

Abstract
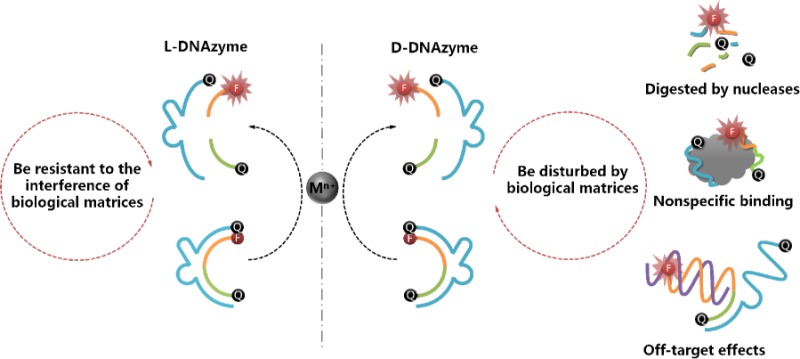
DNAzymes, an important type of metal ion-dependent functional nucleic acid, are widely applied in bioanalysis and biomedicine. However, the use of DNAzymes in practical applications has been impeded by the intrinsic drawbacks of natural nucleic acids, such as interferences from nuclease digestion and protein binding, as well as undesired intermolecular interactions with other nucleic acids. On the basis of reciprocal chiral substrate specificity, the enantiomer of D-DNAzyme, L-DNAzyme, could initiate catalytic cleavage activity with the same achiral metal ion as a cofactor. Meanwhile, by using the advantage of nonbiological L-DNAzyme, which is not subject to the interferences of biological matrixes, as recognition units, a facile and stable L-DNAzyme sensor was proposed for sensing metal ions in complex biological samples and live cells.
DNAzymes, with special DNA molecular structures and enzymatic activities, can specifically catalyze a diverse range of reactions, including RNA/DNA cleavage, ligation, or phosphorylation.1−3 DNAzymes have wide applications in the fields of biotechnology and biomedicine.4−7 In particular, metal ions are necessary cofactors enabling most DNAzymes to accelerate or inhibit catalytic reactions. As a result, DNAzyme-based systems are ideal choices for the development of metal ion sensors.8,9 By adjusting an activated loop, allosteric DNAzymes have also been designed to detect noncofactor targets.10,11 Currently, DNAzymes have shown the ability to recognize a broad range of targets, including metal ions, small molecules, proteins, viruses, and even bacteria.12
However, the practical application of DNAzymes, especially in biological matrixes, has been impeded by their intrinsic drawbacks, including nuclease degradation,13,14 protein binding,15 and off-target effects from partial complementarity of the DNAzyme to other DNA/RNAs,16 leading to a nonfunctional DNAzyme. Meanwhile, the challenge in developing an interference-free DNAzyme in biological fluids has been partly addressed. For example, Lu et al. reported the cellular uptake of a nuclease-resistant gold nanoparticle/DNAzyme conjugate, thus enabling the detection of endosomal uranyl.17 This is an encouraging report that has inspired the subsequent imaging of other cofactors by using different protective nanomaterials,18−20 albeit with complicated design and engineering procedures. Thus, a facile and stable DNAzyme for sensing metal ions in biological samples is highly desired.
The concept of enantiomer in chemistry involves stereoisomers that are mirror images of each other but not identical, which has been hypothesized and verified since enantiomorphic crystals were first described by Pasteur more than 150 years ago.21−23 On the basis of this idea, Kent et al. synthesized the D and L forms of the enzyme HIV-1 protease,24 which have the same molecular weight and identical covalent structure but opposite optical activity. The hexapeptide analogue of a GAG cleavage site was then used as a substrate to test the reactivity of the enantiomers. The results demonstrated that both enzymes were equally active yet exhibited reciprocal chiral specificity in that the L-enzyme could cleave only an L-substrate, whereas the corresponding D-enzyme could cleave only the D-substrate. Following Kent’s report, this principle was used to design mirror-image enantiomers of nucleic acids, such as spiegelmers that bind and inhibit target molecules and spiegelzymes that are able to recognize complementary enantiomeric substrates and hydrolyze, ligate, or polymerize them, thus facilitating new findings and applications.25−38
This concept inspired us to design a novel L-DNAzyme to circumvent the drawbacks of D-DNAzyme, as noted above. On the basis of reciprocal chiral substrate specificity, as explained above, D-enzyme is able to catalyze D-substrate in the presence of an achiral metal ion. Correspondingly, the mirror-image enantiomer of D-DNAzyme, L-DNAzyme, can perform the same function with the help of the same achiral metal ion cofactor. Importantly, the nonbiological L-DNA (Figure S1) is sufficiently stable to resist nuclease digestion and nonspecific binding to natural proteins, and it cannot hybridize with any D-nucleic acids.16,39−42 Therefore, by taking Cu2+- and Pb2+-dependent DNAzymes as two examples, the reported D-DNAzyme sequences were adopted to systematically synthesize and investigate the corresponding L-DNAzymes. It was found that the L-DNAzyme possessed catalytic activity similar to that of D-DNAzyme in the presence of the same achiral metal ion cofactors but with the extra merit of biostability in such biological matrixes (Figure 1). Therefore, the L-DNAzyme was proposed for detection of metal ions, both in serum and living cells, without interferences from biological matrixes.
