

Abstract
Background & objectives:
Alpha-thalassaemias are genetic disorders with high prevalence in northern Thailand. However, common genotypes and current data on the prevalence of α-thalassaemias have not been reported in this region. Therefore, the objective of the present study was to determine the prevalence of α-thalassaemia genotypes in pregnant women in northern Thailand.
Methods:
Genomic DNA was extracted from blood samples of pregnant women who came to Maharaj Nakorn Chiang Mai University Hospital during July 2009 to 2010. The common deletion and point mutation genotypes of α-thalassaemia were evaluated by gap- polymerase chain reaction (PCR) and PCR with restriction fragment length polymorphism (RFLP).
Results:
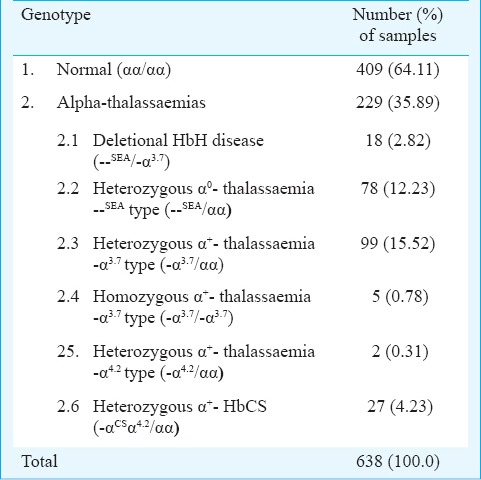
Genotypes of 638 pregnant women were: 409 samples (64.11%) being normal subjects (αα/αα) and 229 samples (35.89%) with α-thalassaemias. These 229 samples could be classified into deletional HbH disease (--SEA/-α3.7) for 18 samples (2.82%); heterozygous α0-thalassaemia --SEA type (--SEA/αα)) for 78 (12.23%); heterozygous α+-thalassaemia - α3.7 type (-α3.7/αα) for 99 (15.52%); homozygous α+-thalassaemia - α3.7 type (-α3.7/- α3.7) for five (0.78%); heterozygous α+-thalassaemia - α4.2 type (-α4.2/αα) for two (0.31%); and heterozygous HbCS (αCSα/αα) for 27 (4.23%) cases.
Interpretation & conclusions:
The prevalence of α-thalassaemias in pregnant women in northern Thailand was high. This finding supports the implementation of the prevention and control of this common genetic disorder by screening for α-thalassaemia genotypes.
Keywords: Alpha-thalassaemia, northern Thailand, polymerase chain reaction, pregnant women, prevalence
Alpha-thalassaemias are genetic disorders caused by deficient synthesis of α-globin chains1. The majority of α-thalassaemias are caused by deletion of one (-α, α+-thalassaemia) or both (--, α0-thalassaemia) α-globin genes on the same chromosome. The α+-thalassaemia occurs at a much higher frequency across the tropical belt2. The carriers’ frequency of α-thalassaemia ranged from 2-70 per cent2, such as in India it varied from 3.84-18 per cent depending on the regions3,4,5, was 4.5 per cent in south China6, in certain parts of Africa and Polynesia it was reported up to 30 per cent7. The carriers’ frequencies in southern Nepal and northern coast of Papua New Guinea were 80 per cent or more2. The two most common α+-thalassaemias are the -α3.7 and the -α4.2 types, which are caused by unequal homologous recombination with the loss of 3.7 and 4.2 kb of DNA, respectively7. The two most common α0-thalassaemias are --SEA and --MED which are found mostly in Southeast Asia and the Mediterranean basin, respectively7. In addition to these deletional α-thalassaemias, the less frequent non-deletional α-thalassaemias are caused by point mutations (αTα) or other small alterations in the structural genes such as Hb Constant Spring (HbCS), Hb Paksé, Hb Quong Sze (HbQS) and Hb Adana. The most common of all these non-deletional types is HbCS which is also associated with the more severe form of haemoglobin H (HbH) disease7. The main area of HbCS distribution is Southeast Asia8, especially in northeast Thailand (up to 10.6%)9.
