

Abstract
Objective
Myasthenia gravis (MG) is an autoimmune condition in which neurotransmission is impaired by binding of autoantibodies to acetylcholine receptors (AChR) or, in a minority of patients, to muscle specific kinase (MuSK). There are differences in the dominant IgG subclass, pathogenic mechanisms, and treatment responses between the two MG subtypes (AChR or MuSK). The antibodies are thought to be T‐cell dependent, but the mechanisms underlying their production are not well understood. One aspect not previously described is whether defects in central and peripheral tolerance checkpoints, which allow autoreactive B cells to accumulate in the naive repertoire, are found in both or either form of MG.
Methods
An established set of assays that measure the frequency of both polyreactive and autoreactive B cell receptors (BCR) in naive populations was applied to specimens collected from patients with either AChR or MuSK MG and healthy controls. Radioimmuno‐ and cell‐based assays were used to measure BCR binding to AChR and MuSK.
Results
The frequency of polyreactive and autoreactive BCRs (n = 262) was higher in both AChR and MuSK MG patients than in healthy controls. None of the MG‐derived BCRs bound AChR or MuSK.
Interpretation
The results indicate that both these MG subtypes harbor defects in central and peripheral B cell tolerance checkpoints. Defective B cell tolerance may represent a fundamental contributor to autoimmunity in MG and is of particular importance when considering the durability of myasthenia gravis treatment strategies, particularly biologics that eliminate B cells.
Introduction
Myasthenia gravis (MG) is a chronic autoantibody‐mediated disorder of the neuromuscular junction characterized by muscle weakness and fatigability.1 In the majority of patients the autoantibodies target the acetylcholine receptor (AChR),2 while a smaller population is defined by autoantibodies targeting muscle specific kinase (MuSK).3 Although the immunopathology is directly mediated by these autoantibodies, their mechanisms differ. In AChR MG the autoantibodies are primarily of the IgG1 subclass4 and lead to the loss of AChR through internalization and by localized complement‐mediated postsynaptic damage. The majority of autoantibodies that recognize MuSK are of the IgG4 subclass and do not cause internalization or fix complement. Instead, MuSK autoantibodies inhibit the agrin‐LRP4‐MuSK‐AChR clustering pathway by preventing agrin‐activated LRP4 binding to MuSK, leading to dispersal of the AChRs.5, 6 The two subtypes of MG also differ in clinical presentation and in response to immunotherapies.7 For example, B cell depletion therapy has shown encouraging results in MuSK MG but appears less effective in AChR MG.8, 9 This observation suggests that there might be differences in the underlying B cell populations that give rise to the autoantibodies.
To evade the development of an immune response against self, two separate tolerance mechanisms counter‐select B cells during their development.10 The first is a central tolerance checkpoint in the bone marrow between the early immature and immature B cell development stages, which removes a large population of B cells that express self‐reactive antibodies.11 The second checkpoint selects against self‐reactive new emigrant/transitional B cells before they enter the mature naive B cell compartment. Deficiencies in the integrity of these tolerance mechanisms can be demonstrated through quantifying the frequency of both polyspecific and autoreactive B cells downstream of each checkpoint. A number of autoimmune diseases include central and peripheral B cell checkpoints that fail to enforce tolerance.12, 13, 14
Despite the detailed understanding of MG pathophysiology, there are no reports of whether defects in autoimmune regulation, namely tolerance, contribute to this disease. Here we asked whether the naive B cell repertoire in MG shows evidence of compromised tolerance, due to checkpoint defects, leading to the accumulation of autoreactive B cells in the naive compartment. Given the divergent immunobiology underlying AChR and MuSK MG, we included study subjects representing both disease subtypes to determine whether there are differences in B cell tolerance integrity between the two forms of the disease.
Materials and Methods
Study subjects
Patients were recruited from the Yale Myasthenia Gravis Clinic or the University of Kansas, Department of Neurology. Peripheral blood was obtained from healthy controls and patients with MG after providing informed consent. Peripheral blood mononuclear cells (PBMC) were isolated from the blood, aliquoted and cryopreserved in liquid nitrogen until use. The MG patients were primarily immunotherapy naive (Table 1). All patients had clinical and electrodiagnostic features consistent with MG and were tested for either AChR or MuSK autoantibodies using a commercial radioimmunoassay (RIA) as part of their clinical evaluation. Collected clinical data included demographics, duration of disease, immunosuppressive medications, thymoma/thymectomy status and Myasthenia Gravis Foundation of America (MGFA) Clinical Classification (Table 1). The healthy controls had no reported history of autoimmune disease, or malignancies and no acute or chronic infections. These controls were matched to the patients as closely as possible with regard to age and gender (Table 1). Patients and healthy donors were further clinically assessed for the possibility of co‐morbid autoimmunity; no such co‐morbidity was present. Additionally, serological antibody screening for subclinical autoimmune disease included measurement of autoantibodies against nuclear antigens (ANA panel) or extractable nuclear antigens (ENA panel). These tests were performed, using commercial assays, by the Department of Laboratory Medicine at Yale New Haven Hospital (Table 1). Direct sequencing of the PTPN22 R620W polymorphism region was performed on all subjects in the study.15
Table 1.
Clinical demographics.
| Subject ID | Autoantibody status (titer1) | Gender | Age (yrs) | Disease duration (yrs) | Diagnosis/Disease status (MGFA) | Symptoms | Treatment | Thymus status | Autoimmune comorbidity (ANA and ENA assays) |
|---|---|---|---|---|---|---|---|---|---|
| MG‐AChR‐1 | AChR (16.6 nmol/L) | F | 66 | <1 | Generalized MG (crisis/exacerbation)‐grade IIIb | Dysarthria, dysphagia, ptosis, diplopia, fatigue, | None | No thymectomy, no thymoma | neg, neg |
| MG‐AChR‐2 | AChR (2.54 nmol/L) | M | 60 | <1 | Generalized MG (crisis/exacerbation)‐grade IIIa | Ptosis, dysphagia, dyspnea | None | No thymectomy, no thymoma | neg, neg |
| MG‐AChR‐3 | AChR (0.09 nmol/L) | M | 17 | 1–2 | Ocular MG‐grade I | Ptosis | None | No thymectomy, no thymoma | neg, neg |
| MG‐MuSK‐1 | MuSK | M | 49 | 25 | IIIb | Diplopia | Low dose prednisone every other day, pyridostigmine bromide | No thymectomy, no thymoma | neg, neg |
| MG‐MuSK‐2 | MuSK | F | 28 | 8 | IIIb | Diplopia, dysarthria | Low dose prednisone every other day | No thymectomy, no thymoma | neg, neg |
| HD‐1 | – | F | 30 | – | – | – | – | – | neg, neg |
| HD‐2 | – | M | 47 | – | – | – | – | – | neg, neg |
Titers were available for AChR only1. Three AChR MG patients, two MuSK MG patients and two healthy control donors (HD) were recruited for the study. AChR MG patients were immunotherapy naive. MuSK MG patients were treated with low dose prednisone every other day and/or with pyridostigmine bromide (Mestinon®). All patients were non‐carriers of the PTPN22 R620W polymorphism. Under thymus status, “no thymoma”, indicates that a thymoma was not detected by chest imaging. ANA, anti‐nuclear antibody; ENA, extractable nuclear antigen antibodies (six antigen preparations were tested: Smith, Smith/RNP, SS‐A (Ro), SS‐B (La), Scl‐70 (topoisomerase I) and Jo‐1).
