

Abstract
Understanding the process that underlies multi-drug recognition and efflux by P-glycoprotein (ABCB1) remains a key biological challenge. Structural data has recently become available for the murine and C. elegans homologues of ABCB1; however all structures were obtained in the absence of nucleotide. A feature of these structures was the presence of a central cavity that is inaccessible from the extracellular face of the protein. To determine the conformational dynamics of this region several residues in transmembrane helices TM6 (331, 343 and 354) and TM12 (980) were mutated to cysteine. Based upon structural predictions these residues are proposed to line, or reside proximal to, the central cavity. The mutant isoforms were labelled with a paramagnetic probe enabling the application of electron paramagnetic resonance (EPR) spectroscopic methods. Power saturation EPR spectra were recorded in the presence of hydrophobic (O2) or hydrophilic (NiEDDA) quenching agents to study the local environment of each residue. ABCB1 was trapped in both its nucleotide bound and post-hydrolytic conformations and EPR spectra were again recorded in the presence and absence of quenching agents. The EPR line shapes provide information on the movements of these residues within TM6 and TM12 during ATP hydrolysis. Rationalisation of the data with molecular dynamic simulations indicate that the cavity is converted to a configuration open to the aqueous phase following nucleotide binding, thereby suggesting alternating access to the cavity on opposite sides of the membrane during translocation.
Keywords: Multi-drug resistance, P-glycoprotein, ABC transporter, membrane transport, electron paramagnetic resonance (EPR), cancer chemotherapy, molecular model, ABCB1, molecular dynamics simulation
Introduction
The ATP Binding Cassette (ABC) transporter ABCB1 (known as P-glycoprotein) is an efflux pump that confers resistance to cancer chemotherapy [1]. Further, it is also localised to numerous sanctuary and excretory sites within the body and, thereby, influences the absorption, distribution and elimination of a wide range of drugs [2]. This broad specificity of ABCB1 remains an enigmatic property as does its molecular mechanism of action [3, 4].
ABCB1 contains twelve helices within two transmembrane domains (TMDs), and a pair of nucleotide binding domains (NBDs), all part of a single polypeptide chain. Drug translocation requires the coupling of ATP hydrolysis at the NBDs to drug binding/release by the TMDs. Previous structural and biophysical investigations have demonstrated that events during the process of ATP hydrolysis do indeed initiate conformational changes in the NBD domains [5–9]. The simplest demonstration of this coupling is the ability of drug substrates to stimulate the rate of ATP hydrolysis. Furthermore, this communication is bi-directional since the drug binding sites of ABCB1 undergo significant changes in affinity for substrates as the protein moves between intermediates during ATP hydrolysis [6, 10]. Data from these biochemical investigations have shaped the “power stroke” model used to describe drug transport by ABCB1. Similar processes have also been observed for the multidrug efflux pumps ABCC1 and ABCG2 [11, 12]. The regions of ABCB1 mediating this coupling remain unknown. Existing data appear to implicate the cytosolic “loops” between TM helices 4/5 and TM helices 10/11, and the TM helices 6 and 12 [13–16]. The latter helices are directly linked along the polypeptide chain to the NBDs of ABCB1. Previous work has provided a considerable amount of data in support of a role for TM6/TM12 in the coupling process [17–19].
There have been two X-ray crystallographic models of the structure of both murine (3.8 Å resolution) [13] and Caenorhabditis elegans ABCB1 (3.4 Å resolution) [16]. The TM helices form a “clam shell” cavity with the NBDs separated by approximately 30Å. Early electron microscopy-based structural investigations in three conformational states also support the existence of an aqueous filled central cavity [7, 20–23]. However, the low resolution precludes understanding of the molecular details and dynamics of the transition between states. While the two X-ray crystallographic structures [5,6] have shown a central aqueous cavity that is occluded at the extracellular surface by the convergence of several transmembrane helices they also show the NBDs separated by a substantial distance, which would infer the requirement for substantial conformational changes upon ATP binding. The significance of this observation has caused considerable debate (for example [24]). Since these X-ray structures have only been observed for ABCB1 in its basal (i.e. nucleotide and drug free) conformation they represent only a single static snapshot of this dynamic translocation process. This leaves many questions about the mechanism of translocation unanswered. In order to help resolve such questions and characterise the various conformations of ABCB1 in the subsequent stages of translocation, dynamic information is required.