The most severe form of α-thalassaemias is Hb Bart hydrops fetalis syndrome when foetus dies either in utero or soon after birth7. The gene frequencies of α-thalassaemias vary between 30-40 per cent in northern Thailand10. Detection of α-thalassaemias in population is important for management and control of these genetic diseases. Therefore, the prospective thalassaemia screening programme has been implemented in Maharaj Nakorn Chiang Mai Hospital at Chiang Mai, Thailand, since 199411. Previous survey of α-thalassaemias prevalence in pregnant women in 2004 at the same hospital reported only α0-thalassaemia --SEA type but not α+-thalassaemia and HbCS12. Therefore, the objective of the present study was to determine the prevalence of the common genotypes of α-thalassaemia in pregnant women attending a tertiary care government hospital in northern Thailand.
Material & Methods
This study was conducted in the Department of Biochemistry, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand during 2009-2010. The study protocol was approved by the institutional ethics committee. A total of 638 consecutive primigravida who came to attend the antenatal clinic at Maharaj Nakorn Chiang Mai Hospital, a government hospital, available to the people of northern Thailand were included in the study. Of these 638 women, 429 were from Chiang Mai, 26 from Chiang Rai, five from Kamphaeng Phet, 32 from Lampang, 43 from Lamphun, six from Mae Hong Son, 17 from Nan, 14 from Nakhon Swan, 13 from Phayao, 18 from Phitsanulok, 19 from Phrae, one from Sukhothai, three each from Tak, and Uthai Thani, and nine from Uttaradit. On the assumption that the gene frequencies of α-thalassaemias were 30-40 per cent10 with an error of 5 per cent at confidence interval of 95 per cent, a sample of 323-369 eligible subjects was required. All these women were residents in the 15 provinces of northern Thailand. Written informed consent was obtained from all and peripheral venous blood (5 ml) was drawn from each woman.
Haematological analysis: Haematological parameters were analysed in the Haematology Laboratory Unit at Maharaj Nakorn Chiang Mai Hospital using standard protocols. Red blood cell (RBC) count, haemoglobin (Hb) concentration, haematocrit (Hct), mean corpuscular volume (MCV), mean corpuscular haemoglobin (MCH) were measured by an automated complete blood cell count machine (Coulter LH780 Analyzer, USA). Haemoglobin A2 and HbE were determined by microcolumn chromatography, Hb typing by cellulose acetate electrophoresis, and inclusion bodies by brilliant cresyl blue staining.
Preparation of genomic DNA: Genomic DNA was extracted by the salting out method13. The concentration and purity of DNA were determined by measuring the optical density (OD) at 260 and 280 nm. The OD260/280 ratio or purity of DNA more than 1.8 was further subjected for α-thalassaemia genotyping.
Detection of α0-thalassaemia --SEA type: Detection of α-thalassaemia --SEA type was modified from previously described method14. Multiplex gap-PCR reaction tube containing SEA7, 8 and 9 primers (Table I) was performed simultaneously. The PCR reaction mixture was prepared according to the manufacturer's instructions (Finnzymes, Finland). PCR was run in a Perkin Elmer Cetus model 2400 thermal cycler (USA) with the following: initial denaturation at 98°C for 30 sec, followed by 30 cycles of denaturation at 98°C for 10 sec; annealing at 62°C for 30 sec; and extension at 72°C for 30 sec with an additional 10 min extension at 72°C in the final cycle.
Table I.
