

Abstract
Objective
The nicotinamide‐nucleotide adenylyltransferase protein Nmnat1 is a potent inhibitor of axonal degeneration in models of acute axonal injury. Hyperphosphorylation and aggregation of the microtubule‐associated protein Tau are associated with neurodegeneration in Alzheimer's Disease and other disorders. Previous studies have demonstrated that other Nmnat isoforms can act both as axonoprotective agents and have protein chaperone function, exerting protective effects in drosophila and mouse models of tauopathy. Nmnat1 targeted to the cytoplasm (cytNmnat1) is neuroprotective in a mouse model of neonatal hypoxia‐ischemia, but the effect of cytNmnat1 on tauopathy remains unknown.
Methods
We examined the impact of overexpression of cytNmnat1 on tau pathology, neurodegeneration, and brain functional connectivity in the P301S mouse model of chronic tauopathy.
Results
Overexpression of cytNmnat1 preserved cortical neuron functional connectivity in P301S mice in vivo. However, whereas Nmnat1 overexpression decreased the accumulation of detergent‐insoluble tau aggregates in the cerebral cortex, it exerted no effect on immunohistochemical evidence of pathologic tau phosphorylation and misfolding, hippocampal atrophy, or inflammatory markers in P301S mice.
Interpretation
Our results demonstrate that cytNmnat1 partially preserves neuronal function and decreases biochemically insoluble tau in a mouse model of chronic tauopathy without preventing tau phosphorylation, formation of soluble aggregates, or tau‐induced inflammation and atrophy. Nmnat1 might thus represent a therapeutic target for tauopathies.
Introduction
The family of nicotinamide‐nucleotide adenylyltransferase (Nmnat) proteins has been broadly studied for their potent axonal protective effects.1, 2, 3 These enzymes catalyze the conversion of nicotinamide mononucleotide to NAD+ in the final step of the NAD+ salvage pathway. Nmnat enzymes thus contribute to neuronal health via maintenance of cellular NAD+ levels, although they also appear to exert NAD+‐independent cytoprotective effects.2, 4, 5 There are three mammalian Nmnat isoforms: a nuclear form (Nmnat1), a cytoplasmic form (Nmnat2), and a mitochondrial form (Nmnat3). Overexpression of each of these isoforms can prevent axonal degeneration in neurons.3, 6, 7 Targeting of Nmnat1 to the cytoplasm has been shown to exert very potent axonoprotective effects, and to suppress neuronal death in the brain of newborn mice caused by hypoxia‐ischemia.3, 8 Thus, modulation of Nmnat function is an attractive potential target for the treatment of peripheral nerve injuries, and has been postulated as a possible therapeutic target for the treatment of central nervous system degenerative diseases.
The microtubule‐associated protein tau has long been implicated as a major contributor to several age‐related neurodegenerative diseases, including Alzheimer's Disease (AD) and Frontotemporal dementia.9 Intraneuronal aggregates of hyperphosphorylated tau, termed neurofibrillary tangles, are observed in AD, and correlate closely with neurodegeneration. Elevated levels of tau and phospho‐tau are also observed in the cerebrospinal fluid of AD patients and predict the subsequent onset of dementia.10, 11 Mutations in the MAPT gene, which encodes tau protein, cause an adult‐onset neurodegenerative disease characterized by frontotemporal dementia and Parkinsonism.12 Transgenic mice expressing these mutant human tau genes exhibit progressive accumulation of hyperphosphorylated tau aggregates, as well as neuroinflammation and neurodegeneration.13, 14 Thus, strategies which mitigate tau accumulation or toxicity have clear implications for the treatment of AD and other neurodegenerative diseases.
Several studies have described a potential relationship between Nmnat enzymes and tau pathology. In drosophila models of tauopathy, dNmnat overexpression mitigates tau pathology, perhaps by directly targeting phospho‐tau species to the proteasome.15 In mammalian systems, overexpression of Nmnat2 mitigates tau phosphorylation in HEK293 cells in a PP2a‐dependent manner.16 Likewise, Nmnat2 overexpression in a mouse model of accelerated tauopathy (rTg4510) prevented cortical neuron loss and reduced the accumulation of phosphorylated tau, although detailed immunohistochemical examination of tau pathology was not performed.17 In that study, viral overexpression of a nontargeted form of Nmnat1 also mitigated cortical thinning in rTg4510 mice, although the effect on tau aggregation was not assessed. Nmnat1 exerts profound axonal protection in models of axonal degeneration, particularly when it is expressed specifically in the cytoplasm.3 Importantly, cytoplasmic‐targeted Nmnat1 (cytNmnat1) also prevents neurodegeneration in a mouse model of neonatal hypoxic‐ischemic injury, and protects cortical neuronal cultures form NMDA toxicity,8 suggesting that this form of Nmnat might possess neurprotective attributes which go beyond axonal protection. Thus, we sought to examine the impact of cytoplasmic‐targeted Nmnat1 on tau pathology and brain functional connectivity in the transgenic P301S tau mouse model of chronic tauopathy.