Figure 1.

Mirror-image DNAzyme enantiomers. D-DNAzyme can selectively catalyze substrate cleavage in the presence of achiral metal ion cofactors. L-DNAzyme, the mirror form of D-DNAzyme, can perform the same function with the help of the same cofactor. It is well documented that DNAzymes in biological matrixes are subject to nuclease degradation, protein binding, and off-target effects from partial complementarity of the DNAzyme to other DNA/RNAs. In contrast, nonbiological L-DNAzyme is resistant to such interferences of biological matrixes.
Experimental Section
Materials and Reagents
Cu2+- and Pb2+-dependent DNAzyme sequences are shown in Figures S2A and S5A, respectively. Both D- and L-DNAzymes were synthesized on a PolyGen 12-Column DNA synthesizer, and the synthesis reagents were purchased from Chem Gene (Wilmington, MA). Lead(II) acetate, copper(II) nitrate, and all other reagents were of analytical reagent grade, purchased from Sigma-Aldrich Chemical Co. and used without further purification. All solutions were prepared in Milli-Q water (resistance > 18 MΩ cm) from a Millipore system. DNase I and single-stranded DNA binding protein (SSB) were purchased from Takara Biotechnology Co. Ltd. (Dalian, China) and Promega (Madison, WI), respectively.
DNA Synthesis
Both D- and L-DNAzyme were synthesized with the same procedure. The synthesis protocol was set up according to the requirements specified by the reagents’ manufacturers. For Cu2+-dependent L-DNAzyme, an additional Dabcyl CPG was used to synthesize the L-enzyme. The complete DNA sequences were then deprotected in 30% ammonium hydroxide/40% methylamine 1:1 (v/v) for 30 min at 65 °C and purified by high-pressure liquid chromatography. An Agilent 1100 HPLC with a C18 reversed-phase column (solvent A, 0.1 M TEAA; solvent B, 95% acetonitrile; increased solvent B from 4% to 60% in 35 min) was used for probe purification. The Pb2+-dependent substrate (both L- and D-form), which contains adenosine triphosphate, was purified with the following changes. After purification, the product was incubated with DMSO (100 μL) and triethylamine trihydrofluoride (125 μL) for 3 h at 65 °C to remove the 2′-TOM group. Next, 3 M sodium acetate (25 μL) and N-butyl alcohol (1.5 mL) were added, and the product was incubated at −20 °C for 0.5 h to precipitate DNA. The DNA concentration was determined with a NanoDrop spectrometer (Thermo Scientific, Waltham, MA).
Fluorescence Measurement
Fluorescence measurement was carried out on a Fluoromax-2 (HORIBA Jobin Yvon Inc., Edison, NJ) fluorometer (for Pb2+-dependent DNAzyme analysis) or RF-5301-PC (Shimadzu, Japan) fluorescence spectrophotometer (for Cu2+-dependent DNAzyme analysis). On the basis of the fluorescent properties of FAM, the excitation and emission wavelengths were set at 490 and 520 nm, respectively, with a constant bandwidth (5 nm). Pb2+-dependent DNAzyme experiments were carried out in 200 μL of buffer solution (50 mM NaCl, 50 mM KCl, 5 mM MgCl2, and 50 mM HEPES at pH 7.2) containing 50 nM substrate, 60 nM enzyme, and different concentrations of lead acetate (for sensitivity analysis), and the mixture was equilibrated at room temperature for 30 min prior to measurement. For the Cu2+-dependent DNAzyme experiment, the parameters were the same as those for Pb2+, except that the buffer (50 mM HEPES, 3 M NaCl, and 50 μM ascorbate, pH 7.0) was different.