Sequences of primers for PCR genotyping of α-thalassaemias

Detection of α+-thalassaemia -α3.7 and -α4.2 types: Laboratory techniques used for detection of α+-thalassaemia -α3.7 and -α4.2 types were described elsewhere15,16. Briefly, detection of -α3.7 type by gap-PCR was performed in two separate reactions for each DNA sample, the first reaction tube contained normal primers (3.7A and B) while the second tube contained deletion primers (3.7A and C). Determination of -α4.2 type was performed in one tube containing three primers of 4.2G, E and F (Table I).
Detection of stop codon mutation of α2-globin gene (HbCS): HbCS mutation was detected by PCR with RFLP17. Briefly, DNA was amplified using primer CSF and R (Table I) by PCR, the amplicon was further digested with restriction enzyme Mse I (New England Biolabs, England) for the detection of the termination codon mutation.
All PCR products were analyzed by separation in 1.5 per cent agarose gel or 3 per cent Nuseive agarose gel electrophoresis, visualized under UV light and documented by a Bio-Rad gel doc 1000 (USA).
Results
Detection and interpretation of α-thalassaemia genotypes by PCR and gel separation are shown in the Figure. Determination of α0-thalassaemia --SEA type by multiplex gap-PCR generated a 1011 bp amplicon (Fig. A, lane 1) for normal haplotype (αα/αα) and a 660 bp amplicon (Fig. A, lane 2) for α0-thalassaemia --SEA haplotype (--SEA/). Detection of α+-thalassaemia -α3.7 type in two separate pairs of primers generated both a 1766 bp amplicon by the primers 3.7A and B for normal haplotype (αα/) (Fig. B, lanes 1 and 3) but only a 1766 bp amplicon by the primers 3.7A and C for α+-thalassaemia -α3.7 haplotype (-α3.7/) (Fig. B, lanes 2 and 4). Detection of α+-thalassaemia -α4.2 type by multiplex gap-PCR generated a 228 bp amplicon (Fig. C, lane 2) for normal haplotype (αα/) and a 1761 bp amplicon (Fig. C, lane 1) for α+-thalassaemia -α4.2 haplotype (-α4.2/). Detection of HbCS mutation was done in two steps. First, amplification of the α2 specific gene generated a 276 bp amplicon as shown in Fig. D (lanes 1 and 3). Second, the amplicon from the first amplification was differentiated by restriction enzyme cutting. Normal haplotype (αα/) could be completely cut to yield a 165 and a 111 bp fragments (Fig. D, lanes 2 and 4), while HbCS haplotype (αCSα/) could not be cut by the enzyme yielding the remainder of 276 bp fragment (Fig. D, lane 4). Therefore, heterozygous HbCS genotype (αCSα/αα) showed three bands after enzyme cutting (Fig D, lane 4). Fragment sizes 165 and 111 bp resulted from αα/ haplotype and half of the uncut 276 bp belonged to αCSα/ haplotype.
Figure.
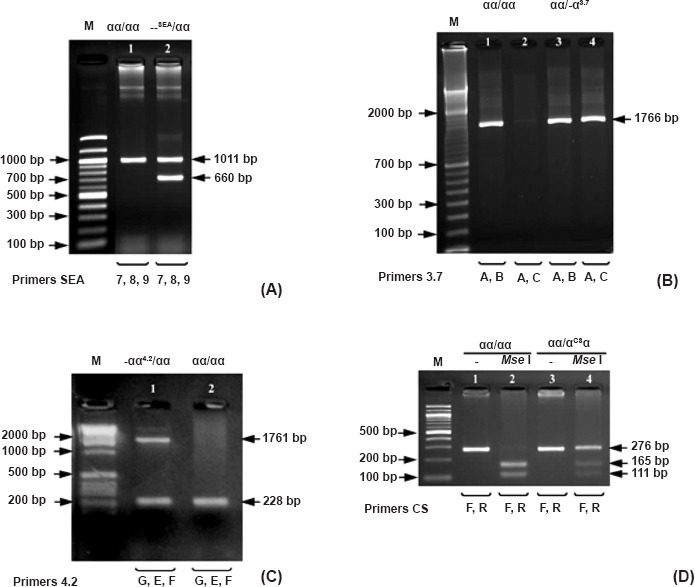
PCR detection of the common genotypes of α-thalassaemias: --SEA type (A), -α3.7 type (B), -α4.2 type (C), and nondeletional type αCSα (HbCS) (D). M, standard DNA molecular weight markers; Mse I, restriction enzyme Mse I.
