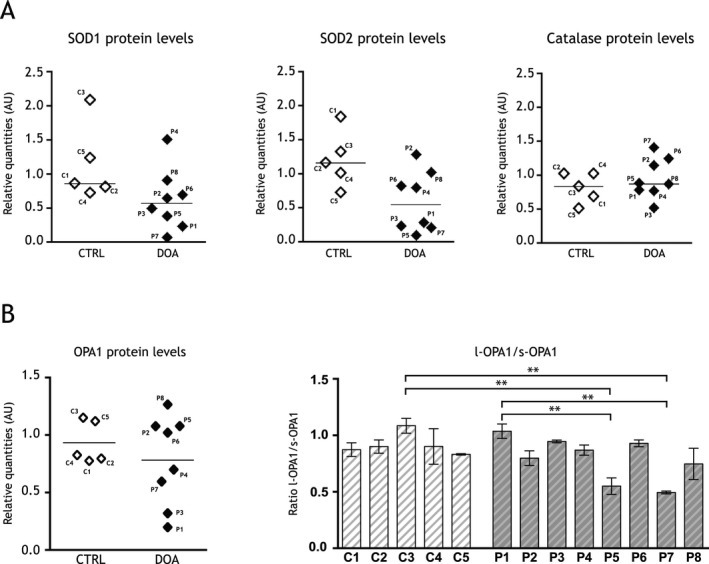
Antioxidant defenses and OPA1 proteins levels in fibroblasts from dominant optic atrophy (DOA) patients. (A) Representative histograms of SOD1, SOD2, and catalase proteins levels relative to actin in five healthy volunteer's skin fibroblasts (CTRL, C1–C5) and eight DOA patient's skin fibroblasts (DOA, P1–P8) ex vivo. SOD1 CTRL: 1.15 ± 0.25 AU; SOD1 DOA: 0.62 ± 0.16 AU; SOD2 CTRL: 1.21 ± 0.19 AU; SOD2 DOA: 0.59 ± 0.16 AU; catalase CTRL: 0.82 ± 0.10 AU; catalase DOA: 0.96 ± 0.10 AU. Results are expressed as mean ± SEM (n = 3–6 per DOA and CTRL fibroblasts). (B) Representative histograms of OPA1 protein levels relative to actin in five healthy volunteer's skin fibroblasts (CTRL, C1–C5) and eight DOA patient's skin fibroblasts (DOA, P1–P8) ex vivo. Histogram representative of the ratio of the long (l‐OPA1) to the short (s‐OPA1) OPA1 isoforms. Results are expressed as mean ± SEM (n = 3–6 per DOA and CTRL fibroblasts) Statistical significance for the ratio of the long to the short OPA1 isoforms was determined by a one‐way analysis of variance (ANOVA), **P < 0.01.
These results show actual heterogeneity of SODs content in patient fibroblasts. To determine if this heterogeneity could be linked to the status of OPA1 protein level in fibroblasts from DOA patients, we investigated the levels of both total OPA1 protein and of the ratio of the long (l‐OPA1) relative to the short (s‐OPA1) OPA1 isoforms (Fig. 4B). P1 and P3 presented a lower quantity of OPA1. P5 and P7 presented differences of the ratio l‐OPA1 over s‐OPA1.
