

Abstract
Myocardial infarction (MI) is the most common cause of cardiac injury, and subsequent reperfusion further enhances the activation of innate and adaptive immune responses and cell death programs. Therefore, inflammation and inflammatory cell infiltration are the hallmarks of MI and reperfusion injury. Ischemic cardiac injury activates the innate immune response via toll-like receptors and upregulates chemokine and cytokine expressions in the infarcted heart. The recruitment of inflammatory cells is a dynamic and superbly orchestrated process. Sequential infiltration of the injured myocardium with neutrophils, monocytes and their descendant macrophages, dendritic cells, and lymphocytes contributes to the initiation and resolution of inflammation, infarct healing, angiogenesis, and ventricular remodeling. Both detrimental effects and a beneficial role in the pathophysiology of MI and reperfusion injury may be attributed to the subset heterogeneity and functional diversity of these inflammatory cells.
Keywords: myocardial infarction, reperfusion injury, inflammation, toll-like receptor, macrophage
Introduction
Myocardial infarction (MI), and subsequent reperfusion injury, is the most common and clinically significant form of acute cardiac injury and results in the ischemic death of cardiomyocytes.1,2 Among the pathological mechanisms underlying myocardial ischemia/reperfusion (MI/R) injury, inflammation and inflammatory cell infiltration, together with the activation of innate and adaptive immune responses, are the hallmark of MI and reperfusion injury.3,4
Ischemic cardiac injury activates the innate immune response via toll-like receptor (TLR)-mediated pathways and upregulates the chemokine and cytokine syntheses in the infarcted heart. TLRs, which are expressed by inflammatory cells and also on endothelial cells and cardiomyocytes, can recognize endogenous danger signals released during cell death following myocardial ischemia and reperfusion.5,6 A growing body of evidence suggests that modulating TLR activation may enhance the benefits and blunt the negative effects of the inflammatory response, providing new therapeutic options for preventing MI/R injury.6 Chemokines stimulate the chemotactic recruitment of inflammatory cells into the infarct. One of the well-studied CC chemokines, CC chemokine ligand 2 (CCL2), is a potent chemoattractant for monocytes, macrophages, T cells, and NK cells; CC chemokine receptor 2 (CCR2), the receptor for CCL2, is mainly expressed by monocytes and macrophages. The CCL2/CCR2 signaling pathway has been implicated in postischemic inflammatory response, and pharmacological inhibition or genetic targeting of the CCL2/CCR2 pathway might represent an attractive approach to blunt excessive inflammation and prevent detrimental ventricular remodeling.7–12 Various cytokines promote adhesive interactions between leukocytes and endothelial cells, resulting in the transmigration of inflammatory cells into the site of injury.
The recruitment of inflammatory cells is a dynamic and superbly orchestrated process comprising sequential infiltration of the injured myocardium with neutrophils, mononuclear cells, dendritic cells (DCs), and lymphocytes.3,4,13 Neutrophils migrate into the infarcted myocardium during the first hours after the onset of ischemia and peak after one day.1 Thereafter, monocytes and their descendant macrophages dominate the cellular infiltration and release inflammatory mediators, reactive oxygen species, and proteolytic enzymes, contributing to the initiation and resolution of inflammation, phagocytosis, proteolysis, angiogenesis, infarct healing, and ventricular remodeling.1,3,14 Meanwhile, DCs and T lymphocytes are recruited into the injured myocardium, contributing to wound healing and ventricular remodeling.15–17 Both detrimental effects and the beneficial role of these inflammatory cells have been documented in the pathophysiology of MI and reperfusion injury, and the diverse and seemingly conflicting roles may be attributable to the subset heterogeneity and functional diversity of the inflammatory cells.3,4
Inflammation in MI and Reperfusion Injury
MI/R triggers a complex inflammatory reaction accompanied by cytokine release and inflammatory leukocyte infiltration into the endangered myocardial region.1,3,4,18 Although the inflammatory response and cytokine elaboration after MI/R are integral to the healing process and contribute to left ventricular (LV) remodeling, excessive inflammatory responses after MI/R injury are detrimental for cell survival and extracellular matrix integrity via an enhanced activation of proapoptotic signaling pathways, with subsequent poor clinical outcome.3,19,20 These findings suggest that an inflammatory reaction is essential for the healing process, and thus, no effective therapeutic strategy against inflammation has been established.4
It has been widely accepted that myocardial infarct healing and post-MI remodeling are processes in which leukocytes, cytokines, and chemokines play both a beneficial role and a detrimental role.3,4,20 Recent studies have demonstrated that recruited monocytes/macrophages persist for days in the infarct zone and contribute to inflammation, phagocytosis, proteolysis, angiogenesis, and collagen deposition. Reduced macrophage infiltration resulted in decreased inflammation, diminished interstitial fibrosis, and attenuated LV remodeling and dysfunction. On the other hand, macrophage participation is integral to wound healing and tissue repair after MI.3,4,21 As shown in Figure 1, these diverse and seemingly contrasting functions might be attributed to macrophage heterogeneity, which is characterized by differential activation, distinct phenotypes, and diverse functions. Hence, the challenge lies in ameliorating the detrimental inflammatory response while not affecting the tissue repair response.