Methods
Animals
P301S mouse breeders (PS19 strain) on a mixed B6C3 background were generously provided by the lab of Dr. Virginia Lee.14 cytNmnat1 transgenic mice were generated in the Milbrandt lab as previously described3 on a C57/B6 background. cytNmnat1 Tg mice were bred with P301S mice, and only F1 generation mice (all on a mixed genetic background) were used in the experiments. Only female mice were included. All experiments were performed in accordance with protocols approved by the Washington Univ. Department of Comparative Medicine.
Optical imaging of functional connectivity
Optical intrinsic signal imaging of resting state functional connectivity (fcOIS) in mice was performed as previously described.18, 19 Briefly, mice were anesthetized with i.p. Ketamine/Xylazine mixture (86.9 mg/kg Ketamine, 13.4 mg/kg Xylazine). Once induced, each animal was placed on a heating pad maintained at 37°C (mTCII, Cell Microcontrols) and its head secured in a stereotactic frame. The scalp fur was shaved, and a midline incision was made along the top of the head and the scalp was reflected, exposing approximately 1 cm2 of the skull. The skull was kept moist with an application of mineral oil. Sequential illumination of the skull surface was provided at four wavelengths by a ring of light‐emitting diodes placed approximately 10 cm above the mouse's head. Images were captured using a cooled, frame‐transfer EMCCD camera (iXon 897, Andor Technologies) which was time synchronized and controlled via computer using custom‐written software (MATLAB, Mathworks). Images were acquired at a frame rate of 120 Hz, and seven to nine 5‐min imaging sessions were performed per mouse. Correlation coefficients were generated between regions of interest (ROIs), which were then subjected to Fisher transformation and normalized to the wt mean. The retrosplenial cortex ROI was chosen a priori in order to minimize the statistical effects of multiple comparisons across several ROIs, as the retrosplenial cortex has previously been shown to be susceptible to loss of functional connectivity in the setting of aging or neuronal injury.19, 20 All analysis of fcOIS imaging data and correlation coefficients was carried out by an investigator who was blinded to genotype.
Quantitative PCR
Brain tissue was homogenized directly in Trizol (Invitrogen) using a motorized grinder. Chloroform (1:5) was added, samples were agitated then centrifuged at 13,000g for 15 min at 4 degrees, and the chloroform layer was removed, diluted 1:1 in 70% ethanol, then purified using RNeasy columns and reagents (Qiagen, Valencia, CA). Reverse transcription was performed using high‐capacity RNA‐cDNA kit (Applied Biosystems (ABI), Carlsbad, CA) with 1 ug RNA per 20 uL reaction. Real‐time qPCR was performed using ABI Taqman primers and reagents on an ABI Prizm 7500 thermocycler according to manufacturer's instructions. All mRNA measurements were normalized to beta‐actin (Actb) mRNA levels. Taqman primer sets were obtained from Life Technologies using their proprietary sequences.
Immunohistochemistry
Mice were anesthetized via i.p. injection of pentobarbital, then transcardiac perfusion was performed for 3 min with ice‐cold Dulbecco's modified PBS (dPBS) containing 3 g/L heparin. One hemisphere was dissected on ice then flash frozen in liquid nitrogen for biochemical analyses, whereas the other hemisphere was fixed in 4% paraformaldehyde for 24 h (4°C), then cryoprotected with 30% sucrose in PBS (4°C) for 48 h, then frozen in powdered dry ice, and cut on a freezing sliding microtome. Serial coronal sections (50 μm thick) were collected from the prefrontal cortex to caudal hippocampus. Sections (each separated by 300 mm) were placed in 12 well plates in mesh inserts for staining. Sections were incubated in 0.3% hydrogen peroxide for 10 min, washed in tris‐buffered saline (TBS), blocked for 30 min TBS‐containing 3% goat and 0.25% triton X‐100, then incubated overnight in TBS + 0.25% triton X‐100 with 1% goat serum containing rabbit AT8 (Thermo Scientific, 1:500) or rabbit MC1 (1:1000) primary antibodies at 4°C. Sections were washed then incubated in TBS‐containing biotinylated goat anti‐rabbit secondary antibody (1:1000) for 1 h at room temperature, then washed again in TBS. Sections were then incubated in TBS‐containing 1:400 dilution of streptavidin‐conjugated HRP (VECTASTAIN ABC Elite, Vector Labs, Burlingame, CA). Following thorough washes in TBS, sections were incubated with diaminobenzidine substrate with hydrogen peroxide and nickel chloride for 8 min. Sections were mounted on glass slides and dried overnight, then subjected to serial dehydration and coverslipping with Cytoseal mounting media. For volume measurement, serial sections 300 μm apart were mounted on gelatin‐coated slides and dried overnight, then subjected to cresyl violet staining as previously described.20 Stained sections were imaged with a NanoZoomer slide scanner (Hamamatsu Photonics).