In the present work EPR spectroscopy of nitroxide spin labels (SL) covalently attached at intervals along TMs 6 and 12 together with molecular dynamics (MD) simulation have been brought together to generate topographical information about ABCB1. This includes data on the hydrophilicity of the central cavity in the basal conformation as well as in its nucleotide bound and post-hydrolytic states. Taken together, this information provides novel insights into the dynamic conformational transitions occurring and the access of hydrophobic versus hydrophilic agents to the transmembrane cavity of ABCB1.
Results
Protein biochemistry and labelling of ABCB1 with MTSL
The site-directed spin-labelling (SDSL) technique uses variant recombinant proteins that contain a single reactive amino acid, usually cysteine, which can be conjugated with a paramagnetic spin-label moiety. In this work, four mutant single cysteine isoforms of ABCB1 were purified using immobilised metal affinity chromatography (IMAC) and subsequently reconstituted into liposomes. As demonstrated in Figure 1 (for the 980C-isoform), the purity was greater than 85% and similar to that previously reported [17–19]. None of the mutations had been shown to affect adversely the rate of ATP hydrolysis or its stimulation by nicardipine [17–19, 25], which was a key factor in their choice for this investigation. To ensure that active protein was used for EPR analysis only reconstituted samples whose nicardipine stimulated (30 μM) ATPase activity exceeded 400 nmol Pi/min/mg were retained, in agreement with the previously reported acceptable values for these isoforms [17–19].
Figure 1. Purification of MTSL-labelled 980C-ABCB1.

Fractions were collected at each stage of the IMAC and PD-10 based purification strategy. The figure corresponds to a 7.5% SDS-PAGE gel stained with PageBlue. FT refers to the flow through following binding stage and represents 0.05% of the total fraction volume. All wash fractions and the elution (E) represent 1% of the total volume, whilst the post PD-10 fraction (GF) corresponds to 0.05% of the volume. Mw refers to the molecular weight markers. All fractions were TCA precipitated prior to the electrophoresis.
It has previously been demonstrated that mutation of the endogenous residue to a cysteine at positions 331, 343, 354 and 980 does not alter the Michaelis-Menten characteristics for ATP hydrolysis by ABCB1 [25–27]. Similarly, the potency and the extent of stimulation by the modulator nicardipine and the substrate vinblastine were unaffected. These investigations also measured the effects of covalently modifying the introduced cysteine on ATPase activity. The addition of coumarin-maleimide (CM) did not alter any of the ATPase parameters for any mutant isoform [19, 28, 29]. CM is significantly larger than MTSL and unlikely to perturb function to a greater extent. Single point assays of ATPase activity in the presence of MTSL did not reveal any modification of the nicardipine stimulated activity.
In parallel, the question of whether the insertion of cysteine residues and their labelling by MTSL had effects on the structural conformation of nucleotide and substrate free ABCB1 (nucleotide and substrate free state) was addressed by MD simulations (Figure S1). The Cα root mean squared deviation (RMSD) of the TMDs of labelled ABCB1 isoforms with respect to the starting membrane-embedded conformation of ABCB1 [24] plateaued between 2-3Å. Thus the MD demonstrated that incorporation of the cysteine mutation and the covalently linked MTSL probe had only minor effects on the structural conformation of the ABCB1 TMDs, consistent with functional data from the biochemical studies.
When ABCB1 is reconstituted into proteoliposomes about 70% is oriented such that the NBD’s are facing the outside of the proteoliposome (data not shown). Hence the protocols used to trap the various pre and post-hydrolytic states should result in conformational change in only these 70% of the protein – assuming the proteolipsomes are non-leaky.
EPR spectral line shapes of nitroxide-labelled ABCB1 isoforms in different conformational states
Representative continuous wave (cw) EPR spectra of the four ABCB1 isoforms, 980C of TM12 and 331C, 343C and 354C of TM6, are shown in Figure 2. Three conformational states were examined: nucleotide free, AMP-PNP bound (pre-hydrolytic) and vanadate-trapped (post-hydrolytic). The overall EPR spectral line shape was rather broad and in the case of the TM helix 6 isoforms, an additional small amount of unresolved sharp signal characteristic of highly mobile, unattached spin label is present due to free label present, probably trapped within the proteoliposomes. In each case the broad nature of the spectral line-shape is characteristic of highly restricted motion. Motions of the SL on the ps-ns time scale result in magnetic relaxation that determines the EPR spectral line shape. Fast protein backbone fluctuations on the ns time scale may also affect the EPR line shape. The line shapes observed arise from highly immobilised labels or are a weighted sum of different immobilised states of the label with slow, or no, inter-conversion between them. There were no significant changes in the observed line shapes when the conformation was changed in any of the ABCB1 isoforms. The cysteine-less isoform, used as a control, gave no EPR spectrum when subjected to the same MTSL labelling protocol (data not shown).