Figure 1.
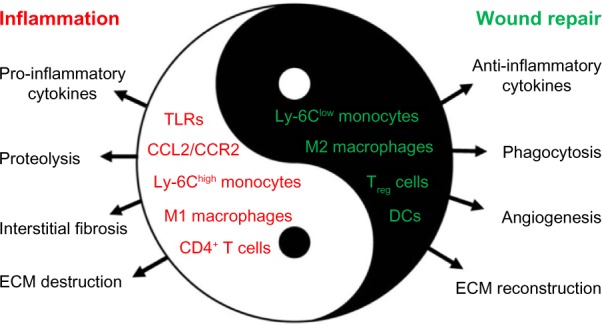
Diverse roles of inflammation and inflammatory cells in the pathophysiology of MI and reperfusion injury. Inflammation is not only involved in reperfusion injury and postinfarction remodeling but is also integral to wound healing and tissue repair after MI. Inflammatory cell subsets and function heterogeneity account for both the detrimental and beneficial roles of monocytes/macrophages, DCs, and lymphocytes in the pathophysiology of reperfusion injury and infarct healing.
Innate Immune System in MI and Reperfusion Injury
The innate immune system is activated after various forms of tissue injury and triggers immune responses in the host.22 The role of innate immune responses in cardiac ischemic injury and tissue repair has been shown to be more pivotal than first thought. The innate immune system contributes importantly to the progression of myocarditis and the remodeling process after MI.23–25 Our understanding of the pathogenesis of MI/R injury became much clearer with the discovery of TLRs. TLRs are expressed by leukocytes and recognize pathogen-associated molecular patterns and endogenous danger signals released during cell death.22 TLRs are also expressed in cells with no direct role in host innate immune responses, such as endothelial cells and cardiomyocytes.26 On activation, TLRs exert their inflammatory response through nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) translocation to the nucleus.5,6,27 Thus, TLRs hold great promise as a therapeutic target within the innate immune system, for cardiac ischemia and other conditions, without affecting host defense or proper scar formation after infarction.
Modulating TLR activation may enhance the benefits, blunt the negative effects of the inflammatory response, and provide new therapeutic options after MI/R injury. This concept is supported by observations in TLR knockout mice.28–31 TLR4-deficient mice sustained smaller infarctions and exhibited less inflammation after MI/R injury.28 Ex vivo experiments showed that TLR2−/− hearts performed better than wild-type hearts after MI/R injury,29 and TLR2−/− mice were protected against endothelial dysfunction after MI/R injury.30 Circulating TLR2 was also demonstrated to mediate MI/R injury. Administration of a TLR2 antagonist just five minutes before reperfusion reduced infarct size and improved cardiac performance and geometry. Furthermore, antagonizing TLR2 reduced inflammation and cell death after infarction.31 Thus, TLR2 has been established as a new therapeutic target for the treatment of acute ischemic and reperfusion injury, even when it is initiated in the late ischemic period.