Image analysis was performed by an investigator blinded to genotype, using a previously described method.21 Images were obtained which included the entire hippocampus. For MC1‐stained sections, all sections were stained together in a single batch, so as to eliminate batch effects. Using ImageJ software, images were converted into grayscale. The lightest and darkest stained sections were selected in a blinded manner, and a threshold was set such that the lightest section was less than 5% saturated, whereas the darkest section was less than 90% saturated. The hippocampus boundaries were then selected as the ROI, and this threshold was then applied to all sections, and percent area of staining was determined. For volumetric analysis, ROIs were drawn around the hippocampus and the cortex immediately above the hippocampus in a consistent manner on five consecutive sections from each mouse, starting with the section 300 μm after the first appearance of the anterior hippocampus. The area of each ROI was determined then multiplied by 300 μm (the depth between sequential sections) to yield a volume. Hippocampal and cortical volumes were determined for each mouse, and normalized as percent wt control.
Serial tau extraction and ELISA
Extraction and tau sandwich ELISA were performed as described previously with minor modifications.21 The cortex of each brain was homogenized in 30 μL/mg of RAB buffer [100 mmol/L MES, 1 mmol/L EDTA, 0.5 mmol/L MgSO4, 750 mmol/L NaCl, 20 mmol/L NaF, 1 mmol/L Na3VO4, supplemented by protease inhibitor (Roche) and phosphatase inhibitor (Roche)]. In brief, the samples were centrifuged at 50,000g for 20 min at 4°C using an Optima MAX‐TL Ultracentrifuge (Beckman). The supernatants were collected as RAB soluble fractions and pellets were resuspended in RIPA buffer [150 mmol/L NaCl, 50 mmol/L Tris, 0.5% deoxycholic acid, 1% Triton X‐100, 0.5% SDS–25 mmol/L EDTA, pH 8.0, supplemented by protease inhibitor (Roche) and phosphatase inhibitor (Roche)], 30 μL/mg and centrifuged at 50,000g for 20 min at 4°C. The supernatants were collected as RIPA soluble fractions. The pellets were further resuspended in 70% formic acid, 10 μL/mg, and centrifuged at 50,000g for 20 min at 4°C. The supernatants were collected as 70% formic acid fractions. All fractions were stored in −80°C until analyzed.
To determine human tau levels, ELISA half 96 well plates (Costar) were coated with Tau5 antibody (20 μg/mL) in carbonate buffer pH 9.6 and incubated at 4°C, overnight on a shaker. ELISA plates were washed five times with PBS with a BioTek ELx405 plate washer and blocked with 4% BSA in PBS for 1 h at 37°C. Plates were then washed five times followed by incubating wells with RAB, RIPA, or 70% Formic acid (FA) biochemically extracted soluble brain tissue fractions diluted in sample buffer (0.25% BSA in PBS, 300 nmol/L Tris pH 7.4 supplemented by protease inhibitor) and incubated at 4°C. FA fractions of 70% were neutralized by diluting 1:20 with 1M Tris pH 11 followed by diluting with sample buffer. The next day, plates were washed eight times with PBS followed by the addition of the biotinylated mouse monoclonal anti‐tau antibody HT7 antibody (0.3 μg/mL, Pierce) in 1% BSA in PBS for 1.5 h at 37°C. Plates were then washed eight times in PBS followed by addition of streptavidin‐poly‐horseradish peroxidase‐40 (1:4000, Fitzgerald), for 1.5 h, in the dark, at room temperature. Plates were then washed eight times with PBS, developed with Super Slow ELISA TMB (Sigma) and absorbance read at 650 nm on BioTek Synergy two plate reader. Recombinant human tau was used to create a standard in each plate. The longest recombinant human (hTau40, 441aa) isoforms produced in the laboratory of Eva‐Maria Mandelkow were used as standards in the ELISA assays.
Results
We bred heterozygous P301S tau transgenic mice with heterozygous cytNmnat1 transgenic mice to generate four groups of female mice: wild type (wt), cytNmnat1 transgenic (Nmnat1), P301S tau transgenic (P301S), and cytNmnat1‐P301S double transgenic (DTg). Our Nmnat1 transgenic mice overexpress Nmnat1 which is targeted to the cytoplasm and has been previously demonstrated to exert optimal protection against axonal injury in vitro and in vivo, as well as neuroprotection from neonatal hypoxia‐ischemia.3, 8 We observed a 41‐fold increase in Nmnat1 mRNA in the cortex of our Nmnat1 transgenic mice, and coexpression of P301S tau had no effect on the degree of overexpression (Fig S1).