Study approval
Specimens originating from patients were collected after informed written consent was obtained under a protocol approved by the Human Research Protection Program at Yale School of Medicine.
Cell staining and sorting
Single B cells were isolated as previously described.16 Briefly, peripheral B cells from the MG patients and healthy controls were isolated by positive selection using CD20 magnetic beads (Miltenyi). Single CD19+CD21loCD10+IgMhiCD27– new emigrant/transitional and CD19+CD21+CD10–IgM+CD27– mature naive B cells were sorted on a FACSAria flow cytometer (BD) into 96‐well PCR plates and immediately frozen on dry ice.
Recombinant antibody production
Reverse transcription of the RNA and RT‐PCR reactions, primer sequences, cloning strategy, expression vectors, antibody expression, and purification were performed as previously described.11, 17 Immunoglobulin sequences were analyzed using the IgBLAST tool18 on the NCBI website (http://www.ncbi.nlm.nih.gov/projects/igblast) or the IMGT/V‐QUEST tool available at the International ImMunoGeneTics information system (IMGT) website (http://www.imgt.org).19 Clones were selected for recombinant expression based on successful amplification of both the heavy and light chain variable region and sequence fidelity with the germline variable region gene segments (Table S1).
ELISA
Cell culture supernatants containing recombinant IgG (rIgG) were tested for polyspecificity using plates coated with dsDNA, LPS or recombinant human insulin (all from Sigma) using a previously described approach.11 HEp‐2 binding was tested using purified rIgG and a commercially available ELISA kit (INOVA) according to the manufacturer's instructions with minor modifications that were previously described.13
MG autoantigen specificity
Binding to AChR or MuSK was tested using purified rIgG and a commercial radioimmunoassay (RIA, RSR Ltd). These results were confirmed by testing for binding to human embryonic kidney cells transfected with DNAs encoding MuSK or the four subunits of the adult isoform of AChR and rapsyn as previously described.20, 21
Statistics and graphing
Differences between patient groups and healthy controls were analyzed for statistical significance with Kruskal‐Wallis tests using Prism software (GraphPad). P‐values of less than 0.05 were considered statistically significant. Power calculations were performed based on the large and reproducible effect size derived from the assay in earlier studies.12, 13, 14,16 The 3‐D plots of the polyreactivity ELISA data were analyzed and plotted using the R statistical computing environment and the R package ggplot2.22, 23
Results
Study design and approach
To determine whether B cell tolerance is maintained in MG patients we utilized a well described approach11, 24, 25, 26 that assesses the fidelity of the two tolerance checkpoints guiding B cell development. The approach is applied to measure the frequency of polyreactive and autoreactive B cell receptors (BCR) in the naive repertoire, which associates with the extent of negative selection thereby providing an assessment of the checkpoint integrity. Specifically, peripheral blood new emigrant/transitional and mature naive B cells, which are each respectively downstream of the central and peripheral B cell tolerance checkpoints, were sorted into single wells. Recombinant antibodies were generated from each single cell, as a means to represent their BCR, which were then tested for reactivity against a panel of defined antigens. The frequency of BCRs from new emigrant/transitional B cells (metric for central tolerance integrity) and the frequency of BCRs from mature naive B cells (metric for peripheral tolerance integrity) expressing poly‐ and/or autoreactive antibodies were determined and compared to healthy controls, where tolerance is properly established. The approach produces a large effect size when tolerance is compromised; therefore we enrolled five MG patients and two healthy controls. We selected our patients based on their well‐defined diagnosis of MG and the absence of aggressive immunotherapy that may alter the tolerance assay results. The MG cohort included two immunotherapy naive generalized AChR MG patients; one immunotherapy naive ocular AChR MG patient and two MuSK MG patients who had received low dose corticosteroids but had not begun aggressive immunotherapy (Table 1). All patients and controls were negative for the PTPN22*R620W variant risk factor allele, which is associated with dysfunctional central tolerance.15 Patients and controls were also negative for co‐morbid autoimmunity; both clinically and based on ANA and ENA screening (Table 1). A total of 109 new emigrant/transitional and 88 mature naive BCRs were cloned from the MG cohort (Table S2) along with 33 new emigrant/transitional and 32 mature naive BCRs from the healthy controls (Table S2). Recombinant antibodies were produced in duplicate from each of these cloned BCRs, totaling 394 MG and 130 control antibodies available for the study.
Central tolerance checkpoint fidelity
The central tolerance checkpoint integrity was measured by determining the frequency of new emigrant/transitional B cells that were characterized as polyreactive through displaying reactivity toward three structurally distinct antigens: dsDNA, lipopolysaccharide (LPS) and insulin. The fraction of BCRs that were positive for binding all three antigens from the healthy controls was compared to the same fraction found in the three AChR and two MuSK MG patients (Fig. 1A and Fig. S1). The median fraction of polyreactive BCRs in the healthy controls was 8.9%, while in the AChR and MuSK MG patients the fraction was 23.3% and 51.7% respectively (Fig. 1B). These values were statistically different (P = 0.029). Collectively, in all five MG patients the mean fraction of polyreactive BCRs was 36.1% ± 14.8 and in the two healthy controls, where central B cell tolerance is established normally, the mean fraction of polyreactive BCRs was 8.9% ± 3.1. These respective values are in agreement with previously reported frequencies of polyreactive new emigrant/transitional BCRs in individuals with autoimmune diseases that include well‐characterized central tolerance abnormalities and in healthy controls.11, 24, 25, 26 These results indicate that the B cell central tolerance checkpoint is dysfunctional in both AChR and MuSK MG patients.
Figure 1.