Of the 638 samples tested, 409 samples (64.11%) were with normal genotypes and 229 (35.89%) with various genotypes of α-thalassaemia as shown in Table II.
Table II.
Prevalence of α-thalassaemia genotypes in Thai pregnant women at Maharaj Nakorn Chiang Mai Hospital, northern Thailand
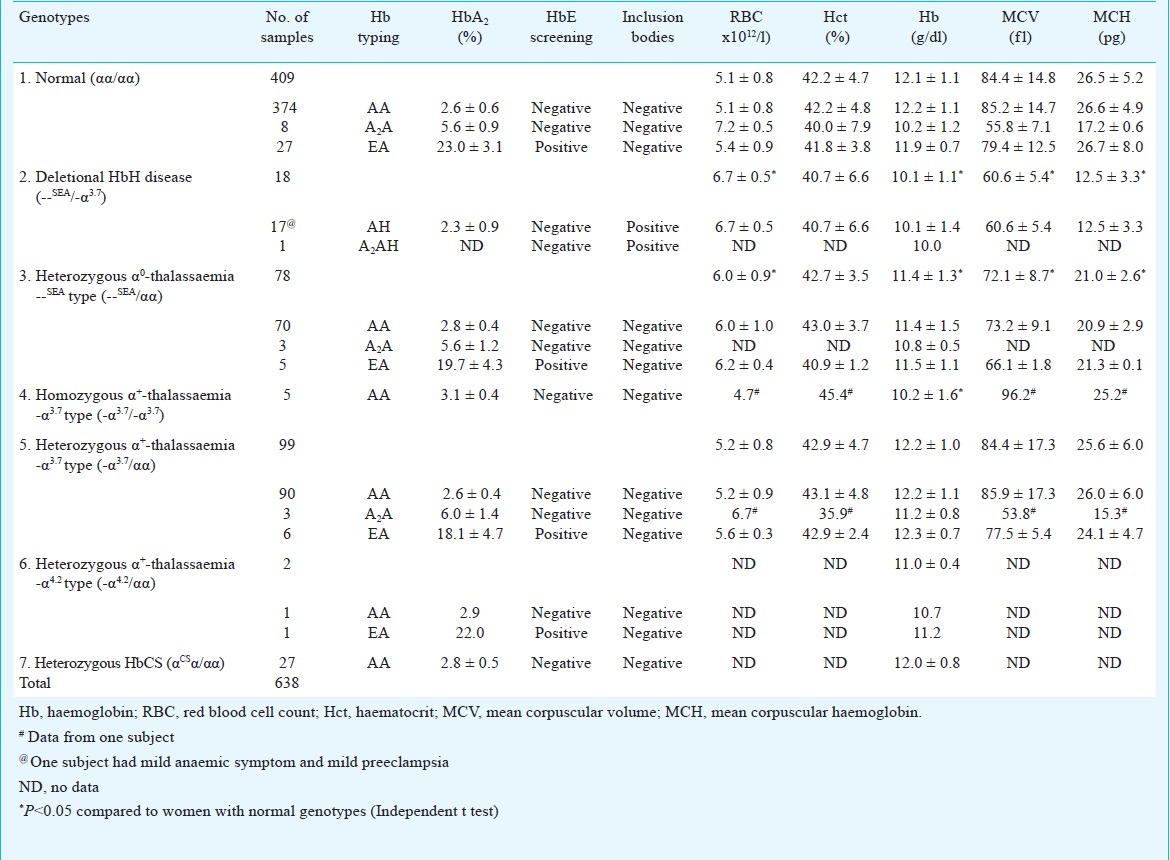
Haematological characteristics of 638 pregnant women are shown in Table III. Haemoglobin levels, MCV and MCH of the deletional HbH disease and heterozygous α0-thalassaemia --SEA type were significantly lower than women with normal genotypes (P<0.05). Haemoglobin levels in homozygous α+-thalassaemia -α3.7 type was also significantly lower than the normal subjects. Inclusion bodies were positive only in deletional HbH disease, while RBC levels in deletional HbH disease and heterozygous α0-thalassaemia --SEA type were significantly higher than normal genotype. Haematological indices of the heterozygous α+-thalassaemia -α3.7 type appeared to be normal. Hb levels in heterozygous α+-thalassaemia -α4.2 type and heterozygous HbCS were in normal range.