In the setting of MI, deficient TLR2 or TLR4 signaling in mice prevented adverse cardiac remodeling, resulting in preserved cardiac function and geometry after MI.24,32 In addition to TLR signaling, activation of the innate immune system through interleukin-1 receptor-associated kinase 4 (IRAK-4) signaling was important for bone marrow-derived DC mobilization and maturation, contributing to post-MI mortality and adverse remodeling. In IRAK-4 knockout mice, attenuation of toll/interleukin-1 receptor signaling resulted in lower expression of cytokines and decreased inflammation through blunted innate immune response.15
As the cause of inappropriate activation of the inflammatory response after MI, an autoimmune reaction is a possible mechanism of unnecessary inflammatory reactions induced secondary to myocardial injury.33,34 It has been demonstrated that cardiac myosin acts as an endogenous ligand for TLR2 and TLR8,35 and the presence of autoimmunity to cardiac myosin and troponin is associated with an adverse clinical outcome after MI.34,36,37 Additionally, lymphocytes obtained from the spleen of rats that have suffered MI can injure normal cardiomyocytes,38 and heart failure was induced by adoptive transfer of splenic lymphocytes from rats after MI.33 Furthermore, mice preimmunized with murine cardiac troponin I displayed greater infarct size, increased more significant fibrosis, higher inflammation score, and more cardiac dysfunction after MI.37 These findings indicate that myocardial damage results in the release of not only endogenous ligands of TLRs but also self-antigens. Taken together, autoimmune responses against myocardial antigens may contribute to secondary myocardial injury after MI and may be a new mechanism of maladaptive LV remodeling after MI.
CCL2/CCR2 Signaling in MI and Reperfusion Injury
Chemokine expression is a prominent feature of the postinfarction inflammatory response, and as an essential player in inflammatory leukocyte trafficking, chemokines are involved in I/R injury, myocardial healing, infarct angiogenesis, and scar formation after MI.39 In addition, chemokines exert important effects on nonhematopoietic cells, such as endothelial cells, smooth muscle, and fibroblasts, and may modulate fibrous tissue deposition and wound angiogenesis.40
One of the extensively studied CC chemokines, CCL2, which was originally named monocyte chemoattractant 1, is a potent chemoattractant for monocytes, T cells, and NK cells and has been implicated in a wide variety of diseases characterized by monocyte-rich leukocyte infiltrates.40 CCL2 upregulation has been observed in murine, rat, and canine models of MI/R.7,41 In the canine model of reperfused infarction, induction of CCL2 mRNA occurred only in ischemic segments within the first hour of reperfusion, peaked at three hours, and was localized by immunostaining on the venular endothelium.7 CCL2 mRNA levels were increased by 40-fold in the noninfarcted LV one day after left coronary artery ligation, and increased levels persisted for 28 days.7 CCL2 expression was also increased in both experimental and clinical heart failures.42,43 Enhanced myocardial CCL2 expression contributed to reperfusion injury, infarct healing, and ventricular remodeling through the following two mechanisms: CCL2-induced infiltration and activation of inflammatory cells, such as monocytes/macrophages and lymphocytes,7,9,10,39 and CCL2-induced transcription factor causing cardiac cell death and ventricular dysfunction.44 CCL2 also promotes the induction of other cytokines, matrix metalloproteinase, and transforming growth factor-β through an autocrine/paracrine mechanism,40,45,46 thus modulating fibrous tissue deposition and wound healing.