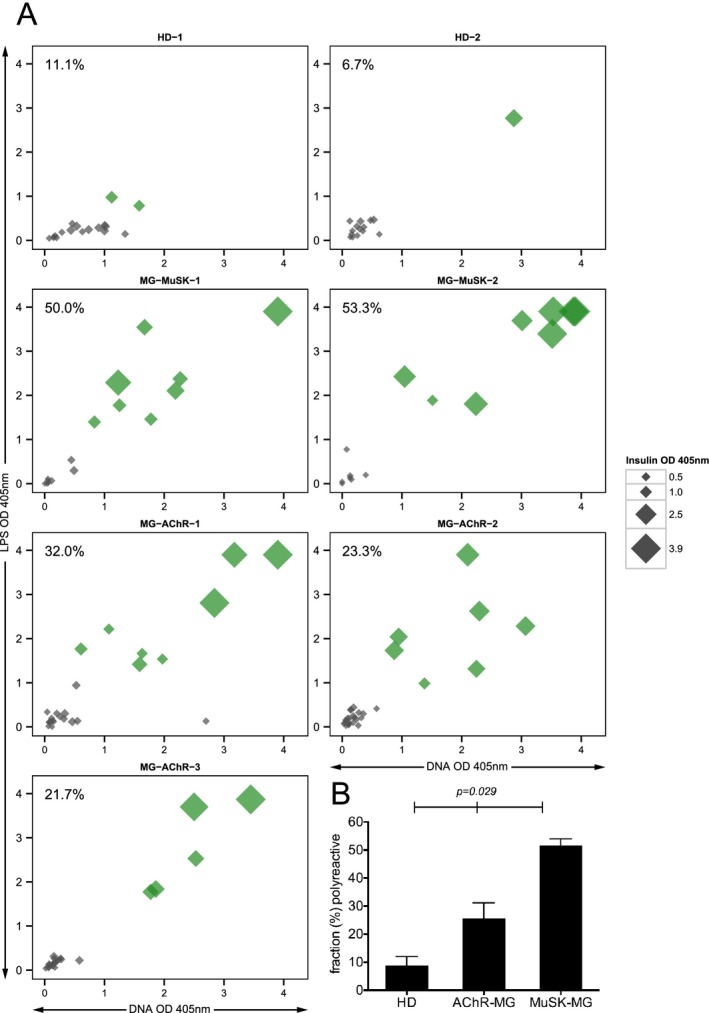
The fidelity of the central tolerance checkpoint is compromised in patients with MG . The integrity of the central B cell tolerance checkpoint was examined through quantifying the fraction of new emigrant/transitional B cells that are polyreactive. Recombinant antibodies, representing the B cell receptors (BCR), were generated from single new emigrant/transitional (CD19+ CD21lo CD10+IgMhi CD27−) B cells derived from three AChR MG patients (MG‐AChR‐1, MG‐AChR‐2, MG‐AChR‐3), two MuSK MG patients (MG‐MuSK‐1, MG‐MuSK‐2) and two healthy controls (HD‐1 and HD‐2). The antibodies were then tested for reactivity with a set of three structurally distinct antigens: dsDNA, lipopolysaccharide (LPS) and insulin by ELISA. The antibodies were tested at 1.0 μg/mL and three additional four‐fold serial dilutions (all shown in Fig. S1). The consolidated three‐dimensional plots (A) summarize the reactivity of each antibody (at the highest concentration of 1.0 μg/mL) from the subjects toward each antigen in the ELISA. The absorbance values for LPS (y‐axis), dsDNA (x‐axis) and insulin (data point size) are plotted together. Green symbols indicate antibodies that were positive for binding all three antigens. The values shown in the top left corner of each graph indicate the fraction of antibodies that were polyreactive. The mean fraction of polyreactive antibodies was plotted (B) for each of the three subject groups; HD and two MG disease subtypes (AChR and MuSK). Statistical differences are shown when significant.
Peripheral tolerance checkpoint fidelity
The peripheral tolerance checkpoint integrity was measured by first determining the frequency of mature naive‐derived BCRs that displayed polyreactivity and secondly by determining the frequency of these cells that displayed autoreactivity toward a human epithelial type 2 (HEp‐2) cell lysate. The median fraction of polyreactive mature naive‐derived BCRs in the healthy controls was 11.7%, while in the AChR and MuSK MG patients the fraction was 30.0% and 27.1% respectively (Fig. 2A, 2B and Fig. S2). These values were statistically different (P = 0.029).
Figure 2.
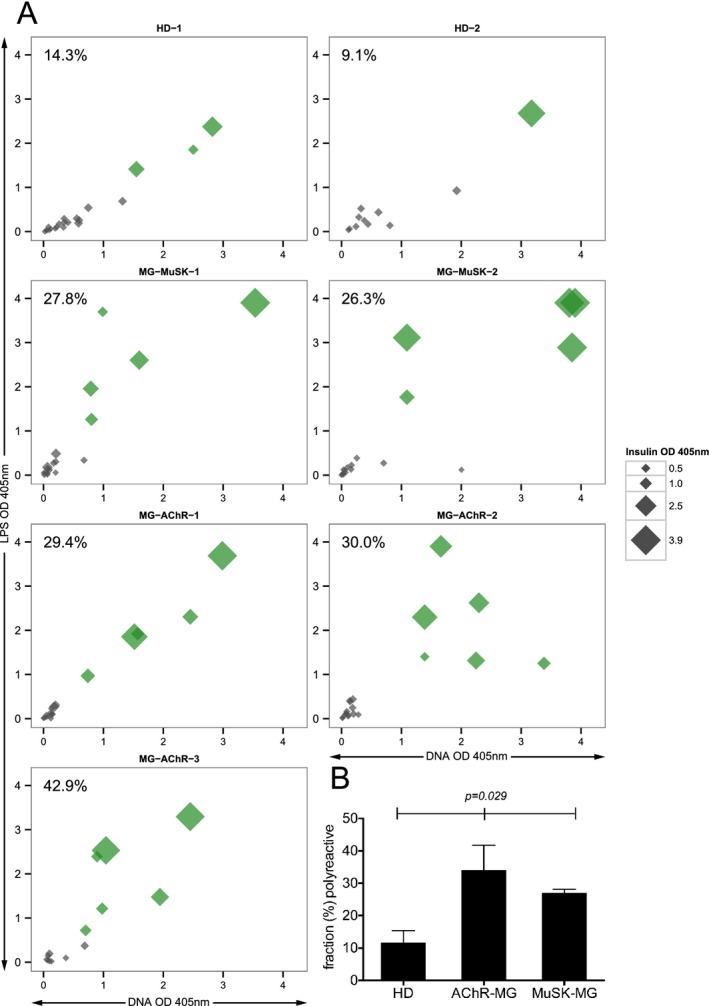
The fidelity of the peripheral tolerance checkpoint is compromised in patients with MG. The integrity of the peripheral B cell tolerance checkpoint was examined through quantifying the fraction of mature naive B cells that are polyreactive or autoreactive. Recombinant antibodies, representing the BCR, were generated from single mature naive B cells (CD19+ CD21+ CD10−IgM+ CD27−) derived from three AChR MG patients (MG‐AChR‐1, MG‐AChR‐2, MG‐AChR‐3), two MuSK MG patients (MG‐MuSK‐1, MG‐MuSK‐2) and two healthy controls (HD‐1 and HD‐2). The antibodies were then tested by ELISA for polyreactivity with a set of three structurally distinct antigens: dsDNA, lipopolysaccharide (LPS) and insulin or for autoreactivity with a HEp‐2 cell lysate. The consolidated three‐dimensional plots (A) summarize the reactivity of each antibody (at the highest concentration of 1.0 μg/mL) from the subjects toward each antigen in the ELISA. The absorbance values for LPS (y‐axis), dsDNA (x‐axis) and insulin (data point size) are plotted together. Green symbols indicate antibodies that were positive for binding all three antigens. The values shown in the top left corner of each graph indicate the frequency (%) of polyreactive antibodies. The mean fraction of polyreactive (B) antibodies was plotted for each of the three subject groups; HD and two MG disease subtypes (AChR and MuSK). Statistical differences are shown when significant. BCR, B cell receptors.