We next aged our four cohorts of female mice to 12 months of age. At this age severe tau pathology and brain atrophy is evident in female P301S tau‐expressing mice. All surviving mice were then subjected to optical intrinsic signal functional connectivity (fcOIS) imaging. This technique involves optical imaging of blood flow in the cerebral cortex of anesthetized mice and allows analysis of correlative activity patterns in connected cortical regions.18 Decreases in resting‐state connectivity between the bilateral retrosplenial cortices have been observed in aged mice, as well as in several models of neurodegeneration.19, 20, 22 We observed a significant decrease in retrosplenial cortex functional connectivity in P301S mice as compared to non–tau‐expressing mice. Overexpression of Nmnat1 partially rescued tau‐induced fcOIS impairments, suggesting that Nmnat1 can protect cortical neuronal function from tau‐mediated injury (Fig. 1).
Figure 1.
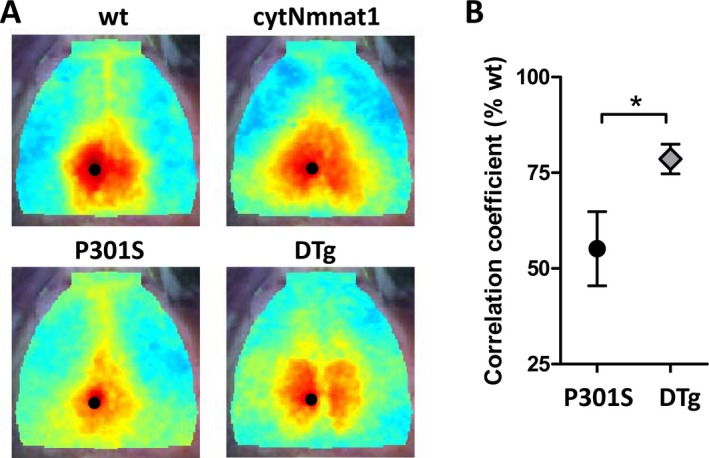
cytNmnat1 overexpression prevents Tau‐induced loss of functional connectivity. Retrosplenial cortex functional connectivity was quantified in anesthetized mice using optical intrinsic signal imaging (fcOIS). (A) Connectivity coefficient maps of the averaged images from n = 5–7 mice/group are shown. The seed region is denoted by the black dot. Correlated cerebral blood flow in the contralateral retrosplenial cortex is shown in wt and cytNmnat1 mice, but is diminished in P301S tau. Warmer colors indicate higher connectivity. cytNmnat1/P301S tau double transgenic (DTg) mice exhibit significant improvement in retrosplenial connectivity. (B) Quantification of retrosplenial connectivity coefficient, present as % of wt. *P < 0.05 by two‐way T‐test.
Based on these observations, we examined tau‐related neuropathology in all four groups of mice. We observed severe pathogenic tau phosphorylation and misfolding in the hippocampi P310S and Nmnat1‐P301S DTg mice, as assessed using immunohistochemistry with the conformation‐specific phospho‐tau antibody MC123 (Fig. 2A). We quantified the degree of MC1 immunoreactivity in the retrosplenial cortex, as well as two hippocampal regions: CA3, which exhibits relatively less tau pathology, and the hilus of the dentate gyrus, which accumulates high levels of pathogenic tau. Although there was considerable variation between mice, we unexpectedly found no significant difference in the degree of hippocampal MC1 immunoreactivity between P310S and DTg mice in any of these regions (Fig. 2B). There was no obvious difference in the distribution or morphology of tau‐reactive structure between P301S and DTg mice, as both genotypes had intracellular neuronal tau deposition and strong neuropil staining. Similarly, staining of cortex and hippocampus with the phosphorylation‐specific tau antibody AT824 also showed no substantial effect of Nmnat1 overexpression on cortical or hippocampal phospho‐tau pathology (Fig. 2C and D).
Figure 2.
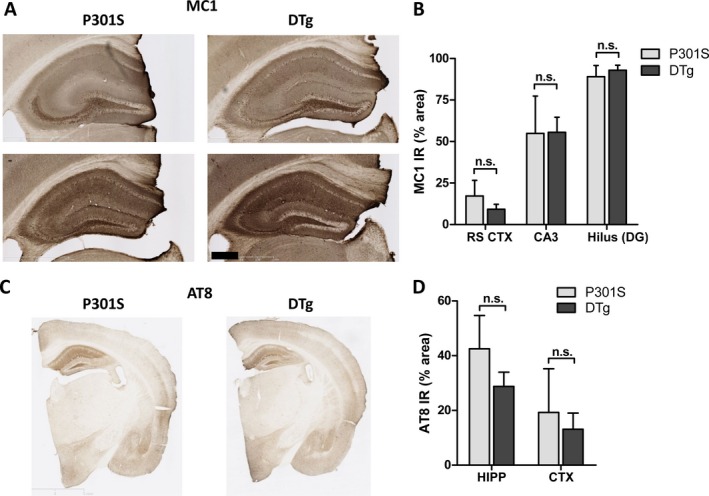
cytNmnat1 overexpression does not prevent accumulation of pathologically phosphorylated and misfolded tau in hippocampus of P301S Tg mice. (A) Representative images showing immunohistochemical analysis of pathogenic tau phosphorylation in the hippocampus of 12‐month female P301S tau Tg and cytNmnat1; P301S double transgenic (DTg) mice using MC1 antibody. Severe tau pathology was observed in both strains of mice. (B) Quantification of MC1 immunoreactivity in retrosplenial cortex (RS CTX) CA3 region and hilus of the dentate gyrus (DG) of the hippocampus of wt, cytNmnat1, P301S, and DTg mice shows similar accumulation of misfolded phospho‐tau in both P301S and DTg hippocampi in these two regions. (C) Representative images showing immunohistochemical detection of phospho‐tau using AT8 antibody in P301S and DTg brains. (D) Quantification of AT8 immunoreactivity in the whole anterior hippocampus (HIPP) and retrospenial cortex (CTX) of both genotypes. N.S: not significant (P > 0.05) by one‐way ANOVA with Bonferroni's posttest. Scale bar = 500 μm.