Table III.
Haematological parameters of α-thalassaemia genotypes in 638 pregnant women
Discussion
Alpha-thalassaemia is a common genetic defect throughout Southeast Asia1. Conventional diagnosis of α-thalassaemia carriers was based on the combination of tests including haemoglobin typing, MCV, MCH, and osmotic fragility test (OFT)18-24. However, these screening tests were not specific only for α-thalassaemias but also for β-thalassaemias. The false positive rate was high up to 25 per cent from iron deficiency anaemia23. Therefore, the definite way of identifying α-thalassaemias was DNA analysis such as PCR.
Low prevalence of HbCS has been previously reported in northern25 and central26 Thailand. However, HbCS when associated with α0-thalassaemia results in more severe form of non-deletional HbH disease. The HbCS also enhanced membrane rigidity of erythrocytes yielding a decrease in red blood cell survival27. Therefore, identification of HbCS was useful for genetic counselling and prenatal diagnosis. Although the other types of α-chain termination mutation of α2-globin gene such as Hb Paksé (TAA→TAT), Hb Koya Dora (TAA→TCA), Hb Seal Rock (TAA→GAA), and Hb Icaria (TAA→AAA)7 could interact with α0-thalassaemias to form non-deletional HbH disease but all these mutation types have not been reported in Thailand except Hb Paksé. Many researchers tried to detect Hb Paksé9,2628 which originated from Lao People's Democratic Republic and found that this abnormal haemoglobin was rare in northern Thailand28.
When the prevalence of α-thalassaemias of the current study was compared with other studies in northern Thailand, the prevalence of α+-thalassaemia -α3.7 and -α4.2 types, and HbCS was not different from the data reported earlier25,29. The prevalence of α0-thalassaemia --SEA type (12.23%) in our study was not different from earlier data12,25. The frequency of α-thalassaemia --SEA type in our study was significantly higher than the result of Hundrieser et al29. It might be due to the migration of people from central Thailand, an area with lower α-thalassaemia frequencies, to the areas of northern Thailand which had been discussed in the previous study25.
The overall prevalence of α0-thalassaemia in this study was high as 15 per cent (12.2% heterozygous α0-thalassaemia+2.8% HbH disease). When compared with Lao pregnant women in Vientiane with 12.3 per cent α0-thalassaemia (12.0% heterozygous α-thalassaemia+0.3% HbH disease)30, there was no significant difference. However, it was higher compared with Chinese pregnant women from Guangdong with 6.63 per cent α0-thalassaemia (6.5% heterozygous α0-thalassaemia+0.13% HbH disease)6.
In 409 samples of normal genotype (αα/αα), there were eight samples having HbA2 moderately higher than normal level but lower than 10 per cent, low levels of haemoglobin, MCV and MCH suggesting that these subjects might carry β-thalassaemia gene or being heterozygous β-thalassaemia. There were 27 samples with high levels of HbA2 and all were positive to HbE suggesting the heterozygous HbE in these subjects.