Using the HEp‐2 cell lysate ELISA, we found that the median fraction of autoreactive mature naive‐derived BCRs in the healthy controls, AChR and MuSK MG patients were 23.2%, 52.9% and 62.0% respectively (Fig. 3A and B). Although trending toward significance, the variance between the groups did not reach statistical significance (P = 0.10). Collectively, in the two healthy control donors, where peripheral B cell tolerance is established normally, the mean fraction of HEp‐2‐reactive mature naive‐derived BCRs was 23.2% ± 5.9. In all five MG patients the mean fraction of HEp‐2‐reactive mature naive‐derived BCRs was 56.2% ± 11.0. These respective values, along with those acquired from the polyreactivity assay, are in agreement with those reported for both healthy individuals and individuals with autoimmune diseases that include well‐characterized peripheral tolerance abnormalities.11, 24, 25, 26 Thus, our findings suggest that AChR and MuSK MG patients fail to remove autoreactive B cells between the new emigrant/transitional and mature naive B cell stages, revealing a second dysfunctional tolerance mechanism in the periphery.
Figure 3.
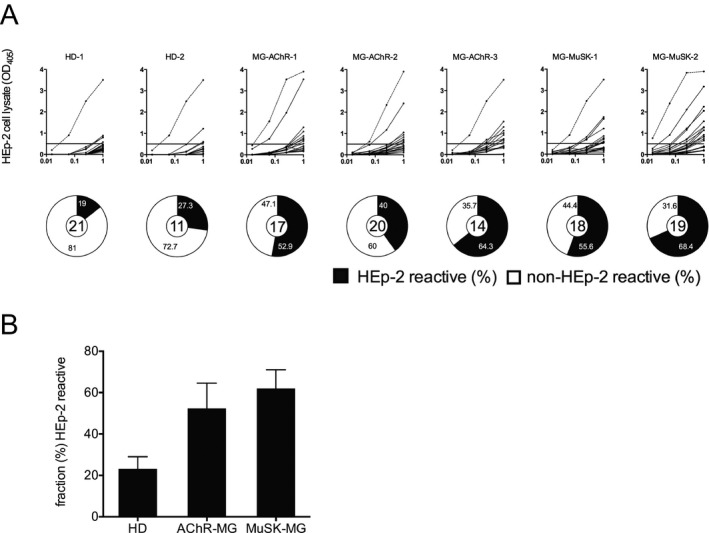
The fidelity of the peripheral tolerance checkpoint is compromised in patients with MG: Peripheral tolerance checkpoint HEp‐2 ELISAs. Recombinant antibodies, representing the B cell receptors (BCR), from single mature naive B cells (CD19+ CD21+ CD10−IgM+ CD27−) derived from three AChR MG patients (MG‐AChR‐1, MG‐AChR‐2, MG‐AChR‐3) and two MuSK MG patients (MG‐MuSK‐1, MG‐MuSK‐2) were cloned, expressed and then tested for reactivity against a HEp‐2 cell lysate by ELISA (A). Dotted lines show the positive control, a monoclonal antibody (ED38) cloned from a VpreB+L+ peripheral B cell that is highly poly‐ and self‐reactive. Solid lines show the binding curve of each cloned recombinant antibody. Horizontal lines at 0.5 OD mark the cutoff for positive reactivity. The control group includes antibodies from two healthy individuals (HD‐1, HD‐2). For each individual subject the fraction (%) of self‐reactive mature naive‐derived BCRs is summarized in the pie charts with the total number of tested clones in the center. Black shading indicates the frequency (%) of HEp‐2 antibodies and white shading indicates the frequency (%) of non‐HEp‐2 reactive antibodies. The mean fraction of HEp‐2‐reactive (B) antibodies was plotted for each of the three subject groups; HD and two MG disease subtypes (AChR and MuSK). Statistical differences are shown when significant.
Reactivity toward MG autoantigens
Given that we demonstrated that the mature naive B cell repertoire in MG is shaped by defective tolerance checkpoints we reasoned that the polyreactive and/or autoreactive BCRs in this compartment might display reactivity toward MG‐specific autoantigens. We tested the mature naive‐derived BCRs for binding to AChR or MuSK using a radioimmunoassay and by using a more sensitive cell‐based assay approach. No binding to AChR or MuSK was detected with either assay format (Fig. S3).
Discussion
Mechanisms of autoimmunity in MG
The two major autoantibody specificities in MG, AChR and MuSK are demonstrably pathogenic as they affect the disease by targeting the AChR and disturbing signal transduction at the neuromuscular junction. Both active and passive transfer of AChR antibodies from humans affect the disease, demonstrating the direct role these molecules play in its pathology.2, 27, 28, 29 Although their production has been well delineated at a descriptive level, the mechanism underlying the production of MG autoantibodies remains unknown. These fundamental aspects of autoimmune pathology represent considerable gaps in our knowledge that need to be addressed by further defining the mechanisms contributing to MG immunopathology.