We also observed atrophy of the hippocampus in P301S tau‐expressing mice, although we again found no significant difference between P310S and DTg mice (Fig. 3A). Cortical thickness was also not statistically different between P301S and DTg genotypes. Finally, we examined the expression of a panel of transcripts related to inflammation and oxidative stress via qPCR in the cortex of all four groups of mice. Again, there was a significant increase in mRNA for several inflammatory markers in tau‐expressing mice, including Tnfa and Gfap, but we observed no significant effect of Nmnat1 overexpression (Fig. 3B). We also quantified transcripts for redox responsive genes, including Hmox1, which encodes the Nrf2‐regulated redox defense protein heme oxygenase 1,25 and Pla2g3, which encodes a phospholipase A2 isoform which has been previous demonstrated to be upregulated in the setting of oxidative stress.26 Both transcripts showed a nonsignificant trend toward upregulation in the hippocampi of tau‐expressing mice, with no significant effect of Nmnat1 coexpression (Fig. S2). Thus, we concluded that although Nmnat1 overexpression prevents tau‐induced loss of functional connectivity, it does not do so by altering accumulation of phospho‐tau, or by mitigating hippocampal atrophy, inflammation, or oxidative stress.
Figure 3.
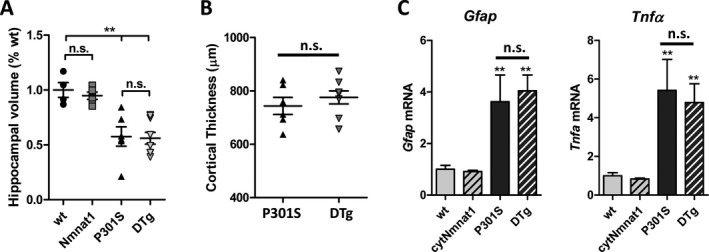
No effect of cytNmnat1 overexpression on brain atrophy or inflammation in P301S tau Tg mice. (A) Volumetric analysis of anterior hippocampus in P301S tau and DTg mice. (B) Quantification of sensorimotor cortex thickness in P301S tau and DTg mice. **P < 0.05 by two‐way ANOVA with Bonferroni's posttest. n.s.: nonsignificant by the same test. (C) qPCR analysis of mRNA levels of Gfap (a marker of astrogliosis) and Tnfa (an inflammatory cytokine) in all four mouse genotypes. **P < 0.05 by one‐way ANOVA as compared to wt. n.s.: nonsignificant.
Previous studies have suggested that Nmnat isoforms possess chaperone function which can accelerate clearance of aggregated species of tau and other proteins.15, 27, 28 We next asked if Nmnat1 exerts any effect on levels of soluble or insoluble (aggregated) tau species. We thus quantified total tau levels by ELISA in cortical samples subjected to serial extraction to look at salt‐soluble, detergent‐soluble, and detergent‐insoluble species.21 We observed no significant difference in the amount of RAB (salt) or RIPA (detergent)‐soluble tau levels between P301S and DTg mice (Fig. 4A and B). However, we found a small but statistically significant decrease in the amount of tau in the detergent‐insoluble (70% formic acid extracted) fraction in DTg mice as compared to P301S mice (Fig. 4C). Thus, Nmnat1 appears to modestly reduce the accumulation of insoluble tau aggregates in the cerebral cortex.
Figure 4.