Given the essential effects of CCL2 signaling on different cell types involved in the postischemia inflammatory response, the pharmacological inhibition or genetic targeting of CCL2/CCR2 signaling might represent an attractive approach to blunt excessive inflammation and decrease monocyte/macrophage infiltration, thereby promoting infarct healing and preventing detrimental ventricular remodeling. In agreement with this concept is the fact that CCL2-deficient mice display a decreased and delayed macrophage infiltration and myofibroblast accumulation associated with a diminished interstitial fibrosis, improvement of LV dysfunction and regional hypocontractility after MI/R.9,10 Similarly, administering a CCL2 competitor reduced inflammatory monocyte recruitment, limited neointimal hyperplasia, and attenuated MI/R injury in mice.12 Moreover, anti-CCL2 gene therapy improved the post-MI survival rate, which was associated with a decreased macrophage recruitment and an attenuated contractile dysfunction, interstitial fibrosis, and LV cavity dilatation.7 Genetic deletion of CCR2 resulted in a decreased macrophage infiltration and a reduced TNF-α and matrix metalloproteinase expressions, which might contribute to the attenuation of LV remodeling after MI.8 Nahrendorf et al showed that pretreatment of mice for three days with a lipid nanoparticle that encapsulated a short interfering RNA targeting CCR2 prior to the induction of MI/R injury resulted in reduced numbers of monocytes and macrophages in the heart and reduced the infarct size by 34%.14
Monocytes/Macrophages in Infarct Healing and Reperfusion Injury
The recruitment of inflammatory cells is a dynamic, well-organized process with sequential infiltration of the injured myocardium with neutrophils, mononuclear cells, DCs, and lymphocytes.3,4,20 A growing number of studies have demonstrated that recruited monocytes/macrophages persist for days in the infarct zone and contribute to inflammation, phagocytosis, proteolysis, angiogenesis, collagen deposition, and ventricular remodeling in the setting of myocardial reperfusion injury and postinfarction healing.7–10,19,20,47–49 On one hand, excessive and prolonged infiltration of inflammatory macrophages into the infarct myocardium is harmful, contributing to excessive inflammatory response, tissue destruction, interstitial fibrosis, cardiac dysfunction, and adverse ventricular remodeling.7–10 On the other hand, a controlled recruitment of macrophages is essential to wound healing and tissue repair through phagocytosis of necrotic cells, facilitating angiogenesis and extracellular matrix reconstruction in the ischemia-injured myocardium and the infarct.48,49 These diverse and seemingly conflicting functions may be attributable to macrophage heterogeneity as characterized by differential activation, distinct phenotypes (M1: classically activated macrophages and M2: alternatively activated macrophages), and diverse functions, both pathogenic and protective.50–52
The subset heterogeneity and function diversity also hold true for the monocytes. As reported by Nahrendorf et al.14, infarcted hearts modulate their chemokine expression profile over time, and they sequentially and actively recruit Ly-6Chigh and Ly-6Clow monocytes via CCR2 and CX3CR1, respectively. Ly-6Chigh monocytes dominate early and exhibit phagocytic, proteolytic, and inflammatory functions. Ly-6Clow monocytes dominate later. Ly-6Chigh monocytes digest damaged tissue, whereas Ly-6Clow monocytes promote healing via myofibroblast accumulation, angiogenesis, and collagen deposition.3,4,14 Thus, a therapeutic strategy targeting CCR2+ monocyte/macrophage migration is a promising approach for treating numerous inflammatory diseases without disrupting inflammation resolution, which is associated with noninflammatory monocytes and alternatively activated macrophages. Consistent with this concept, recent comprehensive analysis of mouse cardiac macrophage subsets in a steady state and during inflammation have revealed that proinflammatory macrophages, which comprise half of all Ly-6C+ monocytes and MHCIIhigh CD11chigh CCR2high macrophages, specifically express CCR2.53 This finding might explain why short interfering RNA, targeting the CCR2 axis that blocks the monocyte/macrophage lineage, was successful in preventing the ischemic cardiac injury.14
The role of monocytes/macrophages in MI/R injury and postinfarction healing is a double-edged sword, as illustrated in Figure 1. Monocyte/macrophage recruitment is integral to the infarct healing; on the other hand, an uncontrolled inflammatory cell infiltration may exacerbate reperfusion injury and compromise the reparative functions mediated by these cells. Thus, the challenge is how to ameliorate the detrimental effects of proinflammatory monocytes/macrophages, while sparing the beneficial roles of the reparative or regulatory macrophages. The existence of monocyte/macrophage subset heterogeneity and their biphasic recruitment in response to ischemic injury provide a possible solution. A recent study has demonstrated that knocking down interferon regulatory factor 5, which is a critical transcription factor favoring M1 polarization, decreased infiltration of M1 and ameliorated inflammation following MI, thereby improving infarct healing.54 Additionally, nuclear receptor subfamily 4, group a, member 1, was demonstrated to be essential to Ly-6Clow monocyte production. In the absence of nuclear receptor subfamily 4, group a, member 1, Ly-6Chigh monocytes expressed increased levels of CCR2 on their surface, avidly infiltrated the myocardium, and differentiated to proinflammatory macrophages, resulting in defective healing and compromised heart function.55 These findings suggest that therapeutics targeting distinct macrophage lineages by genetic manipulation of polarity-determining genes may serve as new treatments for cardiovascular diseases.