Figure 2. cw-EPR spectra of MTSL-labelled ABCB1 in different conformational states.

EPR spectral line shapes of ABCB1 containing the nitroxide side chain at the indicated sites in different conformational states: basal (black trace), AMP-PNP bound (grey trace) and ADP/Vi bound (red trace). Traces have been normalised for each isoform.
The Basal State: Comparison of cw EPR power saturation results and MD simulations
The extent of solvent exposure of a cysteine-attached nitroxide spin label can be determined by measuring its collision frequency with either a non-polar spin relaxation agent (oxygen) or a water soluble polar spin relaxant (NiEDDA). Soluble NiEDDA is easily accessible to aqueous protein surfaces (including within water accessible cavities) but not those in the protein interior or those exposed to the lipid bilayer. Oxygen is poorly soluble in aqueous solutions, but its solubility and diffusion coefficient are high in lipid bilayers and within a protein interior. The NiEDDA accessibility decreases within the lipid bilayer and is complementary to an increase in O2 accessibility. When combined with conformational trapping and structural data, the accessibility parameter, Π, and the derived polarity index, ɸ, isoforms (331C, 343C, 354C and 980C) have a SL that is exposed to the lipid phase, orientated towards the protein interior or accessible to an aqueous cavity.
Figure S2 demonstrates the representative power saturation curves of the spin labels to both the non-polar relaxation agent O2 and the polar relaxant NiEDDA for each isoform of ABCB1 in three key conformational states; nucleotide free, nucleotide bound and post-hydrolytic. EPR accessibility profile data from these curves are summarised in Table 1 (and Figure S3). In the nucleotide free conformation, isoforms 980C and 331C have relatively low Π values for O2 and NiEDDA implying low O2 and NiEDDA collision frequencies, respectively, indicating that these residues neither protrude into the hydrophobic core of the lipid bilayer nor into a solvent accessible cavity, but instead are predicted to be deeply buried in the protein interior excluding access of both O2 and NiEDDA.
Table 1. EPR accessibility profiles of MTSL-labelled ABCB1 isoforms.
| ISOFORM | STATE | ACCESSIBILITY PROFILE Π(O2) (x10-3) | ACCESSIBILITY PROFILE Π(NiEDDA) (x10-3) | POLARITY INDEX Փ |
|---|---|---|---|---|
| 980C | Basal | 9.61±1.06 | 2.90±0.97 | 1.20±0.106 |
| AMP-PNP | 17.05±2.09 | 8.73±0.51 | 0.67±0.070 | |
| ADP/Vi | 11.36±2.23 | 1.24±0.72 | 2.21±0.067 | |
| 331C | Basal | 9.83±0.84 | 6.40±0.60 | 0.43±0.082 |
| AMP-PNP | 31.84±3.30 | 10.90±0.42 | 1.07±0.038 | |
| ADP/Vi | 12.54±1.29 | 4.54±0.47 | 1.02±0.053 | |
| 343C | Basal | 21.33±9.36 | 6.31±3.71 | 1.22±0.217 |
| AMP-PNP | 32.33±4.29 | 49.76±0.27 | -0.43±0.205 | |
| ADP/Vi | 26.70±3.95 | 18.17±1.16 | 0.39±0.110 | |
| 354C | Basal | 32.34±1.21 | 21.06±1.93 | 0.43±0.064 |
| AMP-PNP | 29.17±4.75 | 7.24±0.50 | 1.39±0.044 | |
| ADP/Vi | 27.67±2.73 | 7.59±2.43 | 1.29±0.092 | |
The accessibility profile Π of the nitroxide spin label, attached to the indicated sites of ABCB1, to non-polar relaxation agent O2 and polar relaxant NiEDDA in different states: Basal, AMP-PNP and ADP/Vi. All measurements were from a minimum of three independent protein preparations to give an average value for Π. The Polarity Index, Փ, was calculated by:
Examination of the MD trajectories of 331C-MTSL ABCB1 indicates that 331C-MTSL is in a tightly packed region of the protein, located near the extracellular membrane-water interface, agreeing with EPR spectral line shapes. For example, the angular motion of the MTSL pyrrole nitrogen and the z-axis of the coordinate system fluctuated between just 40 and 43° (Figure S4d). This small angular fluctuation is consistent with the incorporation of MTSL in a region that has neighbouring steric constraints along the z-axis, such as the adjacent residues of an α-helix. A similar conclusion applies to the MTSL probe attached to 980C, for which the modelling orients the probe away from the translocation cavity but facing the protein interior and the vicinity of the cavity occlusion (Figure 3-4). Figure 3 shows the geometric description of the analysis of the MTSL-labelled cysteine at residue 980 and similar analyses were undertaken for all mutant isoforms, with the primary data presented in Figure S4.