There were 18 samples of deletional HbH disease (--SEA/-α3.7), all positive for inclusion bodies. Their haemoglobin levels, MCV and MCH were significantly lower than normal, however, their RBC levels were significantly higher than normal. There were 78 women of heterozygous α0-thalassaemia --SEA type (--SEA/αα) with haemoglobin levels, MCV, and MCH significantly lower than normal, however, the RBC level was significantly higher than normal. In this group, there were three women having moderate levels of HbA2 suggesting them as β-thalassaemia carriers and five with high level of HbA2 and also positive to HbE indicating to be heterozygous HbE. There were five women with homozygous α+-thalassaemia -α3.7 type, with Hb typing AA. Their Hb levels were significantly lower than normal. It was noted that deletional HbH disease with defective three α-globin genes, heterozygous α0-thalassaemia --SEA type and homozygous α+-thalassaemia -α3.7 type with two defective α-genes showed significant abnormality in phenotypes of the haematological characteristics.
In 99 women with heterozygous α+-thalassaemia - α3.7 type, the Hb levels, MCV, MCH, Hct, and RBC levels appeared to be normal. However, three had moderate level of HbA2 and six with high level of HbA2 with HbE positivity suggesting them to be β-thalassaemia gene carriers and heterozygous HbE, respectively. Of the two samples of heterozygous α+-thalassaemia -α4.2 type having high level of HbA2, one was positive for HbE. There were 27 samples with heterozygous HbCS with normal values of HbA2 and haemoglobin levels. All pregnant women with α+ thalassaemia 3.7 kb deletion, 4.2 kb deletion and HbCS had no complication during their pregnancy until delivery.
The significant findings of this study were the high prevalence of α0-thalassaemia and detection of deletional HbH disease in the northern Thai pregnant women. The limitation of this study was determining only the common genotypes of α-thalassaemia due to the problem of each genotype requiring specific primers. Therefore, uncommon genotypes of α-thalassaemia could not be detected.
This study revealed high prevalence of α-thalassaemias in pregnant women in northern Thailand. Though the prospective screening programme has been implemented at Maharaj Nakorn Chiang Mai Hospital since 199411, more effective screening programme coupled with genetic counselling should be considered for prevention and control of these genetic diseases.
Acknowledgment
This study was supported by the National Center for Genetic Engineering and Biotechnology, National Science and Technology Development Agency, Thailand, and the Faculty of Medicine Endowment Fund, Faculty of Medicine, Chiang Mai University, Thailand.
Footnotes
Conflicts of Interest: None.
References
- 1.Weatherall DJ. The thalassemias. In: Stamatoyannopoulos G, Majerus PW, Perlmutter RM, Varmus H, editors. The molecular basis of blood diseases. 3rd ed. Pennsylvania: W.B. Saunders Company; 2001. pp. 183–226. [Google Scholar]
- 2.Higgs DR, Weatherall DJ. The alpha thalassaemias. Cell Mol Life Sci. 2009;66:1154–62. doi: 10.1007/s00018-008-8529-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sen R, Chakrabarti S, Sengupta B, De M, Haldar A, Poddar S, et al. Alpha-thalassemia among tribal populations of Eastern India. Hemoglobin. 2005;29:277–80. doi: 10.1080/03630260500310711. [DOI] [PubMed] [Google Scholar]