Stem cell therapy has been considered as the promising therapeutic in treating cardiovascular diseases, and many preclinical studies and clinical trials reported beneficial effects of stem cell therapy on infarct healing, although the underlying mechanism remains poorly understood. A recent study has provided an insightful explanation that the cardioprotective effects of mesenchymal stromal cell treatment might be attributed to the cross talk between injected stem cells and macrophages.56 Mesenchymal stromal cell treatment reshaped the macrophage response by favoring M2 polarization, resulting in increased numbers of M2 and changed cytokine profile of macrophages, thereby improving postinfarction healing.56
DCs in MI and Reperfusion Injury
DCs and their precursors are considered sentinels of the immune system, and they circulate through the blood and nonlymphoid peripheral tissues, where they become resident cells over time.57 After pathogen invasion or tissue injury, DC precursors accumulate rapidly in the local infected or injured tissues.57 As early as 1993, it was demonstrated that DCs infiltrated the injured myocardium and participated in the activation of lymphocytes after MI.58 However, both detrimental effects and beneficial or regulatory roles of DCs in the postinfarction healing and remodeling were observed.15,16,59 It has been demonstrated that G-CSF improved early postinfarction LV remodeling through decreased DCs’ infiltration and suppression of DC-mediated immunity.59 Bone marrow-derived DCs’ mobilization, mediated via IRAK-4 signaling, contributed to postinfarction myocardium apoptosis, Th1 cytokine expression, and interstitial fibrosis, leading to an increased mortality and an adverse LV remodeling.15 By contrast, in transgenic mice expressing diphtheria toxin receptor on DCs, which allowed the investigator to specifically deplete DCs by injecting diphtheria toxin, DCs were demonstrated to be a potent immunoprotective regulator during the postinfarction healing process via control of monocyte/macrophage homeostasis.16 In addition, DCs were suggested to regulate the development of autoimmune heart failure through the recognition of heart-specific peptides.60
Lymphocytes in MI and Reperfusion Injury
In addition to neutrophils, monocytes/macrophages, and DCs, lymphocytes are also present in the ischemic and reperfused myocardia and the infarct. CD4+ T lymphocytes accumulate in the infarct zone early during reperfusion, and the infarct-sparing effect of adenosine A2A receptor activation is primarily due to inhibition of CD4+ T cells’ infiltration and activation in the reperfused heart.61 Hofmann et al.17 demonstrated that CD4+ T cells proliferated in draining lymph nodes shortly after ischemic injury, and CD4+ T-cell-deficient mice displayed higher total numbers of leukocytes and proinflammatory monocytes and increased LV dilation as determined by serial echocardiography up to day 56 after MI. Additionally, Foxp3+ CD4+ regulatory T cells (Treg) contributed to inflammation resolution and beneficially influenced infarct healing by modulating monocyte/macrophage differentiation. Mechanistically, Treg cell depletion was associated with M1-like polarization, characterized by decreased expression of inflammation-resolving and healing-promoting factors. Therapeutic Treg cell activation induced an M2-like differentiation within the healing myocardium, associated with myofibroblast activation and increased expression of monocyte/macrophage-derived proteins, fostering wound healing.62
Apart from T lymphocytes, the interaction between mature B lymphocytes and monocytes was also involved in myocardial ischemic injury and maladaptive LV remodeling. Mature B lymphocytes selectively produce CCL7 and induce Ly6Chigh monocyte mobilization and recruitment to the heart, leading to an enhanced tissue injury and deterioration of myocardial function. Genetic or antibody-mediated depletion of mature B lymphocytes impeded CCL7 production and monocyte mobilization, attenuated myocardial injury, and improved cardiac function.63 Collectively, these findings suggest that therapeutic modulation of lymphocytes constitutes a new approach to improve infarct healing post-MI.