Figure 3. Geometric descriptions of the MTSL angular motion shown for 980C-MTSL.

a) The angle between the MTSL pyrrol nitrogen vector (calculated with respect to the origin) and the z-axis.
b) The angle between the MTSL pyrrol nitrogen (calculated with respect to the Cα of the covalently bound cysteine residue) and the cysteine backbone normal vector through Cα.
c) The relative rotational angle of the vector between the MTSL pyrrol nitrogen and the cysteine Cα as its orientation changes throughout an MD simulation.
The initial orientation of 980C-MTSL is given as a solid CPK colouring and the orientation of 980CMTSL after 30ns in simulation run 3 is shown in transparent CKP colouring. The lipid headgroups are shown in liquorice CPK colouring. TMD1 is shown in bronze and TM helix 12 of TMD2 is shown in silver. The lipid tails and the remainder of TMD2 have been hidden for clarity.
Figure 4. Location of the four cysteine-linked MTSL probes.

The location and orientation of the cys-MTSL moiety incorporated into specific positions in the MD equilibrated membrane-embedded ABCB1 (starting conformation: adapted from PDB 3G5U-a) are shown in panels (a-d). The N- and C-terminal domains of ABCB1 are in bronze and grey ribbons, respectively. MTSL labelled 331C (red), 343C (green), 354C (blue) and 980C (purple) are shown in spacefill. The POPC headgroups are in liquorice with CPK colouring. The NBDs have been removed from the structures in panels (b-d) for clarity.
a) Structure of the MD equilibrated ABCB1 (PDBid: 3GU-a) viewed along the plane of the membrane.
b) View of MD equilibrated ABCB1 from the extracellular side of the membrane.
c) Side profile of MD equilibrated ABCB1.
d) The MD equilibrated ABCB1 TMD cavity, viewed from the intracellular side of the membrane.
Although these two isoforms are predicted to have a very low accessibility to the aqueous phase in the basal state, isoform 331C is more accessible to water than isoform 980C (Table 1). This is also reflected by the modelling, the solvent accessible surface area (SASA), reported here as the number of water contacts made, being relatively high for the 331C-MTSL isoform. There were 11.7±4.1 water molecules within a 0.4 nm contact radius of 331C-MTSL, compared to 6.2±1.5 for the 980C-MTSL isoform.
In the basal conformation, isoforms 343C and 354C have a greater accessibility to O2 than isoforms 980C and 331C, indicating that these residues are likely to be inserted into the hydrophobic core of the lipid bilayer. However, isoform 354C also has a large Π(NiEDDA) suggesting that it is also readily accessible to the aqueous phase presumably on a helical interface between the hydrophobic core and the aqueous central cavity.
These results correspond well with the MD simulation results for membrane-embedded MTSL-354C ABCB1, which lies in the vicinity of the intracellular lipid-water interface and the protein backbone around the residue has a high degree of flexibility. When averaged over three 30ns simulations, the angle between the MTSL pyrrole nitrogen and the z-axis of the coordinate system was maintained at a constant angle of between 52 and 55° (Figure S4b). Rotational angles around the 354C Cα of up to 60° were obtained for the MTSL pyrrole nitrogen. Examination of the hydrophilic SASA shows 354C-MTSL to be freely accessible to solvent, corresponding to the experimentally determined Π(NiEDDA). The average SASA was 0.41±0.03 nm2 and there was an average of 14.7±3.8 water molecules within a 0.4nm contact radius of 354C-MTSL across the three simulations.