- 4.Rudra T, Chakrabarti S, Sengupta B. Alpha thalassemia: experience of referral cases in Kolkata, India. Int J Hum Genet. 2008;8:357–60. [Google Scholar]
- 5.Nadkarni A, Phanasgaonkar S, Colah R, Mohanty D, Ghosh K. Prevalence and molecular characterization of alpha-thalassemia syndromes among Indians. Genet Test. 2008;12:177–80. doi: 10.1089/gte.2007.0080. [DOI] [PubMed] [Google Scholar]
- 6.Yin A, Li B, Luo M, Xu L, Wu L, Zhang L, et al. The prevalence and molecular spectrum of alpha- and beta-globin gene mutations in 14,332 families of Guangdong Province, China. PLoS One. 2014;9:e89855. doi: 10.1371/journal.pone.0089855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weatherall DJ, Clegg JB. The thalassemia syndromes. 4th ed. Oxford: Blackwell Science Ltd; 2001. [Google Scholar]
- 8.Singsanan S, Fucharoen G, Savongsy O, Sanchaisuriya K, Fucharoen S. Molecular characterization and origins of Hb Constant Spring and HbPakse’ in Southeast Asian populations. Ann Hematol. 2007;86:665–9. doi: 10.1007/s00277-007-0310-x. [DOI] [PubMed] [Google Scholar]
- 9.Tritipsombut J, Sanchaisuriya K, Phollarp P, Bouakhasith D, Sanchaisuriya P, Fucharoen G, et al. Micromapping of thalassemia and hemoglobinopathies in different regions of northeast Thailand and Vientiane, Laos People's Democratic Republic. Hemoglobin. 2012;36:47–56. doi: 10.3109/03630269.2011.637149. [DOI] [PubMed] [Google Scholar]
- 10.Fucharoen S, Winichagoon P. Haemoglobinopathies in Southeast Asia. Indian J Med Res. 2011;134:498–506. [PMC free article] [PubMed] [Google Scholar]
- 11.Tongsong T, Wanapirak C, Sirivatanapa P, Sanguansermsri T, Sirichotiyakul S, Piyamongkol W, et al. Prenatal control of severe thalassaemia: Chiang Mai strategy. Prenat Diagn. 2000;20:229–34. [PubMed] [Google Scholar]
- 12.Wanapirak C, Muninthorn W, Sanguansermsri T, Dhananjayanonda P, Tongsong T. Prevalence of thalassemia in pregnant women at Maharaj Nakorn Chiang Mai Hospital. J Med Assoc Thai. 2004;87:1415–8. [PubMed] [Google Scholar]
- 13.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowden DK, Vickers MA, Higgs DR. A PCR-based strategy to detect the common severe determinants of alpha thalassaemia. Br J Haematol. 1992;81:104–8. doi: 10.1111/j.1365-2141.1992.tb08180.x. [DOI] [PubMed] [Google Scholar]
- 15.Smetanina NS, Huisman TH. Detection of alpha-thalassemia-2 (-3.7 kb) and its corresponding triplication (alpha)(alpha)(alpha) (anti-3.7 kb) by PCR: an improved technical change. Am J Hematol. 1996;53:202–3. doi: 10.1002/(SICI)1096-8652(199611)53:3<202::AID-AJH11>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 16.Baysal E, Huisman TH. Detection of common deletional alpha-thalassemia-2 determinants by PCR. Am J Hematol. 1994;46:208–13. doi: 10.1002/ajh.2830460309. [DOI] [PubMed] [Google Scholar]
- 17.Makonkawkeyoon L, Sanguansermsri T, Asato T, Nakashima Y, Takei H. Rapid detection of chain termination mutations in the alpha 2 globin gene. Blood. 1993;82:3503–4. [PubMed] [Google Scholar]
- 18.Pranpanus S, Sirichotiyakul S, Srisupundit K, Tongsong T. Sensitivity and specificity of mean corpuscular hemoglobin (MCH): for screening alpha-thalassemia-1 trait and beta-thalassemia trait. J Med Assoc Thai. 2009;92:739–43. [PubMed] [Google Scholar]