Perspective
Postinfarction immunoinflammation, as characterized by inflammatory cell infiltration and the activation of innate and adaptive immune responses, is essential to cardiac injury and repair. The inflammatory cascade may provide unique opportunities for interventions aimed at reducing cardiomyocyte injury while optimizing the healing response and attenuating adverse remodeling. Timely resolution of the inflammatory infiltrate and spatial containment of the inflammatory and reparative response into the infarcted area are essential for optimal infarct healing. For instance, targeting proinflammatory leukocyte subsets, to dampen detrimental inflammation while sparing the wound healing roles of the anti-inflammatory monocytes/macrophages, may reduce cardiomyocyte injury and adverse remodeling. Despite the challenges ahead, we are hopeful that new therapies for myocardial ischemia and reperfusion will soon be integrated into clinical practice.
Abbreviations
- CCL2
CC chemokine ligand 2
- CCR2
CC chemokine receptor 2
- DCs
dendritic cells
- IRAK-4
Interleukin-1 receptor-associated kinase-4
- LV
left ventricular
- M1
classically activated macrophages
- M2
alternatively activated macrophages
- MI
myocardial infarction
- MI/R
myocardial ischemia reperfusion
- MMP
matrix metalloproteinase
- Nr4a1
nuclear receptor subfamily 4, group a, member 1
- TLRs
toll-like receptors
- Tre.g.
Foxp3+ CD4+ regulatory T cells
Footnotes
ACADEMIC EDITOR: Thomas E. Vanhecke, Editor in Chief
PEER REVIEW: Seven peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1406 words, excluding any confidential comments to the academic editor.
FUNDING: This article was supported by the National Natural Science Foundation of China (81000088). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Wrote the first draft of the manuscript: JLiu. Contribution to the writing and the revision of the manuscript: HW, JLi. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357(11):1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 2.Ibáñez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–71. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 3.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121(22):2437–45. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339(6116):161–6. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marchant DJ, Boyd JH, Lin DC, Granville DJ, Garmaroudi FS, McManus BM. Inflammation in myocardial diseases. Circ Res. 2012;110(1):126–44. doi: 10.1161/CIRCRESAHA.111.243170. [DOI] [PubMed] [Google Scholar]
- 6.Coggins M, Rosenzweig A. The fire within: cardiac inflammatory signaling in health and disease. Circ Res. 2012;110(1):116–25. doi: 10.1161/CIRCRESAHA.111.243196. [DOI] [PubMed] [Google Scholar]
- 7.Hayashidani S, Tsutsui H, Shiomi T, et al. Anti-mcp-1 gene therapy attenuates left ventricular remodeling and failure after experimental MI. Circulation. 2003;108(17):2134–40. doi: 10.1161/01.CIR.0000092890.29552.22. [DOI] [PubMed] [Google Scholar]
- 8.Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol. 2004;165(2):439–47. doi: 10.1016/S0002-9440(10)63309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dewald O, Zymek P, Winkelmann K, et al. CCL2/MCP-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96(8):881–9. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 10.Frangogiannis NG, Dewald O, Xia Y, et al. Critical role of MCP-1 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–92. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- 11.Leuschner F, Dutta P, Gorbatov R, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29(11):1005–10. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liehn EA, Piccinini AM, Koenen RR, et al. A new monocyte chemotactic protein-1/chemokine CC motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J Am Coll Cardiol. 2010;56(22):1847–57. doi: 10.1016/j.jacc.2010.04.066. [DOI] [PubMed] [Google Scholar]
- 13.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110(1):159–73. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–47. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maekawa Y, Mizue N, Chan A, et al. Survival and cardiac remodeling after myocardial infarction are critically dependent on the host innate immune interleukin-1 receptor-associated kinase-4 signaling: a regulator of bone marrow-derived dendritic cells. Circulation. 2009;120(14):1401–14. doi: 10.1161/CIRCULATIONAHA.109.865956. [DOI] [PubMed] [Google Scholar]