Gel Electrophoresis
For Cu2+-dependent L-DNAzyme activity analysis, six tubes of reaction buffer solution (50 mM HEPES, 3 M NaCl, and 50 μM ascorbate, pH 7.0) containing 5 μM substrate and 6 μM enzyme (D-E/D-S with or without Cu2+, L-E/L-S with or without Cu2+, D-E/L-S with Cu2+, and L-E/D-S with Cu2+, respectively) were incubated at room temperature. After 90 min reaction, the mixed solution were transferred into 15 μL of saturated urea to quench the cleavage reaction. Then the reaction substrates were separated by 15% denaturing polyacrylamide gel electrophoresis and analyzed by a fluorescence image scanner (FLA-3000G; Fuji, Tokyo, Japan) using 473 nm excitation. To monitor FBS digestion of DNAzyme, nine tubes of 5 μM L-S in 10 μL of FBS were prepared and incubated for 0, 0.5, 1, 2, 4, 12, 24, 48, and 72 h, respectively, at 37 °C. Afterward, the samples were incubated at 95 °C for 10 min, and then 15 μL of saturated urea were added into the sample to deactivate. A 7.5/15% gradient denaturing polyacrylamide gel was prepared using 1× TBE buffer (pH 8.3). The gel was run at 120 V for about 1 h in 1× TBE buffer and analyzed by a fluorescence image scanner.
Cell Culture, Transfection, and Confocal Imaging
HeLa cells were cultured in DMEM medium with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37 °C in 95% air, 5% CO2. Transfection experiment was carried out in light of the manufacturer’s protocol. Briefly, L-DNAzymes were annealed by heating to 85 °C for 5 min and cooling to room temperature. A volume of 5 μL of Lipofectamine 2000 and 1 nmol of annealed L-DNAzymes were incubated separately in Opti-MEM media containing 50 μM ascorbate for 5 min. After 25 min incubation at room temperature, the samples was added to HeLa cells for 10 h. Then, 0.2 mM Cu2+ was added, and the results were observed under fluorescence imaging.
Confocal fluorescence imaging was carried out on a FV1000 confocal laser-scanning microscope. The images were collected with different objective lens (40×, UplanApo N.A. 0.95, for Cu2+-DNAzyme; 60×, UplanApo N.A. 1.4, for Pb2+-DNAzyme, Olympus).
Results and Discussion
The sensor design is shown in Figure S2A. The Cu2+-dependent DNAzyme is a deoxyribonucleotide-based complex that cleaves separate DNA substrates in the presence of Cu2+.43−45 In order to accurately estimate the catalytic activity of the enantiomeric L-DNAzymes, we measured the fluorescence of DNAzyme catalysis using a molecular beacon according to Lu’s report.46 As shown in Figure S2B, the presence of Cu2+ results in irreversible cleavage of the substrate, leading to fluorescence enhancement.
Circular Dichroism and Thermodynamics of Enantiomer DNAzymes
To verify the structure of the two enantiomers, circular dichroism (CD) spectra were first measured. As shown in Figure 2A, a typical profile for a right-handed DNA helix was observed from D-DNAzyme with a negative peak at 246 nm and a positive peak at 278 nm, whereas the nearly inverted mirror spectrum was observed from L-DNAzyme, indicating that L-DNAzyme was formed with the left-handed helical property, as expected. In addition, the thermal stability of the two enantiomers was also investigated. As shown in Figure 2B, the melting temperature indicated no apparent difference between L- and D-DNAzyme, both at approximately 42–44 °C. These results suggested that the L-DNAzyme was formed as the mirror image of D-DNAzyme.
Figure 2.

(A) Circular dichroism spectra of L- and D-DNAzyme are inverted images for the two enantiomer DNAzymes. For D-DNAzyme, a negative peak is around 246 nm, and a positive peak is around 278 nm, while for L-DNAzyme, a positive peak is around 246 nm, and a negative peak is around 278 nm. (B) The two enantiomers have similar DNA melting temperatures at approximately 42–44 °C. FI = fluorescence intensity.