Discussion
In the present work, we clearly demonstrate that OPA1 dysfunction induces a decline in endogenous mitochondrial respiration in primary cultured neurons with a maximal mitochondrial respiration that does not exceed basal mitochondrial respiration. Defects in mitochondrial respiration have also been observed in several cell lines upon OPA1 knockdown. In agreement with our results, OPA1‐depleted MEFs also showed a severe reduction in endogenous respiration, no effect after uncoupling, and a decrease in oxygen consumption mediated by complexes I, II, and IV.32 Studies using fibroblasts from DOA patients support the observation of a coupling defect of oxidative phosphorylation due to a reduction in complex IV at the protein level and heightened complex V activity, with no change in ATP production in either case.12 Defective oxidative phosphorylation with lowered ATP production has been found in DOA patient tissue analyzed by MR spectroscopy.36 However, the existence and the nature of the mitochondrial oxidative phosphorylation dysfunction are still controversial since other studies did not find respiratory defects in lymphoblastoid lines31 or in muscle biopsies of patients with DOA.8 Along these lines, cardiomyocytes derived from Opa1 +/− mice also do not display any respiratory defects although mitochondrial morphology is altered.37
Mitochondrial respiration impairment due to OPA1 loss could originate from either downregulation/deregulation of MRC, or disorganization of the mitochondrial inner membrane structure,38 or both. Indeed, we have previously shown that the protein levels of certain subunits of MRC were decreased in siOPA1‐transfected neurons.21 Besides, NDUFB8 and NDUFA9 from complex I, Core 1 and Core 2 from complex III, and Cox I and Cox VIc from complex IV decreased by 30–50%. Moreover, Zanna and colleagues have suggested a direct interaction between OPA1 and the respiratory chain complexes I, II, and III.14 On the other hand, a decrease in respiration in siOPA1‐treated neurons could also damage the spatial organization of the MRC.39, 40 The organization of respiratory chain complexes into supercomplexes (RCS) is essential for the assembly and stability of each complex41, 42, 43 and may influence the efficiency of electron transfer.44, 45, 46 Therefore, it is likely, as recently suggested, that the respiratory deficiency that we observed upon OPA1 downregulation in neurons results from disruption of RCS.47
Our studies using primary cortical neurons did not reveal any significant changes in ATP or ADP levels. This may indicate that either OPA1 knockdown is not sufficient to induce an energetic failure or any decrease in mitochondrial ATP synthesis is offset by a decrease in global ATP consumption. In addition, we found an increase in mitochondrial ROS production revealed by a decrease in aconitase activity in OPA1‐depleted neurons, consistent with prior findings in OPA1‐mutant flies.48 We demonstrated that this increase in ROS production activates the NRF2 pathway, one of the primary mechanisms of cell detoxification.49 Indeed, downregulation of OPA1 in primary neurons induced relocalization of the NRF2 transcription factor into the nucleus, where it upregulated target genes bearing antioxidant responsive promoter elements, such as catalase. Moreover, upregulation of antioxidant defenses also confirms that OPA1 downregulation promotes higher ROS production. Additional exogenous oxidative stress caused by rotenone reduced neuronal viability. These results are consistent with previous experiments showing hypersensitivity to oxidative stress in C. elegans and in D. melanogaster.17, 18 Our data suggest that the viability impairment of siOPA1‐treated neurons upon acute oxidative stress is due to inefficient antioxidant defenses. SOD1, SOD2, and catalase levels did not change upon acute oxidative stress in siOPA1 rotenone‐treated cells, while SOD1 levels increased in rotenone‐treated control cells. Our experiments performed in Opa1 +/− mutant mice also found decreased aconitase activity and increased antioxidant defenses in Opa1 +/− mouse cortices compared to controls, again suggesting excess mitochondrial ROS production.
Our results thus underscore the critical role of NRF2 signaling in DOA pathogenesis. To note, even if we observed a difference in the NRF2‐induced target in our different DOA models, this is largely described in the literature (for review see ref. 49) as well as the different answers observed for the antioxidant defenses among the patient's fibroblasts for example for Friedreich Ataxia.50 Because many studies focused on fibroblasts to understand cellular attempts common to different cells and particularly neurodegenerative diseases,7, 14, 50 we decided in this study to compare the effect of OPA1 dysfunction in DOA patients fibroblasts and neuronal models. In all our models, the NRF2 pathway is associated with activation of SOD1 or SOD2 or catalase, but not GSTP1 and NQO1. Indeed, these last two enzymes remain constant for all the patients as well as in neurons treated by siOPA1 or siCtrl, which supports distinguishing SOD1 or SOD2 or catalase as reliable markers.
Our results also show that DOA pathogenesis may be related to both the quantity of OPA1 protein and the relative levels of the five isoforms of OPA1. P1 and P3 present a decrease in OPA1 protein levels, suggesting that, in these two cases, haploinsufficiency may contribute to DOA pathogenesis. P5 and P7 present lower ratios of l‐OPA1 to s‐OPA1, which might also contribute to the severity of this disease. Changes in the ratio of l‐OPA1 to s‐OPA1 were shown to impact OPA1 functions,10 and in a recent work, the level of l‐OPA1 was presented as a possible therapeutic target for ischemia–reperfusion injury‐related diseases at least for the retina because it could restore mitochondrial morphology.51 These results suggest that the protein profile of OPA1, beyond merely its genetic status, should be considered in future research.