- 16.Anzai A, Anzai T, Nagai S, et al. Regulatory role of DCs in postinfarction healing and left ventricular remodeling. Circulation. 2012;125(10):1234–45. doi: 10.1161/CIRCULATIONAHA.111.052126. [DOI] [PubMed] [Google Scholar]
- 17.Hofmann U, Beyersdorf N, Weirather J, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125(13):1652–63. doi: 10.1161/CIRCULATIONAHA.111.044164. [DOI] [PubMed] [Google Scholar]
- 18.Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med. 2011;17(11):1391–401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and post-myocardial infarction remodeling. Circ Res. 2004;94(12):1543–53. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 20.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58(2):88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinberger T, Schulz C. Myocardial infarction: a critical role of macrophages in cardiac remodeling. Front Physiol. 2015;6:107. doi: 10.3389/fphys.2015.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 23.Marty RR, Dirnhofer S, Mauermann N, et al. MyD88 signaling controls autoimmune myocarditis induction. Circulation. 2006;113(2):258–65. doi: 10.1161/CIRCULATIONAHA.105.564294. [DOI] [PubMed] [Google Scholar]
- 24.Shishido T, Nozaki N, Yamaguchi S, et al. TLR2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;108(23):2905–10. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- 25.Valeur HS, Valen G. Innate immunity and myocardial adaptation to ischemia. Basic Res Cardiol. 2009;104(1):22–32. doi: 10.1007/s00395-008-0756-6. [DOI] [PubMed] [Google Scholar]
- 26.Frantz S, Kobzik L, Kim YD, et al. TLR4 expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999;104(3):271–80. doi: 10.1172/JCI6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akira S, Takeda K. TLR signaling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 28.Oyama J, Blais C, Jr, Liu X, et al. Reduced myocardial ischemia-reperfusion injury in TLR4-deficient mice. Circulation. 2004;109(6):784–9. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 29.Sakata Y, Dong JW, Vallejo JG, et al. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;292(1):H503–9. doi: 10.1152/ajpheart.00642.2006. [DOI] [PubMed] [Google Scholar]
- 30.Favre J, Musette P, Douin-Echinard V, et al. TLR2–deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27(5):1064–71. doi: 10.1161/ATVBAHA.107.140723. [DOI] [PubMed] [Google Scholar]
- 31.Arslan F, Smeets MB, O’Neill LA, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti–toll-like receptor-2 antibody. Circulation. 2010;121(1):80–90. doi: 10.1161/CIRCULATIONAHA.109.880187. [DOI] [PubMed] [Google Scholar]
- 32.Timmers L, Sluijter JP, van Keulen JK, et al. TLR4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res. 2008;102(2):257–64. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 33.Maisel A, Cesario D, Baird S, Rehman J, Haghighi P, Carter S. Experimental autoimmune myocarditis produced by adoptive transfer of splenocytes after myocardial infarction. Circ Res. 1998;82(4):458–63. doi: 10.1161/01.res.82.4.458. [DOI] [PubMed] [Google Scholar]
- 34.Moraru M, Roth A, Keren G, George J. Cellular autoimmunity to cardiac myosin in patients with a recent myocardial infarction. Int J Cardiol. 2006;107(1):61–6. doi: 10.1016/j.ijcard.2005.02.036. [DOI] [PubMed] [Google Scholar]
- 35.Zhang P, Cox CJ, Alvarez KM, Cunningham MW. Cutting edge: cardiac myosin activates innate immune responses through TLRs. J Immunol. 2009;183(1):27–31. doi: 10.4049/jimmunol.0800861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eriksson S, Hellman J, Pettersson K. Autoantibodies against cardiac troponins. N Engl J Med. 2005;352(1):98–100. doi: 10.1056/NEJM200501063520123. [DOI] [PubMed] [Google Scholar]
- 37.Göser S, Andrassy M, Buss SJ, et al. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation. 2006;114(16):1693–702. doi: 10.1161/CIRCULATIONAHA.106.635664. [DOI] [PubMed] [Google Scholar]
- 38.Varda-Bloom N, Leor J, Ohad DG, et al. Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J Mol Cell Cardiol. 2000;32(12):2141–9. doi: 10.1006/jmcc.2000.1261. [DOI] [PubMed] [Google Scholar]