Understanding the fidelity of B cell tolerance represents one such fundamental aspect of autoimmunity that has been unexplored in this prototypical autoimmune disease. We applied a well‐established assay platform to measure the frequency of autoreactive BCRs in the naive repertoire. In all AChR and MuSK MG patients examined in this study the frequency of new emigrant/transitional B cells and mature naive B cells that expressed polyreactive and/or autoreactive BCRs was significantly increased over normal controls, revealing both defective central and peripheral tolerance mechanisms. The frequency of polyreactive new emigrant BCRs in our MuSK MG patients was conspicuously higher than that in our AChR cohort. These data are not likely to reflect a quantitative difference in the central tolerance checkpoint defect we found in both cohorts. Rather, we reason that this represents natural variability among our subjects to the antigens used in this assay. Such variability has been previously observed in subjects with clearly identifiable defective tolerance checkpoints.12, 13, 14
Counter‐selection of polyreactive and autoreactive developing B cells is not absolute in healthy individuals.11 Both polyreactivity and autoreactivity was measured in BCRs derived from our healthy control cohort, consistent with other reports.12, 13, 14 The occurrence of these low affinity, polyreactive and autoreactive B cells in the healthy naive repertoire is thought to indicate an overlap between autoreactive and protective repertoires. The purpose of which is to provide protection through the production of natural antibodies and/or provides an initial response to newly encountered pathogens.30
Our analysis of the BCR immunogenetics data (not shown) did not reveal sequence characteristics that have been associated with polyspecificity and autoreactivity. These characteristics included variable region gene segment usage, D gene reading frame preference and heavy chain CDR3 features including charge, hydrophobicity and length. Our data however, do not contradict previous studies that report detectable differences in these features between subjects where tolerance is normally and abnormally established. Rather, the absence of measurable differences may be due to the limited size of our sequence library. However, unique BCR repertoire characteristics are expected to be conspicuous in a B cell compartment that is shaped without proper tolerance regulation. Next‐generation sequencing (NGS) of the B cell repertoire, where unrestricted sequence depth can be achieved, is well suited to accurately establish the repertoire characteristics that associate with tolerance defects and thus should be applied in future studies.
MG is a complex and heterogeneous disease. One limitation of our study was that the MG cohort was not strictly homogeneous in terms of clinical demographics. Indeed, a considerably larger study would be required to evaluate the contribution of B cell tolerance defects to disease subsets. We leave open the possibility that differences in such defects may associate with MG clinical subtypes and that our data may not represent MG disease subtypes not included in our study. However, our data demonstrate that the immune dysregulation in the AChR and MuSK MG patients we studied includes the accumulation of autoreactive BCRs in the naive repertoire, as a consequence of compromised fidelity in tolerance checkpoints. This suggests that defective B cell tolerance may be a fundamental feature of the disease. Further highlighting this point is that our cohort included patients with recent diagnosis along with those who had long‐standing disease. How this immune dysregulation contributes to mechanisms of MG pathology is not evident. Specifically, it is not clear whether the pool of poly‐ and auto‐reactive B cells that populate the naive compartment serve as a reservoir supplying the mature population of B lineage cells that produce disease associated MG autoantibodies. Our data showing the lack of specificity toward MG autoantigens by the self‐reactive BCRs prohibited us from establishing a direct link between the self‐reactive naive repertoire and the development of MG autoantibodies. We reason that such a link is likely to exist, but the rarity of antigen‐specific B cells, especially those in the naive compartment with expected lower affinity, prohibits their easy identification. While our data may suggest that tolerance checkpoint defects in MG may be independent of specific antibody production, we favor a model that includes poly‐ and/or autoreactive naive B cells serving as precursors to mature pathogenic autoantibody‐producers.
Contributors to MG tolerance defects
While the development of antibodies that target self in MG point toward a breach in tolerance, the pre‐germinal center mechanisms underpinning this inability to properly regulate autoimmunity have not been formally demonstrated. Our data indicate that tolerance dysfunction precedes the development of autoimmunity that may occur during antigen‐driven affinity maturation. This supports the notion that autoreactivity in MG develops prior to disease manifestation. While this fundamental incapability to establish B cell tolerance may manifest in the development of MG autoantibodies that target AChR or MuSK, other manifestations are apparent. The first is that MG can simultaneously co‐exist with other autoimmune diseases known to include tolerance defects. The frequency of a second autoimmune disease occurring in AChR MG patients is reported to be 13–22%.31 The most frequently associated autoimmune disease is autoimmune thyroid disease followed by SLE and RA. The second is that some patients with non‐thymomatous MG harbor autoantibody specificities, in addition to AChR, that are directed toward a number of muscle components, including low‐density lipoprotein receptor‐related protein 4 (LRP4),32 titin, the ryanodine receptor (RyR)33 and cortactin.34 Given that comorbidities in MG include autoimmune diseases mediated, in part, by autoantibodies and that autoantibodies in addition to AChR and MuSK are present in MG, collectively point toward a failure to establish B cell tolerance to self. Thus, the loss of tolerance in MG may manifest immunologically through the production of several autoantibody specificities, while the AChR and MuSK autoantibodies result in the clinical manifestation of the disease.
Distinct mechanisms contribute to B cell selection at each checkpoint. Central tolerance is associated with intrinsic BCR and toll‐like receptor (TLR) signals,35 while extrinsic signals, delivered by Tregs, are essential for the establishment and for the maintenance of peripheral tolerance.36 Thus, an additional factor influencing tolerance checkpoint defects may be linked to MG: Defects in the ability of Tregs to suppress responder cells. Tregs derived directly from the thymus of patients with MG are present at normal frequencies, but their ability to suppress is defective37 and T responders also derived from MG thymus resist Treg mediated‐suppression.38 In the periphery, Treg‐mediated suppression of responder T cells is impaired in MG patients, but can be restored using Tregs isolated from healthy controls.39
Immune dysregulation in AChR and MuSK MG
This study included both AChR and MuSK MG. Our cohort of patients from both disease subtypes included compromised B cell tolerance checkpoint integrity, indicating that they share a common immune dysregulation. This is intriguing given how dissimilarly the autoantibody‐mediated pathology propagates in the respective subtypes. In AChR MG the autoantibodies are primarily of the IgG1 and IgG3 subclass,4, 40 both of which are associated with Th1 responses. These IgGs are thought to target the receptor through antigenic modulation by crosslinking AChR molecules, complement activation and functional AChR blockage.40, 41, 42, 43, 44 The pathogenic autoantibodies that recognize MuSK are mainly of the IgG4 subclass,45 which are associated with a Th2 response. This subclass is inefficient at fixing complement and directing antibody‐dependent cell‐mediated cytotoxicity (ADCC). Rather, MuSK IgG4 antibodies disrupt agrin‐stimulated MuSK signaling by inhibiting the association between LRP4 and MuSK.46 Furthermore, serum‐derived IgG4, but not IgG1‐3, transfer from MuSK patients induced severe muscle weakness in mice indicating that MuSK IgG4 autoantibodies are directly pathogenic.45 It is now understood that IgG4 recombines half‐antibodies with other IgG4 antibodies (“Fab arm‐exchange”) generating hetero‐bi‐specific antibodies. These IgG4 become functionally monovalent and, accordingly, are unable to cross‐link antigens.47 Fab arm‐exchange has been demonstrated in experimental MuSK MG.48 A phenomenon such as Fab arm‐exchange, which occurs independently of immune cell influence, might suggest that MuSK MG could arise in the absence of a dysregulated cellular immune system. That is, hetero bi‐specificity could represent a mechanism of autoreactivity that is causatively exclusive of other autoimmune mechanisms. However, our study suggests that MuSK MG also includes a more fundamental immune dysregulation that occurs upstream of antibody production and thereby implies that a number of elements are likely to contribute to the disease pathology.