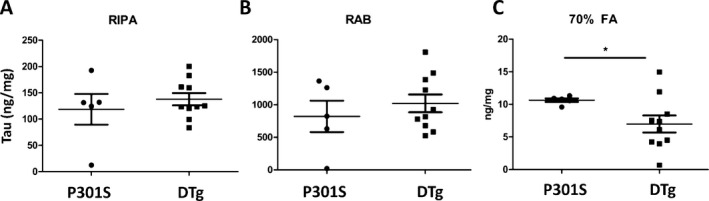
Reduction in accumulation of insoluble tau aggregates in cerebral cortex by cytNmnat1. Cerebral cortex tissue samples were subjected to serial extraction in RIPA, RAB, and 70% Formic acid (FA) buffers. Total tau was then quantified in each set of fractions via ELISA. No significant differences were observed in soluble forms of tau (RIPA and RAB fractions, panels A and B). However, a significant decrease in insoluble, aggregated tau (70% formic acid fraction) was observed in DTg cortex as compared to P301S cortex. *P < 0.05 by two‐tailed T‐test.
Discussion
Tau pathology is closely correlated with cognitive impairment in Alzheimer's Disease and is increasingly implicated as a critical mediator of neurodegeneration in a growing number of diseases.9 Thus, identification of pathways which mitigate tau toxicity has tremendous implications for understanding or treating neurodegenerative diseases. Nmnat proteins have been well established as potent protective factors in the setting of axonal injury, although the specific mechanisms mediating this protection are still under investigation.1, 2, 7 Drosophila Nmnat has been shown to be protective in a fly model of tauopathy,15 whereas viral overexpression of Nmnat1 or Nmnat2 can protect cortical neurons from tau‐induced cell death in rTg4510 Tau transgenic mice.17 However, no previous studies have evaluated cytNmnat1 in a tau model, or have they provided detailed immunohistochemical analysis of the effect of Nmnat overexpression on pathologic forms of tau. Herein, we present data which suggest that overexpression of cytNmnat1 in a mouse model of chronic tauopathy (human P301S tau transgenic mice) fails to prevent accumulation of detergent‐soluble misfolded and phosphorylated tau, or downstream inflammation, oxidative stress, and hippocampal atrophy. However, Nmnat1 does preserve cortical functional connectivity, and mitigates the accumulation of insoluble tau aggregates in the brain. These results suggest that optimization of Nmnat1 activity in the brain might help to preserve neuronal function in the setting of severe tau pathology, such as that seen in the hippocampus and cortex of Alzheimer's disease patients.
Several previous studies have examined the effect of Nmnat isoforms on tau pathology. In a drosophila model of tauopathy, dNmnat overexpression prevented neuronal vacuolar degeneration and reduced levels of phospho‐tau (including AT8‐positive species) in fly heads.15 In this model, Nmnat appeared to interact directly with pathogenic tau species and promote their ubiquitin‐mediated degradation by the proteasome. Indeed, dNmnat also suppresses neuronal toxicity associated with the toxic protein SCA2 in drosophila via a proteasome‐dependent chaperone‐like mechanism.27 Studies in yeast suggest that this chaperone function may be an NAD+‐independent property of Nmnat.28 A previous mouse experiment showed an Nmnat2‐mediated decrease in phospho‐tau and misfolded tau in the brains of the r4510 model of tauopathy by Western blotting with MC1 and CP‐13 antibodies.17 We observed no effect of cytoplasmic Nmnat1 on phospho‐tau pathology in our study, suggesting that Nmnat1 may not possess the same ability as Nmnat2 to act as a chaperone to facilitate degradation of phospho‐tau. It is notable that the previous study also examined MC1 levels in much younger (3.5‐month) mice, which could also contribute to this discrepancy with our findings in a different model (P301S) at a much older age (12 months). Our observation that Nmnat1 overexpression leads to a specific decrease in detergent‐insoluble tau aggregates suggests that Nmnat1 may somehow target certain insoluble tau species for degradation, thus leaving the soluble tau pool unaffected. Thus, it is possible that different Nmnat isoforms have unique affinities for different tau species, although this hypothesis has not yet been formally tested. Although we did not observe any effect of cytNmnat1 overexpression on overall proteasome activity in transfected cells (data not shown), future detailed analysis of the effect of different Nmnat isoforms on proteolytic pathways (including autophagy and the proteasome) and chaperone systems would be valuable.
We also observed no effect of Nmnat1 overexpression on tau‐induced brain atrophy in our study, in contrast to the only other previous murine study employing AAV‐expressed Nmnat1 or Nmnat2 in the rTg4510 tau model.17 This may again be due to the different mouse tauopathy models used in these two experiments, as rTg4510 mice overexpress human tau with a different mutation (P301L) and have a more accelerated neurodegenerative course, suggesting that different cytotoxic mechanisms may be at play. Again, we looked at much older mice (12 months vs. 4 months) with more severe disease. Future studies could examine cytNmnat1 overexpression or deletion in the rTg4510 mouse tauopathy model, or examine earlier time points in the disease progression of P301S mice to address these discrepancies. It is still unclear to what extent the chaperone activity of Nmnat isoforms contributes to their relative neuroprotective effects in vivo. It is possible that the preservation of function connectivity that we have observed is related to the ability of Nmnat1 to mitigate the accumulation of detergent‐insoluble tau species, although a more detailed understanding of the specific tau species which mediate neuronal dysfunction, as well as the impact of Nmnat1 expression on the accumulation of these specific molecules is needed to formally reach this conclusion.