- 39.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost. 2007;97(5):738–47. [PubMed] [Google Scholar]
- 40.White GE, Iqbal AJ, Greaves DR. CC chemokine receptors and chronic inflammation – therapeutic opportunities and pharmacological challenges. Pharmacol Rev. 2013;65(1):47–89. doi: 10.1124/pr.111.005074. [DOI] [PubMed] [Google Scholar]
- 41.Kumar AG, Ballantyne CM, Michael LH, et al. Induction of MCP-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation. 1997;95(3):693–700. doi: 10.1161/01.cir.95.3.693. [DOI] [PubMed] [Google Scholar]
- 42.Behr TM, Wang X, Aiyar N, et al. MCP-1 is upregulated in rats with volume: overload congestive heart failure. Circulation. 2000;102(11):1315–22. doi: 10.1161/01.cir.102.11.1315. [DOI] [PubMed] [Google Scholar]
- 43.Aukrust P, Ueland T, Müller F, et al. Elevated circulating levels of C-C chemokines in patients with congestive heart failure. Circulation. 1998;97(12):1136–43. doi: 10.1161/01.cir.97.12.1136. [DOI] [PubMed] [Google Scholar]
- 44.Zhou L, Azfer A, Niu J, et al. MCP-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ Res. 2006;98(9):1177–85. doi: 10.1161/01.RES.0000220106.64661.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto T, Eckes B, Mauch C, et al. MCP-1 enhances gene expression and synthesis of matrix metalloproteinase-1 in human fibroblasts by an autocrine IL-1 alpha loop. J Immunol. 2000;164(12):6174–9. doi: 10.4049/jimmunol.164.12.6174. [DOI] [PubMed] [Google Scholar]
- 46.Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and TGF-beta1 gene expression by MCP-1 via specific receptors. J Biol Chem. 1996;271(30):17779–84. doi: 10.1074/jbc.271.30.17779. [DOI] [PubMed] [Google Scholar]
- 47.Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130(2):147–58. doi: 10.1016/j.ijcard.2008.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischaemic heart diseases. Cardiovasc Res. 2014;102(2):240–8. doi: 10.1093/cvr/cvu025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujiu K, Wang J, Nagai R. Cardioprotective function of cardiac macrophages. Cardiovasc Res. 2014;102(2):232–9. doi: 10.1093/cvr/cvu059. [DOI] [PubMed] [Google Scholar]
- 50.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 51.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Epelman S, Lavine KJ, Beaudin AE, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40(1):91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Courties G, Heidt T, Sebas M, et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol. 2014;63(15):1556–66. doi: 10.1016/j.jacc.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hilgendorf I, Gerhardt LM, Tan TC, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114(10):1611–22. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ben-Mordechai T, Holbova R, Landa-Rouben N, et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol. 2013;62:1890–901. doi: 10.1016/j.jacc.2013.07.057. [DOI] [PubMed] [Google Scholar]
- 57.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 58.Zhang J, Yu ZX, Fujita S, Yamaguchi ML, Ferrans VJ. Interstitial dendritic cells of the rat heart: quantitative and ultrastructural changes in experimental myocardial infarction. Circulation. 1993;87(3):909–20. doi: 10.1161/01.cir.87.3.909. [DOI] [PubMed] [Google Scholar]
- 59.Naito K, Anzai T, Sugano Y, et al. Differential effects of GM-CSF and G-CSF on infiltration of dendritic cells during early left ventricular remodeling after myocardial infarction. J Immunol. 2008;181(8):5691–701. doi: 10.4049/jimmunol.181.8.5691. [DOI] [PubMed] [Google Scholar]
- 60.Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH. Dendritic cell induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med. 2003;9(12):1484–90. doi: 10.1038/nm960. [DOI] [PubMed] [Google Scholar]
- 61.Yang Z, Day YJ, Toufektsian MC, et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114(19):2056–64. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 62.Weirather J, Hofmann UD, Beyersdorf N, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115(1):55–67. doi: 10.1161/CIRCRESAHA.115.303895. [DOI] [PubMed] [Google Scholar]
- 63.Zouggari Y, Ait-Oufella H, Bonnin P, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19(10):1273–80. doi: 10.1038/nm.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]