Treatment strategies that contend with intrinsic tolerance defects
The current standard treatment for MG consists of symptomatic therapy with acetylcholinesterase inhibitors and generalized immunomodulatory agents such as corticosteroids, cyclosporin and azathioprine, intravenous immunoglobulin as well as thymectomy and plasmapheresis.49, 50 Two of our patients had received low dose corticosteroids, but still had remarkably high frequencies of polyreactive and autoreactive BCRs. This suggests that steroid treatment is unlikely to alter B cell tolerance defects and further suggests that such defects represent fundamental aspects of autoimmunity. This point of view is supported by recent studies demonstrating that neither B cell depletion nor methotrexate and/or anti‐TNFα therapy corrects the elevated frequencies of polyreactive and autoreactive BCRs, which persist after such treatment.16, 51
In response to the need for more effective treatment, biologics have been applied to MG management. Several recent studies8, 9 have demonstrated the benefits of rituximab‐mediated B cell depletion in refractory MG, especially in MuSK MG. Rituximab led to a sustained clinical improvement in parallel to a reduction or discontinuation of corticosteroid therapy and plasma exchange treatments. The observed positive effect of rituximab in patients with MG is promising and suggests that further investigation of this agent in MG is warranted; both in terms of a clinical trial and in understanding how it interrupts the autoimmune mechanisms. Our study highlights conceivable associations between the autoimmune mechanisms of MG and B cell depletion. Particularly, the durability of B cell depletion in MG may be fragile given that the repopulated naive repertoire is expected to carry the intrinsic defects that contribute to tolerance checkpoint breaches.51 This is highlighted in the case of AChR MG where the patient response is usually weaker and short lasting, despite improvement shortly after rituximab treatment. Although the extraordinary and long‐lasting response to rituximab of MuSK MG is encouraging, the benefit may not be permanent given the breach in B cell tolerance fidelity.
Conclusion and future perspectives
There is strong evidence that MuSK and AChR MG are distinct disease entities with different etiological and pathological mechanisms. Our study revealed defective central and peripheral tolerance mechanisms in both MG subgroups. These defective central and peripheral tolerance checkpoints in MG may indirectly contribute to the accumulation of autoreactive antibodies through serving as precursors for autoantibody‐producing B cells. Understanding the exact pathological mechanisms that account for autoantibody production and identifying additional elements that are involved in tolerance mechanisms are the next steps toward developing durable B cell targeted therapies for MG.
Author contributions
The study was directed by KO and the study was initiated and designed by JL and KO. JL, PS, RN and KO designed the experiments. Experiments were performed and data were collected by JL, PS, SG, JS, EC, CL, LJ and AV. Data were analyzed and interpreted by JL, PS, SG, LQ, JB, HM, JVH, AV, RN and KO. JL and KO wrote the manuscript with key contributions by PS, RN, PW and LQ and editing by all authors. RN, RB and MD provided clinical specimens, associated clinical data and interpretation of the clinical data.
Conflicts of Interest
PW and AV hold patents for antibody assays and have received royalties. The other authors declare no competing financial interests.
Supporting information
Figure S1. The fidelity of the central tolerance checkpoint is compromised in patients with MG: Central tolerance checkpoint polyreactivity ELISAs. The BCR from single new emigrant/transitional B cells (CD19 + CD21loCD10 + IgMhiCD27–) derived from three AChR MG patients (MGAChR‐1, MG‐AChR‐2, MG‐AChR‐3) and two MuSK MG patients (MG‐MuSK‐1, MG‐MuSK‐2) were cloned, expressed as recombinant antibodies and then tested for reactivity against dsDNA, insulin and LPS by ELISA. Dotted lines show the positive control, a monoclonal antibody (ED38) cloned from a VpreB+L+ peripheral B cell that is highly poly‐ and self‐reactive. Solid lines show the binding curve of each cloned recombinant antibody. Horizontal lines at 0.5 OD405 nm marks the cut‐off for positive reactivity. Negative control group includes antibodies from two healthy individuals (HD‐1, HD‐2). For each individual subject the proportion of polyreactive and non‐polyreactive new emigrant B cells is summarized in the pie charts with the total number of tested clones in the center. Black shading indicates the frequency (%) of polyreactive antibodies and white shading indicates the frequency (%) of non‐polyreactive antibodies.
Figure S2. The fidelity of the peripheral tolerance checkpoint is compromised in patients with MG: Peripheral tolerance checkpoint polyreactivity ELISAs. The BCR from single mature naive B cells (CD19 + CD21 + CD10–IgM+CD27–) derived from three AChR MG patients (MG‐AChR‐1, MGAChR‐ 2, MG‐AChR‐3) and MuSK MG patients (MG‐MuSK‐1, MG‐MuSK‐2) were cloned, expressed as recombinant antibodies and then tested for reactivity against dsDNA, insulin and LPS by ELISA. Dotted lines show the positive control, a monoclonal antibody (ED38) cloned from a VpreB+L+ peripheral B cell that is highly poly‐ and self‐reactive. Solid lines show the binding curve of each cloned recombinant antibody. Horizontal lines at 0.5 marks the cut off OD405 nm for positive reactivity. The control group includes antibodies from two healthy individuals (HD‐1, HD‐2). For each individual subject the frequency (%) of polyreactive and non‐polyreactive mature naive B cells is summarized in the pie charts with the total number of tested clones in the center. Black shading indicates the frequency (%) of polyreactive antibodies and white shading indicates the frequency (%) of non‐polyreactive antibodies.
Figure S3. Mature naive B cell‐derived BCRs reactivity toward AChR and MuSK. The BCR from single mature naive B cells (CD19 + CD21 + CD10–IgM+CD27–) derived from three AChR MG patients (MG‐AChR‐1, MG‐AChR‐2, MG‐AChR‐3), two MuSK MG patients (MGMuSK‐1, MG‐MuSK‐2) and two healthy individuals (HD‐1, HD‐2) were cloned, expressed as recombinant antibodies (rIgG) and then tested for reactivity against AChR or MuSK. A radioimmunoassay (RIA) was first used to evaluate the specificity of the rIgG to AChR (A) or MuSK (B). Recombinant antibody (2.5 μg) was diluted to 50 μL in Hartmann's solution and mixed with 1 μL normal human serum and 50 μl of radioactive antigen (either AChR or MuSK from RSR Ltd). These mixtures were incubated overnight at 4°C. Polyreactive goat anti‐human Ig (25 μL) was incubated with the mix for 1 h to allow precipitation. The pellet was centrifuged and washed twice and counted on an automatic gamma counter. Cell‐based assays (CBA) were used further evaluate the same recombinant antibodies using transfected human embryonic kidney (HEK) 293T
Table S1. Single cell PCR efficiency.