As we showed here for DOA, dysfunctions in ROS metabolism may also underlie Leber's hereditary optic neuropathy,52 glaucoma,53 and other neurodegenerative pathologies such as Alzheimer's and Parkinson's diseases.54 In our model, the redox state imbalance induced by OPA1 haploinsufficiency could escalate into an oxidative stress state upon exposure to factors such as UV light, blue light, smoke, aging, or diet. Interestingly, some studies described an increase in sensitivity of RGCs to blue light stimulation and to hypoxia.55, 56, 57 Additionally, within the retina, mitochondrial DNA lesions reach remarkable levels after exposure to endogenous ROS generated by the mitochondria themselves or from photosensitizers58 (for review see ref. 59). In this framework, it is noteworthy that numerous studies suggest that increasing mitochondrial antioxidant defenses may rescue optic nerve defects.53, 60, 61 Cellular systems that protect against oxidants include antioxidant defenses enzymes and oxidant scavengers. In addition to direct antioxidants, a number of antioxidant compounds are weak pro‐oxidants with indirect antioxidant properties.62 Acting as weak pro‐oxidants, these molecules have limited ability to cause oxidative damage, yet they are potent enough to activate the NRF2‐mediated adaptive response, thereby protecting against existing or future high‐level stresses.63 Given that our findings that OPA1 downregulation activates NRF2 pathway, the pro‐oxidant compounds could be insufficient or dangerous therapeutic approaches for some DOA patients. Instead, direct testing of antioxidant compounds could offer a complementary pharmacological treatment for DOA. Accordingly, a recent study focused on idebenone, a benzoquinone that transfers electrons to complex III bypassing complex I, as a potential curative compound for DOA patients.64 Thus, it is of primary importance to identify patients able to induce an efficient NRF2 response. Our work could thus have a direct impact on both treatment and care of DOA patients. Indeed, the low expression of some antioxidant enzymes in several DOA patients could support their role as modifier factors for DOA.
Conflict of Interest
None declared.
Supporting information
Figure S1. Protein levels of MFN1 and mitofilin in OPA1 downregulated neurons. Representative immunoblots and histograms showing protein levels of mitofilin and mitofusin 1 (MFN1) relative to actin in siOPA1‐ (gray bars) and siCtrl‐transfected (white bars) neurons. Results are expressed as mean ± SEM (n = 5). Statistical significance was determined by Student's paired t‐test.
Figure S2. DOA patient's fibroblasts and control's fibroblasts antioxidant proteins levels. (A) Representative immunoblots showing protein levels of SOD1, SOD2, catalase, GSTP1, NQO1, and actin, and histograms representing GSTP1 and NQO1 proteins quantities, in five healthy volunteer's (CTRL) fibroblasts (C1–C5) and in eight DOA patient's fibroblasts (P1–P8). (B) Representative immunoblots showing protein levels of OPA1 and actin.
Figure S3. DOA patients' and controls' fibroblasts antioxidant proteins levels relative to their age at the moment of the biopsies. Antioxidant proteins levels (SOD1, SOD2, catalase, GSTP1, and NQO1) relative to the age of patients and controls' fibroblasts: the quantity of each protein is not correlated to the age of the persons.
Table S1. Clinical symptoms and mutations of DOA patients.
Acknowledgments
We thank Brice Ronsin for technical assistance with Image J software and technical assistance in confocal analysis on the Toulouse RIO imaging platform. We thank the animal core facility at the Center of Developmental Biology. This project was supported by grants from the Centre National de la Recherche Scientifique, the Université Paul Sabatier, the “Rétina‐France,” the “Union Nationale Des Aveugles et Déficients Visuels,” the “Gueules Cassées Sourire quand même,” and the “Association contre les Maladies Mitochondriales.” A. Millet was funded by the French Ministry for Research and Education and the “Rétina‐France” for Ph.D. studies.