Several studies have suggested that in addition to their known effect on axons that Nmnat isoforms can also prevent neuronal cell death caused by toxic protein overexpression in flies (polyglutamine‐expanded SCA2 or mutant human tau) or by hypoxic‐ischemic injury in mice.8, 15, 27 Our data suggest that cytNmnat1 does not prevent neuronal loss in the P301S chronic tauopathy model, as no diminution of tau‐induced hippocampal or cortical atrophy was observed. One interpretation of our data is that although Nmnat1 is unable to prevent pathogenic tau phosphorylation and subsequent brain atrophy and inflammation, it may be able to preserve axonal function in remaining neurons, leading to preservation of functional connectivity. This would be in keeping with the known axonoprotective function of cytNmnat1. Although axonal degeneration is a major pathologic concern in the spinal cord and peripheral nervous system, its role in chronic neurodegenerative diseases of the brain has also been described.29 Axonal injury and degeneration are present in post mortem brains from patients with AD, and both amyloid‐beta and tau‐expressing mouse models of AD exhibit axonal damage which likely contributes to observed cognitive deficits.30, 31, 32, 33, 34, 35 Thus, targeting Nmnat1 in AD and other chronic neurodegenerative diseases of the CNS could potentially have beneficial effects by preserving axonal function in injured neurons, although more detailed examination of both the mechanisms of axonal injury in tau models, as well as of the effect of Nmnat isoforms on this pathology, is needed in the future.
In summary, our data demonstrate that cytNmnat1 can preserve the functional connectivity of cortical neurons in vivo in the setting of severe tau‐mediated neurodegeneration without significantly reducing phospho‐tau pathology or brain atrophy. Further investigation into the mechanisms by which different tau isoforms interact with various classes of aggregates tau species, as well as development of novel methods to optimize Nmnat expression in the aging brain will help to illuminate Nmnat proteins as potential therapeutic targets for age‐related neurodegenerative diseases.
Authors Contribution
E. S. M. and D. M. H. conceived and designed the experiments. E. S. M., D. D. X., T. P., Y. S., A. Q. B., Y. W., S. L. F., and R. S. performed the experiments and assisted in data analysis. E. S. M., D. D. X., T. P., and D. M. H. wrote the manuscript. J. P. C. and J. M. assisted with data interpretation and provided editorial input.
Conflict of Interest
The authors declare that they have no conflict of interest.
Supporting information
Figure S1. cytNmnat expression is not significantly impacted by P301S transgene expression. Nmnat1 mRNA was quantified by qPCR in cortex samples. *P < 0.05 by one‐way ANOVA with Bonferroni's posttest as compared to wt.
Figure S2. Inflammation and oxidative stress response in P301S tau mice are not altered by cytNmnat1 overexpression. Cortex samples from wt, cytNmnat1, P301S, and DTg mice were subjected to qPCR analysis of mRNA expression of Il6 (inflammatory cytokine), Hmox1 and Pla2g3 (oxidative stress responsive genes), and Hspa1a (a subunit of the chaperone hsp70). A trend toward increase in all markers was observed in tau‐expressing mice. However, no significant difference between P301S and DTg mice was observed for any of these markers. **P < 0.05 by one‐way ANOVA as compared to wt. n.s. nonsignificant.
Acknowledgments
E.S.M was funded by NINDS grant K08NS079405. D.M.H was funded by the Tau Consortium and The JPB Foundation. J.M was funded by NIH grants NS087632 and AG013730. The authors thank Dr. Peter Davies for MC1 antibody and Dr. Virginia Lee for P301S tau Tg mice.