Isoform 343C has a low Π(NiEDDA) but a high Π(O2) implying that faces into the protein interior. Inspection of the membrane-embedded ABCB1 structure confirms the EPR data; residue 343C is located near the centre of TM helix 6, in the vicinity of the transmembrane cavity occlusion (Figure 4c) and the MTSL probe faces away from the transmembrane cavity, into the protein interior. This orientation was maintained throughout three independent 30 ns simulations. During the simulations, 343C-MTSL was coordinated by protein-protein contacts and did not contact the lipid bilayer making a constant angle of between 43 and 45° with the z-axis (Figure S4c). While 343C-MTSL was coordinated by protein-protein contacts, the rotational motion (up to 30° in the first 25ns) suggests that it has more degrees of freedom than 980C-MTSL. The low Π(NiEDDA) value is also supported by the SASA, which is only 0.14 ± 0.05 nm2 across the three simulations. Analysis of the MD trajectories shows that there are on average 4.6 ± 2.6 water molecules within a 0.4nm contact radius. 343C-MTSL has the lowest solvent accessible of the four MTSL labelled isoforms.
Using EPR spectroscopy of the nitroxide-labelled ABCB1 to probe conformational changes elicited by nucleotide binding and hydrolysis
Table 1 (Figure S3) contains the polarity Index Φ of all four ABCB1 isoforms in each of the three conformational states studied. In the nucleotide-free conformation, all four isoforms have Φ>0 i.e. the collision frequency with O2 is greater than with NiEDDA, which is an indication that these residues interact more strongly with the hydrophobic core of the lipid bilayer/protein interior than with the aqueous phase.
Nucleotide-bound conformation
In the nucleotide bound (AMP PNP) conformation, the accessibility to O2 and NiEDDA of both isoforms 980C and 331C increases. However, the fold change in Π(O2) of isoform 331C is greater than Π(NiEDDA), hence the 331C isoform Φ value also increases as the environment becomes less polar as the residue becomes embedded in the hydrophobic core of the lipid bilayer. By contrast, the Π(NiEDDA) for the 980C isoform increases more than the Π(O2) and consequently, the Φ value decreases as the environment of this residue becomes more polar and accessible to the aqueous phase. The overall accessibility of the 980C isoform is still relatively low suggesting that its insertion is relatively shallow, in comparison to the 331C isoform, or that it is still somewhat buried within the protein.
For isoform 343C, there is a dramatic increase in Π(NiEDDA) with Π(O2) only increasing marginally with the resulting negative Φ value implying that the environment of the residue has become very polar. This would suggest that the spin-probe is highly accessible to the aqueous phase of the translocation cavity. The overall increase in accessibility of the 343C isoform could indicate that it is becoming less buried within the protein. The mobility of this residue remains restricted in the nucleotide bound (AMP PNP) conformation (Figure 2 and Table 1) and it is highly associated with both the hydrophobic core of the bilayer and the aqueous phase of the translocation cavity/extracellular space. This indicates that the nitroxide spin label must now be facing along the interface between these two different environments.
For isoform 354C, the decrease in Π(NiEDDA) without any increase in Π(O2) would indicate that this residue has become more buried within the protein interior rather than being lipid embedded.
Vanadate-trapped conformation
In the vanadate-trapped (ADP/Vi) conformation, the environment of the spin-label in isoform 354C is almost identical to the environment in the nucleotide bound (AMP-PNP) conformation and the Φ value doesn’t alter significantly. For isoforms 980C and 331C, the accessibility to both paramagnetic reagents reverts back to the low values seen in their nucleotide free (basal) conformations suggesting that these two residues either approach the phospholipid head groups of the membrane interface again or become more buried within the protein. The respective Φ values clearly imply that they are more hydrophobic than in their nucleotide free (basal) conformations. The Φ value of isoform 343C signifies that it has become less polar than in the nucleotide bound (AMP-PNP) conformation but less hydrophobic than in the nucleotide free (basal) conformation. The overall accessibility decreases but does not revert back to the nucleotide free (basal) conformation since 343C (ADP/Vi) is still more accessible to both paramagnetic reagents but with an increased Π(NiEDDA).