References
- 1. Amati‐Bonneau P, Milea D, Bonneau D, et al. OPA1‐associated disorders: phenotypes and pathophysiology. Int J Biochem Cell Biol 2009;41:1855–1865. [DOI] [PubMed] [Google Scholar]
- 2. Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin‐related protein, is mutated in dominant optic atrophy [In Process Citation]. Nat Genet 2000;26:207–210. [DOI] [PubMed] [Google Scholar]
- 3. Cohn AC, Toomes C, Potter C, et al. Autosomal dominant optic atrophy: penetrance and expressivity in patients with OPA1 mutations. Am J Ophthalmol 2007;143:656–662. [DOI] [PubMed] [Google Scholar]
- 4. Yu‐Wai‐Man P, Griffiths PG, Burke A, et al. The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology 2010;117:1538–1546, 46 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amati‐Bonneau P, Valentino ML, Reynier P, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus' phenotypes. Brain 2008;131(Pt 2):338–351. [DOI] [PubMed] [Google Scholar]
- 6. Zeviani M. OPA1 mutations and mitochondrial DNA damage: keeping the magic circle in shape. Brain 2008;131(Pt 2):314–317. [DOI] [PubMed] [Google Scholar]
- 7. Carelli V, Musumeci O, Caporali L, et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann Neurol 2015;78:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spinazzi M, Cazzola S, Bortolozzi M, et al. A novel deletion in the GTPase domain of OPA1 causes defects in mitochondrial morphology and distribution, but not in function. Hum Mol Genet 2008;17:3291–3302. [DOI] [PubMed] [Google Scholar]
- 9. Mackey DA, Trounce I. Genetics: optic nerve genetics–more than meets the eye. Nat Rev Neurol 2010;6:357–358. [DOI] [PubMed] [Google Scholar]
- 10. Belenguer P, Pellegrini L. The dynamin GTPase OPA1: more than mitochondria? Biochim Biophys Acta 2013;1833:176–183. [DOI] [PubMed] [Google Scholar]
- 11. Olichon A, Guillou E, Delettre C, et al. Mitochondrial dynamics and disease, OPA1. Biochim Biophys Acta 2006;1763:500–509. [DOI] [PubMed] [Google Scholar]
- 12. Chevrollier A, Guillet V, Loiseau D, et al. Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann Neurol 2008;63:794–798. [DOI] [PubMed] [Google Scholar]
- 13. Alavi MV, Fuhrmann N, Nguyen HP, et al. Subtle neurological and metabolic abnormalities in an Opa1 mouse model of autosomal dominant optic atrophy. Exp Neurol 2009;220:404–409. [DOI] [PubMed] [Google Scholar]
- 14. Zanna C, Ghelli A, Porcelli AM, et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2008;131(Pt 2):352–367. [DOI] [PubMed] [Google Scholar]
- 15. Yu‐Wai‐Man P, Shankar SP, Biousse V, et al. Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies. Ophthalmology 2011;118:558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Landes T, Leroy I, Bertholet A, et al. OPA1 (dys)functions. Semin Cell Dev Biol 2010;21:593–598. [DOI] [PubMed] [Google Scholar]
- 17. Kanazawa T, Zappaterra MD, Hasegawa A, et al. The C. elegans Opa1 homologue EAT‐3 is essential for resistance to free radicals. PLoS Genet 2008;4:e1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang S, Le PK, Tse S, et al. Heterozygous mutation of Opa1 in Drosophila shortens lifespan mediated through increased reactive oxygen species production. PLoS One 2009;4:e4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shahrestani P, Leung HT, Le PK, et al. Heterozygous mutation of Drosophila Opa1 causes the development of multiple organ abnormalities in an age‐dependent and organ‐specific manner. PLoS One 2009;4:e6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alavi MV, Bette S, Schimpf S, et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 2007;130(Pt 4):1029–1042. [DOI] [PubMed] [Google Scholar]
- 21. Bertholet AM, Millet AM, Guillermin O, et al. OPA1 loss of function affects in vitro neuronal maturation. Brain 2013;136(Pt 5):1518–1533. [DOI] [PubMed] [Google Scholar]
- 22. Chevrollier A, Cassereau J, Ferre M, et al. Standardized mitochondrial analysis gives new insights into mitochondrial dynamics and OPA1 function. Int J Biochem Cell Biol 2012;44:980–988. [DOI] [PubMed] [Google Scholar]
- 23. Heiduschka P, Schnichels S, Fuhrmann N, et al. Electrophysiological and histologic assessment of retinal ganglion cell fate in a mouse model for OPA1‐associated autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci 2010;51:1424–1431. [DOI] [PubMed] [Google Scholar]