Table S2. Sequence and reactivity tables for the MG and HD study subjects.
Acknowledgments
The authors are sincerely appreciative for the guidance and consultation graciously provided by Dr. Eric Meffre of the Yale School of Medicine who was a principal developer of the B cell tolerance assay.
References
- 1. Vincent A. Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol 2002;2:797–804. [DOI] [PubMed] [Google Scholar]
- 2. Lindstrom JM, Engel AG, Seybold ME, et al. Pathological mechanisms in experimental autoimmune myasthenia gravis. II. Passive transfer of experimental autoimmune myasthenia gravis in rats with anti‐acetylcholine recepotr antibodies. J Exp Med 1976;144:739–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoch W, McConville J, Helms S, et al. Auto‐antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 2001;7:365–368. [DOI] [PubMed] [Google Scholar]
- 4. Rodgaard A, Nielsen FC, Djurup R, et al. Acetylcholine receptor antibody in myasthenia gravis: predominance of IgG subclasses 1 and 3. Clin Exp Immunol 1987;67:82–88. [PMC free article] [PubMed] [Google Scholar]
- 5. Niks EH, van Leeuwen Y, Leite MI, et al. Clinical fluctuations in MuSK myasthenia gravis are related to antigen‐specific IgG4 instead of IgG1. J Neuroimmunol 2008;195:151–156. [DOI] [PubMed] [Google Scholar]
- 6. Koneczny I, Cossins J, Waters P, et al. MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1‐3 can disperse preformed agrin‐independent AChR clusters. PLoS ONE 2013;8:e80695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Querol L, Illa I. Myasthenia gravis and the neuromuscular junction. Curr Opin Neurol 2013;26:459–465. [DOI] [PubMed] [Google Scholar]
- 8. Diaz‐Manera J, Martinez‐Hernandez E, Querol L, et al. Long‐lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012;78:189–193. [DOI] [PubMed] [Google Scholar]
- 9. Nowak RJ, Dicapua DB, Zebardast N, et al. Response of patients with refractory myasthenia gravis to rituximab: a retrospective study. Ther Adv Neurol Disord 2011;4:259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meffre E, Wardemann H. B‐cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol 2008;20:632–638. [DOI] [PubMed] [Google Scholar]
- 11. Wardemann H, Yurasov S, Schaefer A, et al. Predominant autoantibody production by early human B cell precursors. Science 2003;301:1374–1377. [DOI] [PubMed] [Google Scholar]
- 12. Samuels J, Ng YS, Coupillaud C, et al. Impaired early B cell tolerance in patients with rheumatoid arthritis. J Exp Med 2005;201:1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yurasov S, Wardemann H, Hammersen J, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med 2005;201:703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kinnunen T, Chamberlain N, Morbach H, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest 2013;123:2737–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Menard L, Saadoun D, Isnardi I, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest 2011;121:3635–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Menard L, Samuels J, Ng YS, et al. Inflammation‐independent defective early B cell tolerance checkpoints in rheumatoid arthritis. Arthritis Rheum 2011;63:1237–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meffre E, Schaefer A, Wardemann H, et al. Surrogate light chain expressing human peripheral B cells produce self‐reactive antibodies. J Exp Med 2004;199:145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ye J, Ma N, Madden TL, et al. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 2013;41:W34–W40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brochet X, Lefranc MP, Giudicelli V. IMGT/V‐QUEST: the highly customized and integrated system for IG and TR standardized V‐J and V‐D‐J sequence analysis. Nucleic Acids Res. 2008;36:W503–W508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008;131:1940–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jacob S, Viegas S, Leite MI, et al. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 2012;69:994–1001. [DOI] [PubMed] [Google Scholar]
- 22. Wickham H. ggplot2: elegant Graphics for Data Analysis. New York: Springer‐Verlag, 2009. [Google Scholar]
- 23. R_Core_Team . R: A language and environment for statistical computing. Vienna, Austria. 2015; Available at: https://www.R-project.org (accessed March 2016)
- 24. Herve M, Isnardi I, Ng YS, et al. CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J Exp Med 2007;204:1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Isnardi I, Ng YS, Srdanovic I, et al. IRAK‐4‐ and MyD88‐dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity 2008;29:746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sauer AV, Morbach H, Brigida I, et al. Defective B cell tolerance in adenosine deaminase deficiency is corrected by gene therapy. J Clin Invest 2012;122:2141–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Viegas S, Jacobson L, Waters P, et al. Passive and active immunization models of MuSK‐Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol 2012;234:506–512. [DOI] [PubMed] [Google Scholar]
- 28. Oda K, Korenaga S, Ito Y. Myasthenia gravis: passive transfer to mice of antibody to human and mouse acetylcholine receptor. Neurology 1981;31:282–287. [DOI] [PubMed] [Google Scholar]
- 29. Sterz R, Hohlfeld R, Rajki K, et al. Effector mechanisms in myasthenia gravis: end‐plate function after passive transfer of IgG, Fab, and F(ab’)2 hybrid molecules. Muscle Nerve 1986;9:306–312. [DOI] [PubMed] [Google Scholar]
- 30. Wardemann H, Nussenzweig MC. B‐cell self‐tolerance in humans. Adv Immunol 2007;95:83–110. [DOI] [PubMed] [Google Scholar]
- 31. Nacu A, Andersen JB, Lisnic V, et al. Complicating autoimmune diseases in myasthenia gravis: a review. Autoimmunity 2015;48:362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Higuchi O, Hamuro J, Motomura M, et al. Autoantibodies to low‐density lipoprotein receptor‐related protein 4 in myasthenia gravis. Ann Neurol 2011;69:418–422. [DOI] [PubMed] [Google Scholar]
- 33. Skeie GO, Aarli JA, Gilhus NE. Titin and ryanodine receptor antibodies in myasthenia gravis. Acta Neurol Scand Suppl 2006;183:19–23. [DOI] [PubMed] [Google Scholar]
- 34. Gallardo E, Martinez‐Hernandez E, Titulaer MJ, et al. Cortactin autoantibodies in myasthenia gravis. Autoimmun Rev 2014;13:1003–1007. [DOI] [PubMed] [Google Scholar]