Discussion
Previous studies of ABCB1 from our group had focussed on evaluating the accessibility of cysteine residues within TM6 and TM12 to three large fluorescent probes with distinct physico-chemical properties (e.g. hydrophobic, hydrophilic or amphiphilic) [17–19]. The approach provided considerable information on the general topography of two key helices involved in drug binding and possible interdomain coupling, however such accessibility to a covalent modification is only in part determined by the local chemical microenvironment. Steric hindrance by local amino-acid side chains and neighbouring domains will also influence the apparent accessibility to the large thiol-reactive fluorescent probes. Consequently, in the present study, the powerful EPR spectroscopic technique was applied to analysing the residue microenvironment in significantly greater detail. A series of TM domain positions on TM helices 6 and 12 were selected which demonstrate accessibility to thiol-modification without perturbation of function. The smaller paramagnetic probe, MTSL was attached and the EPR spectra used to ascertain its freedom of movement and more especially provide an accurate assessment of the local environment. This was achieved through the use of quenching agents that specifically target either hydrophobic (O2) or hydrophilic environments (Ni(II)EDDA).
At the extracellular face, residues 331 and 980 are situated at an interfacial region within a highly constricted environment. The extracellular proximal sections of TM6 and TM12 lie within a protein dense region of ABCB1 as revealed by the two X-ray structural models. Both these helices undergo conformational change following nucleotide binding as demonstrated previously by an altered cross-linking profile of TM6/TM12 in the absence or presence of nucleotide [30]. The EPR results presented here clearly indicate that this region reverts to the basal conformation immediately post-nucleotide hydrolysis. Both structural data and homology models predict that the 343C residue is located above the junction point (i.e. V345) of TM helices 5, 6, 11 and 12 helices. This location may be interpreted as the point at which the central cavity is occluded in the basal conformation of ABCB1 and hence the central cavity of ABCB1 is in an inward facing conformation. Following nucleotide binding, the 343C-MTSL moiety demonstrates the most prominent change (of all the residues examined) in EPR accessibility and this high accessibility to the aqueous phase above the predicted pivot at V345, suggests that the central cavity is no longer occluded or has altered its conformation. Moreover, the modelling predicts that following nucleotide binding the extracellular region of the cavity has greater aqueous accessibility, which would be in agreement with pharmacology data [6, 10]. In the post-hydrolytic state, the EPR accessibility implies a further change in environment. However, it does not revert back to the basal state and likely reflects a transition state. At the intracellular face (i.e. as reported by 354C-MTSL) MD simulations suggest that large movements in the protein backbone are possible in this region of TM6 permitting the accessibility to a large hydrophilic fluorescein-maleimide probe (FM) as described previously [19]. The 354C-MTSL is located on an extended coil region of TM helix 6 in the homology model and thus, higher conformational flexibility is possible due to the relative lack of residue-residue interactions. Following nucleotide binding, 354C-MTSL revealed a substantial decrease in accessibility to the aqueous phase, presumably due to the closing of the central cavity following nucleotide-“sandwich” formation by the NBDs. In the post-hydrolytic state, the EPR data indicate that there is no change from the nucleotide-bound conformation, suggesting that the aqueous cavity remained closed, presumably until ADP is released [6, 10].
A central (potentially aqueous filled cavity) was first postulated by early electron microscopy driven structural models of ABCB1 [22] and subsequently confirmed by cross-linking data [31–33]. More recently, the two high resolution structures of ABCB1 [13, 16] have revealed a substantial central cavity that is occluded at the extracellular surface by the apposition of TM helices. In this EPR investigation a number of residues known to reside near to, or lining, the central cavity were selected [18, 19, 25] and EPR used to generate a dynamic description of the environmental changes that the selected residues underwent during progression of the transport process. The EPR data are best described by the alternate access model [34] where the aqueous cavity closes at the cytosolic face upon nucleotide sandwich formation in the NBDs. Moreover, the helices lining this cavity undergo conformational changes to generate greater aqueous accessibility of the central cavity at the extra-cellular region. In the post-hydrolytic state, the environment of the cytoplasmic end of TM helix 6 does not change in comparison to the nucleotide-bound conformation whereas it seems that the extracellular ends of TM helix 6 and TM helix 12 convert back to the conformation seen in the nucleotide-free state. The local environment of the centre of TM helix 6 is half way between the nucleotide-bound and nucleotide-free states, perhaps due to a twist at the top of the TMDs while the NBDs remain sandwiched.