Catalytic Activity of Enantiomeric DNAzymes
The catalytic cleavage activity of the two enantiomeric DNAzymes was confirmed by gel electrophoresis. As shown in Figure 3A, in the presence or absence Cu2+, the substrate of the D-DNAzyme (D-S) was respectively cleaved (lane 2) or remained intact (lane 1) in accordance with previous reports.43,44 The catalytic activity of the L-DNAzyme is shown in lanes 3 and 4. In the absence of Cu2+ (lane 3), the substrate (L-S) also remained intact, indicating that no nonspecific cleavage had occurred. However, upon the addition of Cu2+, L-S was cleaved, fully confirming L-DNAzyme as the enantiomer of D-DNAzyme and, hence, having a similar ability to catalyze DNA-cleavage with the help of the same metal ion cofactor. A real-time fluorescence assay showed the same results. As illustrated in Figure 3B, using a TaqMan probe with a FAM dye label, unquenched fluorescence was observed from the solution containing 50 nM L-S. However, upon the addition of 60 nM L-enzyme (L-E), an immediate decrease of fluorescence intensity occurred, since the residual fluorescence of FAM was quenched by the nearby quencher, Dabcyl. Next, 30 μM Cu2+ was added into the reaction mixture, resulting in a remarkable increase of fluorescence signal, which indicated a successful catalytic cleavage reaction realized by the L-DNAzyme. Fluorescence titration experiment was also carried out to test the limit of detection. As shown in Figure S3A, fluorescence intensity increased as the concentration of the Cu2+ increased within the range of 0–30 μM, and a 10 nM detection limit was estimated in accordance with previous reports for D-DNAzyme.43,44 Meanwhile, in contrast to Cu2+, no obvious fluorescence change was observed upon the addition of other metal ions (Cd2+, Co2+, Fe3+, Mg2+, Mn2+, Ni2+, Pb2+, and Zn2+) with a 10-fold higher concentration (Figure S3B).
Figure 3.

(A) Denaturing PAGE of the Cu2+-dependent L-/D-DNAzyme in the absence or presence of Cu2+. D-DNAzyme without (lane 1) or with (lane 2) Cu2+; L-DNAzyme without (lane 3) or with (lane 4) Cu2+; D-S/L-E (lane 5) or L-S/D-E (lane 6) with Cu2+. (B) Time course study of Cu2+-dependent L-DNAzyme in the presence of Cu2+.
Biostability and Activity of L-DNAzymes in Biological Fluids
Next, we investigated whether cross-hybridization, such as D-S/L-E and L-S/D-E complexes, would catalyze substrate cleavage. As exhibited in Figure 3A, lanes 5 and 6, in the presence of Cu2+, no cleaved band appeared in either the D-S/L-E or L-S/D-E reaction mixture. This result might be attributed to the inability of D-E (or L-E) to hybridize with L-S (or D-S), respectively,36 as shown in Figure S4A,B, where both D-S and L-S showed unquenched fluorescence signals in the presence of the enantiomeric enzyme. Therefore, since L-DNA could not hybridize with D-DNA, the unexpected off-target effect was avoided. We then demonstrated that the L-DNAzyme could resist the interference of single-stranded DNA binding protein (SSB) and nuclease. As shown in Figure S4C, while D-S exhibited a 3.5-fold increase of anisotropy signal in the presence of SSB, L-S showed no significant change. Finally, the stability of L-DNA was tested in the presence of nuclease. As shown in Figure S4D, after 1.5 h incubation in DNase I, the L-DNAzyme remained intact, while the D-DNAzyme had completely decomposed.
These results all indicate rigid reciprocal chiral substrate specificity, as previously reported.24,35 More importantly, our data also demonstrated that the metal ion, i.e., achiral cofactor, could specifically respond to both DNAzyme enantiomers. Thus, since the biological matrix imposes no restrictions on nonbiological L-DNAzyme, an interference-free DNAzyme may be proposed for biological fluids, thereby circumventing the drawbacks of natural nucleic acids, such as nuclease digestion, protein binding, and undesired intermolecular interactions with other nucleic acids.
To overcome the challenges involving interferences that arise from complex biological matrixes, the L-DNAzyme-based biosensor was first used to detect Cu2+ in fetal bovine serum (FBS), a complex biological matrix in which exogenous natural DNAzymes are vulnerable to enzymatic degradation.47 As exhibited in Figure 4A, in 90% FBS, L-S showed negligible digestion, even after incubation for 72 h at 37 °C, indicating its excellent biostability at physiological temperature. In contrast, D-S was easily hydrolyzed under the same condition after 2 h (Figure 4B). The activity of L-DNAzyme in FBS was also tested in different FBS concentrations using gel electrophoresis. As shown in Figure 4C, by increasing serum concentration from 0 to 50%, the activity of L-DNAzyme remained unchanged in the reaction buffer since negligible reduction of the residual band was observed in the presence of Cu2+. We next applied the stable L-DNAzyme to measure Cu2+ in FBS using a fluorescence titration assay. As demonstrated in Figure 4D, in the presence of different Cu2+ concentrations (0, 0.5–30 μM), the fluorescence intensity of L-DNAzyme increased with elevated Cu2+ concentrations. These results clearly demonstrated that L-DNAzyme could effectively function in complex biological samples, indicating its potential for metal ion detection in clinical trials.