- 24. Devin A, Nogueira V, Averet N, et al. Profound effects of the general anesthetic etomidate on oxidative phosphorylation without effects on their yield. J Bioenerg Biomembr 2006;38:137–142. [DOI] [PubMed] [Google Scholar]
- 25. Colombani AL, Carneiro L, Benani A, et al. Enhanced hypothalamic glucose sensing in obesity: alteration of redox signaling. Diabetes 2009;58:2189–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Galinier A, Carriere A, Fernandez Y, et al. Adipose tissue proadipogenic redox changes in obesity. J Biol Chem 2006;281:12682–12687. [DOI] [PubMed] [Google Scholar]
- 27. Vincent AM, McLean LL, Backus C, Feldman EL. Short‐term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J 2005;19:638–640. [DOI] [PubMed] [Google Scholar]
- 28. Gardner PR, Nguyen DD, White CW. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc Natl Acad Sci USA 1994;91:12248–12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kelly M, Trudel S, Brouillard F, et al. Cystic fibrosis transmembrane regulator inhibitors CFTR(inh)‐172 and GlyH‐101 target mitochondrial functions, independently of chloride channel inhibition. J Pharmacol Exp Ther 2010;333:60–69. [DOI] [PubMed] [Google Scholar]
- 30. Agier V, Oliviero P, Laine J, et al. Defective mitochondrial fusion, altered respiratory function, and distorted cristae structure in skin fibroblasts with heterozygous OPA1 mutations. Biochim Biophys Acta 2012;1822:1570–1580. [DOI] [PubMed] [Google Scholar]
- 31. Van Bergen NJ, Crowston JG, Kearns LS, et al. Mitochondrial oxidative phosphorylation compensation may preserve vision in patients with OPA1‐linked autosomal dominant optic atrophy. PLoS One 2011;6:e21347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen C, Fossar N, Weil D, et al. High frequency trans‐splicing in a cell line producing spliced and polyadenylated RNA polymerase I transcripts from an rDNA‐myc chimeric gene. Nucleic Acids Res 2005;33:2332–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu H, Itoh K, Yamamoto M, et al. Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species‐induced cell injury. FEBS Lett 2005;579:3029–3036. [DOI] [PubMed] [Google Scholar]
- 34. Berndt N, Holzhutter HG, Bulik S. Implications of enzyme deficiencies on mitochondrial energy metabolism and reactive oxygen species formation of neurons involved in rotenone‐induced Parkinson's disease: a model‐based analysis. FEBS J 2013;280:5080–5093. [DOI] [PubMed] [Google Scholar]
- 35. Freestone PS, Chung KK, Guatteo E, et al. Acute action of rotenone on nigral dopaminergic neurons–involvement of reactive oxygen species and disruption of Ca2 + homeostasis. Eur J Neuorsci 2009;30:1849–1859. [DOI] [PubMed] [Google Scholar]
- 36. Lodi R, Tonon C, Valentino ML, et al. Defective mitochondrial adenosine triphosphate production in skeletal muscle from patients with dominant optic atrophy due to OPA1 mutations. Arch Neurol 2011;68:67–73. [DOI] [PubMed] [Google Scholar]
- 37. Piquereau J, Caffin F, Novotova M, et al. Down‐regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res 2012;94:408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Darshi M, Mendiola VL, Mackey MR, et al. ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J Biol Chem 2011;286:2918–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olichon A, Baricault L, Gas N, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 2003;278:7743–7746. [DOI] [PubMed] [Google Scholar]
- 40. Griparic L, van der Wel NN, Orozco IJ, et al. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem 2004;279:18792–18798. [DOI] [PubMed] [Google Scholar]
- 41. Acin‐Perez R, Bayona‐Bafaluy MP, Fernandez‐Silva P, et al. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell 2004;13:805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Diaz F, Fukui H, Garcia S, Moraes CT. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol Cell Biol 2006;26:4872–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li Y, D'Aurelio M, Deng JH, et al. An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. J Biol Chem 2007;282:17557–17562. [DOI] [PubMed] [Google Scholar]
- 44. Acin‐Perez R, Enriquez JA. The function of the respiratory supercomplexes: the plasticity model. Biochim Biophys Acta 2014;1837:444–450. [DOI] [PubMed] [Google Scholar]