- 35. Menard L, Cantaert T, Chamberlain N, et al. Signaling lymphocytic activation molecule (SLAM)/SLAM‐associated protein pathway regulates human B‐cell tolerance. J Allergy Clin Immunol 2014;133:1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kinnunen T, Chamberlain N, Morbach H, et al. Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood 2013;121:1595–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balandina A, Lecart S, Dartevelle P, et al. Functional defect of regulatory CD4(+)CD25 + T cells in the thymus of patients with autoimmune myasthenia gravis. Blood 2005;105:735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gradolatto A, Nazzal D, Truffault F, et al. Both Treg cells and Tconv cells are defective in the Myasthenia gravis thymus: roles of IL‐17 and TNF‐alpha. J Autoimmun 2014;52:53–63. [DOI] [PubMed] [Google Scholar]
- 39. Thiruppathi M, Rowin J, Ganesh B, et al. Impaired regulatory function in circulating CD4(+)CD25(high)CD127(low/‐) T cells in patients with myasthenia gravis. Clin Immunol 2012;145:209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vincent A, Newsom‐Davis J. Acetylcholine receptor antibody characteristics in myasthenia gravis. II. Patients with penicillamine‐induced myasthenia or idiopathic myasthenia of recent onset. Clin Exp Immunol 1982;49:266–272. [PMC free article] [PubMed] [Google Scholar]
- 41. Corey AL, Richman DP, Agius MA, et al. Refractoriness to a second episode of experimental myasthenia gravis. Correlation with AChR concentration and morphologic appearance of the postsynaptic membrane. J immunol 1987;138:3269–3275. [PubMed] [Google Scholar]
- 42. Engel AG, Arahata K. The membrane attack complex of complement at the endplate in myasthenia gravis. Ann N Y Acad Sci 1987;505:326–332. [DOI] [PubMed] [Google Scholar]
- 43. Howard FM Jr, Lennon VA, Finley J, et al. Clinical correlations of antibodies that bind, block, or modulate human acetylcholine receptors in myasthenia gravis. Ann N Y Acad Sci 1987;505:526–538. [DOI] [PubMed] [Google Scholar]
- 44. Pumplin DW, Drachman DB. Myasthenic patients’ IgG causes redistribution of acetylcholine receptors: freeze‐fracture studies. J Neurosci 1983;3:576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klooster R, Plomp JJ, Huijbers MG, et al. Muscle‐specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012;135:1081–1101. [DOI] [PubMed] [Google Scholar]
- 46. Huijbers MG, Zhang W, Klooster R, et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci USA 2013;110:20783–20788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology 2002;105:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van der Neut Kolfschoten M, Schuurman J, Losen M, et al. Anti‐inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007;317:1554–1557. [DOI] [PubMed] [Google Scholar]
- 49. Mantegazza R, Baggi F, Antozzi C, et al. Myasthenia gravis (MG): epidemiological data and prognostic factors. Ann N Y Acad Sci 2003;998:413–423. [DOI] [PubMed] [Google Scholar]
- 50. Sathasivam S. Steroids and immunosuppressant drugs in myasthenia gravis. Nat clin Pract Neurol 2008;4:317–327. [DOI] [PubMed] [Google Scholar]
- 51. Chamberlain N, Massad C, Oe T, et al. Rituximab does not reset defective early B cell tolerance checkpoints. J Clin Invest 2016;126:282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The fidelity of the central tolerance checkpoint is compromised in patients with MG: Central tolerance checkpoint polyreactivity ELISAs. The BCR from single new emigrant/transitional B cells (CD19 + CD21loCD10 + IgMhiCD27–) derived from three AChR MG patients (MGAChR‐1, MG‐AChR‐2, MG‐AChR‐3) and two MuSK MG patients (MG‐MuSK‐1, MG‐MuSK‐2) were cloned, expressed as recombinant antibodies and then tested for reactivity against dsDNA, insulin and LPS by ELISA. Dotted lines show the positive control, a monoclonal antibody (ED38) cloned from a VpreB+L+ peripheral B cell that is highly poly‐ and self‐reactive. Solid lines show the binding curve of each cloned recombinant antibody. Horizontal lines at 0.5 OD405 nm marks the cut‐off for positive reactivity. Negative control group includes antibodies from two healthy individuals (HD‐1, HD‐2). For each individual subject the proportion of polyreactive and non‐polyreactive new emigrant B cells is summarized in the pie charts with the total number of tested clones in the center. Black shading indicates the frequency (%) of polyreactive antibodies and white shading indicates the frequency (%) of non‐polyreactive antibodies.
Figure S2. The fidelity of the peripheral tolerance checkpoint is compromised in patients with MG: Peripheral tolerance checkpoint polyreactivity ELISAs. The BCR from single mature naive B cells (CD19 + CD21 + CD10–IgM+CD27–) derived from three AChR MG patients (MG‐AChR‐1, MGAChR‐ 2, MG‐AChR‐3) and MuSK MG patients (MG‐MuSK‐1, MG‐MuSK‐2) were cloned, expressed as recombinant antibodies and then tested for reactivity against dsDNA, insulin and LPS by ELISA. Dotted lines show the positive control, a monoclonal antibody (ED38) cloned from a VpreB+L+ peripheral B cell that is highly poly‐ and self‐reactive. Solid lines show the binding curve of each cloned recombinant antibody. Horizontal lines at 0.5 marks the cut off OD405 nm for positive reactivity. The control group includes antibodies from two healthy individuals (HD‐1, HD‐2). For each individual subject the frequency (%) of polyreactive and non‐polyreactive mature naive B cells is summarized in the pie charts with the total number of tested clones in the center. Black shading indicates the frequency (%) of polyreactive antibodies and white shading indicates the frequency (%) of non‐polyreactive antibodies.
Figure S3. Mature naive B cell‐derived BCRs reactivity toward AChR and MuSK. The BCR from single mature naive B cells (CD19 + CD21 + CD10–IgM+CD27–) derived from three AChR MG patients (MG‐AChR‐1, MG‐AChR‐2, MG‐AChR‐3), two MuSK MG patients (MGMuSK‐1, MG‐MuSK‐2) and two healthy individuals (HD‐1, HD‐2) were cloned, expressed as recombinant antibodies (rIgG) and then tested for reactivity against AChR or MuSK. A radioimmunoassay (RIA) was first used to evaluate the specificity of the rIgG to AChR (A) or MuSK (B). Recombinant antibody (2.5 μg) was diluted to 50 μL in Hartmann's solution and mixed with 1 μL normal human serum and 50 μl of radioactive antigen (either AChR or MuSK from RSR Ltd). These mixtures were incubated overnight at 4°C. Polyreactive goat anti‐human Ig (25 μL) was incubated with the mix for 1 h to allow precipitation. The pellet was centrifuged and washed twice and counted on an automatic gamma counter. Cell‐based assays (CBA) were used further evaluate the same recombinant antibodies using transfected human embryonic kidney (HEK) 293T
Table S1. Single cell PCR efficiency.
Table S2. Sequence and reactivity tables for the MG and HD study subjects.