Figure 4.

Stability of Cu2+-dependent L-S (A) and D-S (B) in 90% FBS for different times, respectively. (C) Denaturing PAGE of the Cu2+-dependent L-DNAzyme in the absence (0) or presence (1) of Cu2+ in the buffer with 0, 10%, 30%, and 50% FBS. (D) Response of L-DNAzyme to different concentrations (0, 0.5, 0.75, 1, 2.5, 5, 10, 20, and 30 μM) of Cu2+ in 10% FBS.
Application of L-DNAzymes for Intracellular Metal Ion Imaging
After demonstrating that the L-DNAzyme had unchanged activity and sensing performance in FBS, we then challenged its ability to sense Cu2+ in cultured HeLa cells. HeLa cells were first transfected with Cu2+- (Figure 5, left) or Pb2+- (Figure 5, right) dependent L-DNAzymes for 10 h, respectively, followed by the addition of Cu2+ and ascorbic acid. As shown in Figure 5, in the presence of 0.2 mM Cu2+ and 50 μM ascorbic acid, the confocal image showed a bright fluorescence signal. In contrast, only negligible fluorescence intensity change was observed within 45 min after treatment with Pb2+-dependent DNAzyme, indicating that L-DNAzyme could be used for intracellular monitoring of metal ions with high specificity.
Figure 5.
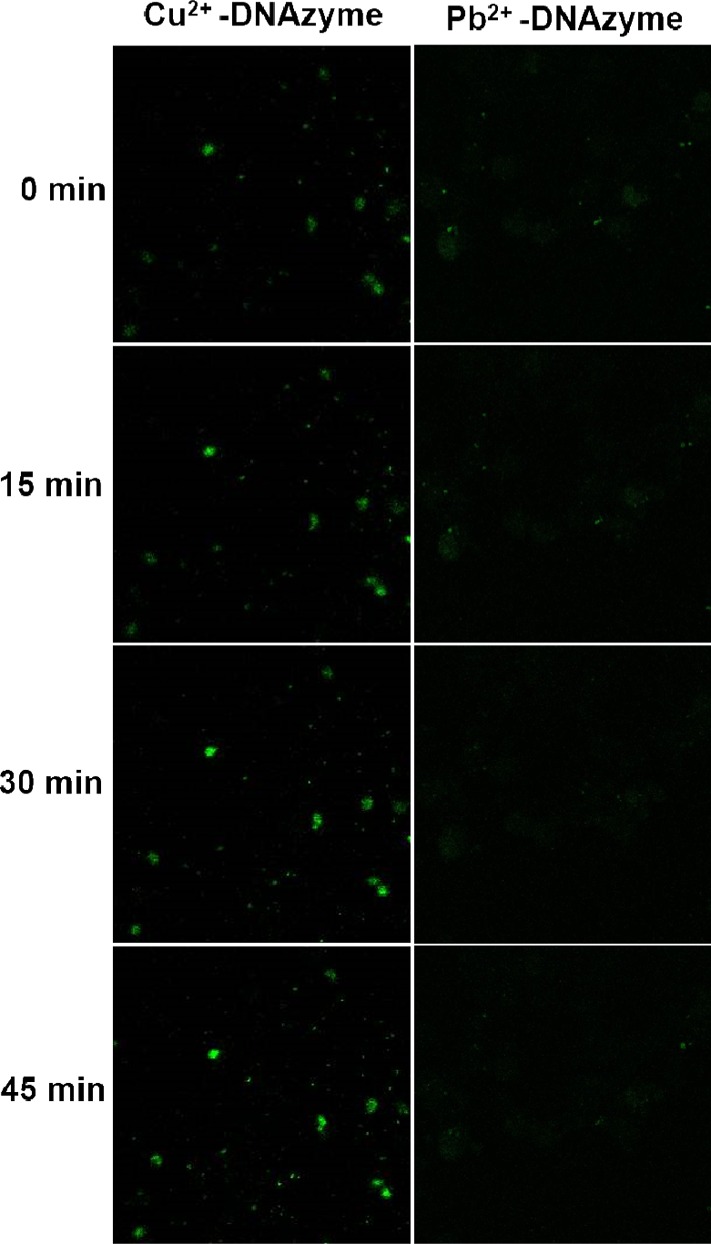
(A) Confocal microscopy images of HeLa cells transfected with Cu2+- (left) or Pb2+- (right) dependent L-DNAzyme for 10 h, followed by addition of 0.2 mM Cu2+ and 50 μM ascorbate.