- 45. Vartak R, Porras CA, Bai Y. Respiratory supercomplexes: structure, function and assembly. Protein Cell 2013;4:582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lenaz G, Genova ML. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv Exp Med Biol 2012;748:107–144. [DOI] [PubMed] [Google Scholar]
- 47. Cogliati S, Frezza C, Soriano ME, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013;155:160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yarosh W, Monserrate J, Tong JJ, et al. The molecular mechanisms of OPA1‐mediated optic atrophy in Drosophila model and prospects for antioxidant treatment. PLoS Genet 2008;4:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 2013;53:401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paupe V, Dassa EP, Goncalves S, et al. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS One 2009;4:e4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sun Y, Xue W, Song Z, et al. Restoration of Opa1‐long isoform inhibits retinal injury‐induced neurodegeneration. J Mol Med (Berl). 2015;94:335–346. [DOI] [PubMed] [Google Scholar]
- 52. Yen MY, Wang AG, Wei YH. Leber's hereditary optic neuropathy: a multifactorial disease. Prog Retin Eye Res 2006;25:381–396. [DOI] [PubMed] [Google Scholar]
- 53. Lee D, Shim MS, Kim KY, et al. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress‐mediated mitochondrial alteration in a mouse model of glaucoma. Invest Ophthalmol Vis Sci 2014;55:993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 2013;62:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bennet D, Kim MG, Kim S. Light‐induced anatomical alterations in retinal cells. Anal Biochem 2013;436:84–92. [DOI] [PubMed] [Google Scholar]
- 56. Ji D, Li GY, Osborne NN. Nicotinamide attenuates retinal ischemia and light insults to neurones. Neurochem Int 2008;52:786–798. [DOI] [PubMed] [Google Scholar]
- 57. Kergoat H, Herard ME, Lemay M. RGC sensitivity to mild systemic hypoxia. Invest Ophthalmol Vis Sci 2006;47:5423–5427. [DOI] [PubMed] [Google Scholar]
- 58. Jarrett SG, Lewin AS, Boulton ME. The importance of mitochondria in age‐related and inherited eye disorders. Ophthalmic Res 2010;44:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sacca SC, Roszkowska AM, Izzotti A. Environmental light and endogenous antioxidants as the main determinants of non‐cancer ocular diseases. Mutat Res 2013;752:153–171. [DOI] [PubMed] [Google Scholar]
- 60. Qi X, Sun L, Hauswirth WW, et al. Use of mitochondrial antioxidant defenses for rescue of cells with a Leber hereditary optic neuropathy‐causing mutation. Arch Ophthalmol 2007;125:268–272. [DOI] [PubMed] [Google Scholar]
- 61. Bessero AC, Clarke PG. Neuroprotection for optic nerve disorders. Curr Opin Neurol 2010;23:10–15. [DOI] [PubMed] [Google Scholar]
- 62. Son TG, Camandola S, Mattson MP. Hormetic dietary phytochemicals. NeuroMol Med 2008;10:236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang Q, Pi J, Woods CG, Andersen ME. A systems biology perspective on Nrf2‐mediated antioxidant response. Toxicol Appl Pharmacol 2010;244:84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Barboni P, Valentino ML, La Morgia C, et al. Idebenone treatment in patients with OPA1‐mutant dominant optic atrophy. Brain 2013;136(Pt 2):e231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Protein levels of MFN1 and mitofilin in OPA1 downregulated neurons. Representative immunoblots and histograms showing protein levels of mitofilin and mitofusin 1 (MFN1) relative to actin in siOPA1‐ (gray bars) and siCtrl‐transfected (white bars) neurons. Results are expressed as mean ± SEM (n = 5). Statistical significance was determined by Student's paired t‐test.
Figure S2. DOA patient's fibroblasts and control's fibroblasts antioxidant proteins levels. (A) Representative immunoblots showing protein levels of SOD1, SOD2, catalase, GSTP1, NQO1, and actin, and histograms representing GSTP1 and NQO1 proteins quantities, in five healthy volunteer's (CTRL) fibroblasts (C1–C5) and in eight DOA patient's fibroblasts (P1–P8). (B) Representative immunoblots showing protein levels of OPA1 and actin.
Figure S3. DOA patients' and controls' fibroblasts antioxidant proteins levels relative to their age at the moment of the biopsies. Antioxidant proteins levels (SOD1, SOD2, catalase, GSTP1, and NQO1) relative to the age of patients and controls' fibroblasts: the quantity of each protein is not correlated to the age of the persons.
Table S1. Clinical symptoms and mutations of DOA patients.