- 19.Giordano PC, Plancke A, Van Meir CA, Janssen CA, Kok PJ, Van Rooijen-Nijdam IH, et al. Carrier diagnostics and prevention of hemoglobinopathies in early pregnancy in The Netherlands: a pilot study. Prenat Diagn. 2006;26:719–24. doi: 10.1002/pd.1490. [DOI] [PubMed] [Google Scholar]
- 20.Sirichotiyakul S, Maneerat J, Sa-nguansermsri T, Dhananjayanonda P, Tongsong T. Sensitivity and specificity of mean corpuscular volume testing for screening for alpha-thalassemia-1 and beta-thalassemia traits. J Obstet Gynaecol Res. 2005;31:198–201. doi: 10.1111/j.1447-0756.2005.00280.x. [DOI] [PubMed] [Google Scholar]
- 21.Akhavan-Niaki H, Youssefi Kamangari R, Banihashemi A, Kholghi Oskooei V, Azizi M, Tamaddoni A, et al. Hematologic features of alpha thalassemia carriers. Int J Mol Cell Med. 2012;1:162–7. [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchaisuriya K, Fucharoen S, Fucharoen G, Ratanasiri T, Sanchaisuriya P, Changtrakul Y, et al. A reliable screening protocol for thalassemia and hemoglobinopathies in pregnancy: an alternative approach to electronic blood cell counting. Am J Clin Pathol. 2005;123:113–8. doi: 10.1309/fuf9evgq24v1pktp. [DOI] [PubMed] [Google Scholar]
- 23.Sirichotiyakul S, Tantipalakorn C, Sanguansermsri T, Wanapirak C, Tongsong T. Erythrocyte osmotic fragility test for screening of alpha-thalassemia-1 and beta-thalassemia trait in pregnancy. Int J Gynaecol Obstet. 2004;86:347–50. doi: 10.1016/j.ijgo.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 24.Fucharoen G, Sanchaisuriya K, Sae-ung N, Dangwibul S, Fucharoen S. A simplified screening strategy for thalassaemia and haemoglobin E in rural communities in south-east Asia. Bull World Health Organ. 2004;82:364–72. [PMC free article] [PubMed] [Google Scholar]
- 25.Lemmens-Zygulska M, Eigel A, Helbig B, Sanguansermsri T, Horst J, Flatz G. Prevalence of alpha-thalassemias in northern Thailand. Hum Genet. 1996;98:345–7. doi: 10.1007/s004390050220. [DOI] [PubMed] [Google Scholar]
- 26.Pichanun D, Munkongdee T, Klamchuen S, Butthep P, Winichagoon P, Fucharoen S, et al. Molecular screening of the Hbs Constant Spring (codon 142, TAA>CAA, alpha2) and Pakse (codon 142, TAA>TAT, alpha2) mutations in Thailand. Hemoglobin. 2010;34:582–6. doi: 10.3109/03630269.2010.526914. [DOI] [PubMed] [Google Scholar]
- 27.Schrier SL, Bunyaratvej A, Khuhapinant A, Fucharoen S, Aljurf M, Snyder LM, et al. The unusual pathobiology of hemoglobin constant spring red blood cells. Blood. 1997;89:1762–9. [PubMed] [Google Scholar]
- 28.Pornprasert S, Panyasai S, Treesuwan K. Unmasking Hb Pakse (codon 142, TAA>TAT, alpha2) and its combinations in patients also carrying Hb Constant Spring (codon 142, TAA>CAA, alpha2) in northern Thailand. Hemoglobin. 2012;36:491–6. doi: 10.3109/03630269.2012.709896. [DOI] [PubMed] [Google Scholar]
- 29.Hundrieser J, Sanguansermsri T, Papp T, Flatz G. Alpha-thalassemia in northern Thailand. Frequency of deletional types characterized at the DNA level. Hum Hered. 1988;38:211–5. doi: 10.1159/000153787. [DOI] [PubMed] [Google Scholar]
- 30.Savongsy O, Fucharoen S, Fucharoen G, Sanchaisuriya K, Sae-Ung N. Thalassemia and hemoglobinopathies in pregnant Lao women: carrier screening, prevalence and molecular basis. Ann Hematol. 2008;87:647–54. doi: 10.1007/s00277-008-0490-z. [DOI] [PubMed] [Google Scholar]