Generality: Pb2+-dependent Enantiomer DNAzymes
Thus far, on the basis of chiral specificity, it has been proved that the Cu2+-dependent mirror-image enantiomer of D-DNAzyme, L-DNAzyme, could catalyze Cu2+-dependent DNA cleavage, thus showing its potential for sensing and imaging of Cu2+ in both serum and living cells. To further illustrate that this chiral specificity is universal in the DNAzyme system, we investigated the applicability of this method to other DNAzymes for the detection of their corresponding target metal ions. The Pb2+-dependent GR-5 DNAzyme was chosen, which selectively employs Pb2+ as the metal cofactor to catalyze and cleave a DNA/RNA chimera substrate at the “rA” site (Figure S5).48−50 The activity of GR-5 L-DNAzyme was also confirmed by gel electrophoresis. As exhibited in the inset of Figure 6A, L-S was cleaved when Pb2+ added, resulting in disappearance of its band mostly, demonstrated that the GR-5 L-DNAzyme has the predicted ability to catalyze Pb2+-dependent substrate cleavage. Fluorescence assay was also used to accurately estimate the catalytic activity, as a result, an 11-fold signal increase was observed after the addition of 5 μM Pb2+ in the buffer (Figure 6B). The stability was also demonstrated by incubation of the GR-5 L-S in DNase I solution for 1.5 h (Figure 6A, lane 3, no digestion of the L-S was observed). Meanwhile, it can also resist undesired intermolecular interactions (Figure S6A,B) and SSB nonspecific binding (Figure S6C). Since Pb2+-dependent GR-5 L-DNAzyme could perform as well as its enantiomer,49 it can be suggested that chiral specificity is universal in the DNAzyme system.
Figure 6.

(A) Denaturing PAGE of the Pb2+-dependent GR-5 L-DNAzyme in the absence (lane 1) or presence (lane 2) of 10 μM Pb2+. Lane 3: GR-5 L-substrate in the presence of 5 U DNase I. (B) Fluorescence spectrum of Pb2+-dependent L-DNAzyme in the absence or presence of 5 μM Pb2+.
Conclusions
In conclusion, the enantiomer of D-DNAzyme, L-DNAzyme, could initiate catalytic cleavage activity with the same achiral metal ion as a cofactor based on reciprocal chiral substrate specificity. By using L-DNAzyme as recognition units, a facile and stable L-DNAzyme sensor was proposed for sensing metal ions in biological samples. Taking Cu2+- and Pb2+-dependent L-DNAzymes as two examples, the results showed that the enantiomer DNAzymes possessed similar catalytic activity. Furthermore, since L-DNAzyme was not influenced by nucleases, natural proteins, or D-nucleic acids, it could be used in complex biological fluids, such as serum and living cells, for quantitative determination of metal ions, indicating that L-DNAzyme can circumvent many challenges associated with metal ion detection in complex biological matrixes. Thus, the ability to recognize metal ions from the enantiomer of D-DNAzyme offers an exciting way to design DNAzymes, which will not only provide new schemes to utilize L-DNAzyme for bioanalysis but also inspire the generation of new DNAzymes for a variety of biomedical applications.
Acknowledgments
This work is supported by the National Key Scientific Program of China (Grant 2011CB911000), NSFC grants (Grants NSFC 21221003, NSFC 21325520, NSFC 21327009, and NSFC 21405039), China National Instrumentation Program 2011YQ03012412 and the Fundamental Research Funds for the Central Universities. The research is also supported by and by the National Institutes of Health (Grants GM079359, GM111386, and CA133086).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.5b04170.