References
- 1. Coleman MP, Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci 2010;33:245–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004;305:1010–1013. [DOI] [PubMed] [Google Scholar]
- 3. Sasaki Y, Vohra BP, Baloh RH, et al. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci 2009;29:6526–6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhai RG, Cao Y, Hiesinger PR, et al. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol 2006;4:e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sasaki Y, Vohra BP, Lund FE, et al. Nicotinamide mononucleotide adenylyl transferase‐mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci 2009;29:5525–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 2010;8:e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci 2006;26:8484–8491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verghese PB, Sasaki Y, Yang D, et al. Nicotinamide mononucleotide adenylyl transferase 1 protects against acute neurodegeneration in developing CNS by inhibiting excitotoxic‐necrotic cell death. Proc Natl Acad Sci U S A 2011;108:19054–19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ballatore C, Lee VM, Trojanowski JQ. Tau‐mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 2007;8:663–672. [DOI] [PubMed] [Google Scholar]
- 10. Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 2009;461:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vos SJ, Xiong C, Visser PJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol 2013;12:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dumanchin C, Camuzat A, Campion D, et al. Segregation of a missense mutation in the microtubule‐associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet 1998;7:1825–1829. [DOI] [PubMed] [Google Scholar]
- 13. Allen B, Ingram E, Takao M, et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 2002;22:9340–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007;53:337–351. [DOI] [PubMed] [Google Scholar]
- 15. Ali YO, Ruan K, Zhai RG. NMNAT suppresses tau‐induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum Mol Genet 2012;21:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheng XS, Zhao KP, Jiang X, et al. Nmnat2 attenuates Tau phosphorylation through activation of PP2A. J Alzheimers Dis 2013;36:185–195. [DOI] [PubMed] [Google Scholar]
- 17. Ljungberg MC, Ali YO, Zhu J, et al. CREB‐activity and nmnat2 transcription are down‐regulated prior to neurodegeneration, while NMNAT2 over‐expression is neuroprotective, in a mouse model of human tauopathy. Hum Mol Genet 2012;21:251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. White BR, Bauer AQ, Snyder AZ, et al. Imaging of functional connectivity in the mouse brain. PLoS ONE 2011;6:e16322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bero AW, Bauer AQ, Stewart FR, et al. Bidirectional relationship between functional connectivity and amyloid‐beta deposition in mouse brain. J Neurosci 2012;32:4334–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Musiek ES, Lim MM, Yang G, et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest 2013;123:5389–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yanamandra K, Kfoury N, Jiang H, et al. Anti‐tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 2013;80:402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bauer AQ, Kraft AW, Wright PW, et al. Optical imaging of disrupted functional connectivity following ischemic stroke in mice. NeuroImage 2014;99:388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weaver CL, Espinoza M, Kress Y, et al. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging 2000;21:719–727. [DOI] [PubMed] [Google Scholar]
- 24. Biernat J, Mandelkow EM, Schroter C, et al. The switch of tau protein to an Alzheimer‐like state includes the phosphorylation of two serine‐proline motifs upstream of the microtubule binding region. EMBO J 1992;11:1593–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vile GF, Basu‐Modak S, Waltner C, et al. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc Natl Acad Sci U S A 1994;91:2607–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martinez‐Garcia A, Sastre I, Recuero M, et al. PLA2G3, a gene involved in oxidative stress induced death, is associated with Alzheimer's disease. J Alzheimers Dis 2010;22:1181–1187. [DOI] [PubMed] [Google Scholar]
- 27. Zhai RG, Zhang F, Hiesinger PR, et al. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 2008;452:887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ocampo A, Liu J, Barrientos A. NAD+ salvage pathway proteins suppress proteotoxicity in yeast models of neurodegeneration by promoting the clearance of misfolded/oligomerized proteins. Hum Mol Genet 2013;22:1699–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Higuchi M, Lee VM, Trojanowski JQ. Tau and axonopathy in neurodegenerative disorders. Neuromolecular Med 2002;2:131–150. [DOI] [PubMed] [Google Scholar]
- 30. Spittaels K, Van den Haute C, Van Dorpe J, et al. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four‐repeat human tau protein. Am J Pathol 1999;155:2153–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Probst A, Gotz J, Wiederhold KH, et al. Axonopathy and amyotrophy in mice transgenic for human four‐repeat tau protein. Acta Neuropathol 2000;99:469–481. [DOI] [PubMed] [Google Scholar]
- 32. Xiao AW, He J, Wang Q, et al. The origin and development of plaques and phosphorylated tau are associated with axonopathy in Alzheimer's disease. Neurosci Bull 2011;27:287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Christensen DZ, Huettenrauch M, Mitkovski M, et al. Axonal degeneration in an Alzheimer mouse model is PS1 gene dose dependent and linked to intraneuronal Abeta accumulation. Front Aging Neurosci 2014;6:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leroy K, Bretteville A, Schindowski K, et al. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am J Pathol 2007;171:976–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wirths O, Weis J, Kayed R, et al. Age‐dependent axonal degeneration in an Alzheimer mouse model. Neurobiol Aging 2007;28:1689–1699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. cytNmnat expression is not significantly impacted by P301S transgene expression. Nmnat1 mRNA was quantified by qPCR in cortex samples. *P < 0.05 by one‐way ANOVA with Bonferroni's posttest as compared to wt.
Figure S2. Inflammation and oxidative stress response in P301S tau mice are not altered by cytNmnat1 overexpression. Cortex samples from wt, cytNmnat1, P301S, and DTg mice were subjected to qPCR analysis of mRNA expression of Il6 (inflammatory cytokine), Hmox1 and Pla2g3 (oxidative stress responsive genes), and Hspa1a (a subunit of the chaperone hsp70). A trend toward increase in all markers was observed in tau‐expressing mice. However, no significant difference between P301S and DTg mice was observed for any of these markers. **P < 0.05 by one‐way ANOVA as compared to wt. n.s. nonsignificant.