Structure of L-DNA and D-DNA; sequence, principle, and analytical performance of L-DNAzyme (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Breaker R. R. Chem. Rev. 1997, 97, 371–390. 10.1021/cr960008k. [DOI] [PubMed] [Google Scholar]
- Breaker R. R. Nat. Biotechnol. 1997, 15, 427–431. 10.1038/nbt0597-427. [DOI] [PubMed] [Google Scholar]
- Wilson D. S.; Szostak J. W. Annu. Rev. Biochem. 1999, 68, 611–647. 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- Santiago F. S.; Lowe H. C.; Kavurma M. M.; Chesterman C. N.; Baker A.; Atkins D. G.; Khachigian L. M. Nat. Med. 1999, 5, 1264–1269. 10.1038/15215. [DOI] [PubMed] [Google Scholar]
- Abdelgany A.; Wood M.; Beeson D. J. Gene Med. 2007, 9, 727–738. 10.1002/jgm.1061. [DOI] [PubMed] [Google Scholar]
- Dass C. R.; Choong P. F.; Khachigian L. M. Mol. Cancer Ther. 2008, 7, 243–251. 10.1158/1535-7163.MCT-07-0510. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Balogh D.; Wang F.; Willner I. J. Am. Chem. Soc. 2013, 135, 1934–1940. 10.1021/ja311385y. [DOI] [PubMed] [Google Scholar]
- Liu J.; Cao Z.; Lu Y. Chem. Rev. 2009, 109, 1948–1998. 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y.; Lu Y. Inorg. Chem. 2014, 53, 1925–1942. 10.1021/ic4019103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei S. H. J.; Liu Z. J.; Brennan J. D.; Li Y. F. J. Am. Chem. Soc. 2003, 125, 412–420. 10.1021/ja0281232. [DOI] [PubMed] [Google Scholar]
- Tram K.; Kanda P.; Salena B. J.; Huan S.; Li Y. Angew. Chem. 2014, 126, 13013–13016. 10.1002/ange.201407021. [DOI] [PubMed] [Google Scholar]
- Liang H.; Zhang X. B.; Lv Y. F.; Gong L.; Wang R. W.; Zhu X. Y.; Yang R. H.; Tan W. H. Acc. Chem. Res. 2014, 47, 1891–1901. 10.1021/ar500078f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama H.; Hirano K.; Kashiwasake-Jibu M.; Taira K. J. Biol. Chem. 1996, 271, 380–384. 10.1074/jbc.271.1.380. [DOI] [PubMed] [Google Scholar]
- Wu C.; Cansiz S.; Zhang L.; Teng I. T.; Qiu L.; Li J.; Liu Y.; Zhou C.; Hu R.; Zhang T.; Cui C.; Cui L.; Tan W. J. Am. Chem. Soc. 2015, 137, 4900–4903. 10.1021/jacs.5b00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. J.; Fang X.; Schuster S. M.; Tan W. Angew. Chem., Int. Ed. 2000, 39, 1049–1052. 10.1002/(SICI)1521-3773(20000317)39:6<1049::AID-ANIE1049>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Yang C. J.; Tan W. Nucleic Acids Res. 2007, 35, 7279–7287. 10.1093/nar/gkm771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P. W.; Hwang K. V.; Lan T.; Lu Y. J. Am. Chem. Soc. 2013, 135, 5254–5257. 10.1021/ja400150v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Feng J.; Fan Y.; Tang B. Anal. Chem. 2015, 87, 4829–4853. 10.1021/acs.analchem.5b00204. [DOI] [PubMed] [Google Scholar]
- Hwang K.; Wu P. W.; Kim T.; Lei L.; Tian S. L.; Wang Y. X.; Lu Y. Angew. Chem., Int. Ed. 2014, 53, 13798–13802. 10.1002/anie.201408333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H. M.; Zhang X. B.; Lv Y. F.; Zhao Z. L.; Wang N. N.; Fu T.; Fan H. H.; Liang H.; Qiu L. P.; Zhu G. Z.; Tan W. H. ACS Nano 2014, 8, 6171–6181. 10.1021/nn5015962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bada J. L. Nature 1995, 374, 594–595. 10.1038/374594a0. [DOI] [PubMed] [Google Scholar]
- Negi S.; Dhanasekaran M.; Hirata T.; Urata H.; Sugiura Y. Chirality 2006, 18, 254–258. 10.1002/chir.20247. [DOI] [PubMed] [Google Scholar]
- Blackmond D. G. Cold Spring Harbor Perspect. Biol. 2010, 2, a002147. 10.1101/cshperspect.a002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton R. C.; Milton S. C.; Kent S. B. Science 1992, 256, 1445–1448. 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- Nolte A.; Klussmann S.; Bald R.; Erdmann V. A.; Furste J. P. Nat. Biotechnol. 1996, 14, 1116–1119. 10.1038/nbt0996-1116. [DOI] [PubMed] [Google Scholar]
- Klussmann S.; Nolte A.; Bald R.; Erdmann V. A.; Furste J. P. Nat. Biotechnol. 1996, 14, 1112–1115. 10.1038/nbt0996-1112. [DOI] [PubMed] [Google Scholar]
- Williams K. P.; Liu X. H.; Schumacher T. N.; Lin H. Y.; Ausiello D. A.; Kim P. S.; Bartel D. P. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 11285–11290. 10.1073/pnas.94.21.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purschke W. G.; Eulberg D.; Buchner K.; Vonhoff S.; Klussmann S. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 5173–5178. 10.1073/pnas.0509663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purschke W. G.; Radtke F.; Kleinjung F.; Klussmann S. Nucleic Acids Res. 2003, 31, 3027–3032. 10.1093/nar/gkg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczepanski J. T.; Joyce G. F. J. Am. Chem. Soc. 2013, 135, 13290–13293. 10.1021/ja406634g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatime L.; Maasch C.; Hoehlig K.; Klussmann S.; Andersen G. R.; Vater A. Nat. Commun. 2015, 6, 6481–6493. 10.1038/ncomms7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberthur D.; Achenbach J.; Gabdulkhakov A.; Buchner K.; Maasch C.; Falke S.; Rehders D.; Klussmann S.; Betzel C. Nat. Commun. 2015, 6, 6923–6933. 10.1038/ncomms7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig B.; Keiper S.; Stuhlmann F.; Jaschke A. Angew. Chem., Int. Ed. 2000, 39, 4576–4579. 10.1002/1521-3773(20001215)39:24<4576::AID-ANIE4576>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Olea C. Jr.; Horning D. P.; Joyce G. F. J. Am. Chem. Soc. 2012, 134, 8050–8053. 10.1021/ja302197x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszko E.; Szymanski M.; Zeichhardt H.; Muller F.; Barciszewski J.; Erdmann V. A. PLoS One 2013, 8, e54741. 10.1371/journal.pone.0054741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehlig K.; Bethge L.; Klussmann S. PLoS One 2015, 10, e0115328. 10.1371/journal.pone.0115328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczepanski J. T.; Joyce G. F. Nature 2014, 515, 440–442. 10.1038/nature13900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q.; Zhou L.; Yuan Y.; Huang Y.; Xiang B.; Chen C.; Nie Z.; Yao S. Biosens. Bioelectron. 2014, 55, 187–194. 10.1016/j.bios.2013.12.019. [DOI] [PubMed] [Google Scholar]
- Hauser N. C.; Martinez R.; Jacob A.; Rupp S.; Hoheisel J. D.; Matysiak S. Nucleic Acids Res. 2006, 34, 5101–5111. 10.1093/nar/gkl671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke G. L.; Wang C. M.; Ge Y.; Zheng N. F.; Zhu Z.; Yang C. J. J. Am. Chem. Soc. 2012, 134, 18908–18911. 10.1021/ja3082439. [DOI] [PubMed] [Google Scholar]
- Kim K. R.; Lee T.; Kim B. S.; Ahn D. R. Chem. Sci. 2014, 5, 1533–1537. 10.1039/C3SC52601A. [DOI] [Google Scholar]
- Lin C. X.; Ke Y. G.; Li Z.; Wang J. H.; Liu Y.; Yan H. Nano Lett. 2009, 9, 433–436. 10.1021/nl803328v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi N.; Balkhi S. R.; Breaker R. R. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 2233–2237. 10.1073/pnas.95.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi N.; Breaker R. R. Bioorg. Med. Chem. 2001, 9, 2589–2600. 10.1016/S0968-0896(01)00035-9. [DOI] [PubMed] [Google Scholar]
- Li H.; Huang X. X.; Kong D. M.; Shen H. X.; Liu Y. Biosens. Bioelectron. 2013, 42, 225–228. 10.1016/j.bios.2012.10.070. [DOI] [PubMed] [Google Scholar]
- Liu J.; Lu Y. J. Am. Chem. Soc. 2007, 129, 9838–9839. 10.1021/ja0717358. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Chen Q.; Huang P. J.; Ding J.; Liu J. Anal. Chem. 2015, 87, 4001–4007. 10.1021/acs.analchem.5b00220. [DOI] [PubMed] [Google Scholar]
- Breaker R. R.; Joyce G. F. Chem. Biol. 1994, 1, 223–229. 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- Lan T.; Furuya K.; Lu Y. Chem. Commun. 2010, 46, 3896–3898. 10.1039/b926910j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Zhang Q.; Cai Y.; Kong D. M.; Shen H. X. Biosens. Bioelectron. 2012, 34, 159–164. 10.1016/j.bios.2012.01.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.