This current investigation has clearly identified and characterised the physical and chemical changes to the local environment of a series of residues proximal to the central cavity. At present, the current data do not permit an accurate determination of the extent of movement of these residues or the helices. Future studies with multiple paramagnetic probes attached will, however be able to determine the degree and magnitude of conformation transition in the helices.
Materials and Methods
Materials
Octyl-β-d-glucoside, C219 antibody and Ni2+–nitrilotriacetic acid His-Bind (NiNTA) Superflow resin were obtained from Merck Chemicals (Nottingham, UK). Dimethylsulfoxide, Na2ATP, AMP-PNP, sodium orthovanadate and cholesterol were purchased from Sigma Aldrich (Poole, UK). Crude Escherichia coli lipid extract was obtained from Avanti Polar Lipids (Alabaster, USA). Insect-Xpress medium was purchased from Lonza (Wokingham, UK) and Excell 405 from SAFC Biosciences (Andover, UK). The nitroxide spin label (1-Oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl) Methanethiosulfonate (MTSL) was purchased from Toronto Research Chemicals (Canada). SM2-BioBeads were purchased from BioRad Laboratories (Hemel Hempstead, UK). The nickel(II)-EDDA complex (NiEDDA) was prepared as previously described [35].
Mutagenesis and expression of ABCB1 isoforms in insect cells
The construction of single cysteine containing, His6-tagged ABCB1 isoforms, and the derivation of recombinant baculoviruses has been fully described previously [25]. ABCB1 isoforms were expressed in Trichoplusia ni (High-Five) insect cells (typically 1-2 × 106 cells) following infection (at a multiplicity of infection of 5). Infected cells were harvested 3 days post-infection by centrifugation (2000g for 10 min). Cell membranes were isolated 72 h post-infection using nitrogen cavitation and ultra-centrifugation as previously described and subsequently stored at -80°C for up to 1 year [25, 36].
Purification, labelling and reconstitution of ABCB1 isoforms
Purification of the ABCB1 isoforms from the High-Five insect cell membranes was achieved using previously published methods with a number of modifications [17, 19]. Membranes (typically 100mg) were solubilised at a total protein concentration of 5mg/ml with octyl-β-d-glucoside (2% (w/v)) supplemented with 0.4% (w/v) crude lipid (4:1 (w/w) E. coli extract:cholesterol). Soluble protein was added to a packed Ni-NTA resin (1ml) with mild agitation for 1hr. The slurry was transferred to glass columns and the flow through collected. Bound protein was sequentially washed in buffer containing 20 bed volumes (bv) of 10, 20 and 30 mM imidazole. All chromatography buffers contained 1.25% (w/v) octyl-β-d-glucoside supplemented with 0.1% (w/v) crude lipid. ABCB1 was labelled with the paramagnetic probe (S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate; MTSL) using an on-column approach, prior to extensive washing to remove residual un-bound MTSL. Specifically, the Ni-NTA resin (containing bound ABCB1) was harvested from the column in buffer containing imidazole (2mM) and MTSL (600μM). The sample was gently mixed overnight at 4°C to ensure complete labelling of the protein. The Ni-NTA resin containing MTSL-labelled ABCB1 was added to glass chromatography columns and the buffer containing unbound MTSL collected. The resin was washed with 15bv of buffer (pH 6.8) containing imidazole (2mM) and then buffer (40bv) with imidazole (2mM) but now at pH 6.8. The protein was then eluted in buffer at pH 6.8 (4bv) which contained 400 mM imidazole. The imidazole and any residual MTSL were removed by buffer exchange using PD-10 columns. The MTSL-labelled ABCB1 was collected in the column void volume in buffer that contained 1.25 % (w/v) octyl-β-d-glucoside supplemented with 0.1 % (w/v) crude lipid (4:1 (w/w) E. coli extract:cholesterol).
Full functional capability of ABCB1 is only observed following reconstitution into a lipid bilayer [37, 38], which was achieved through solid phase adsorption of the detergent on SM-2 Bio-Beads using a previously published procedure [39]. No excess lipid was added to the samples, since the final preparation contained 0.1% (w/v) of a crude mixture. Following reconstitution the samples were subjected to ultra centrifugation (41,000 × g, 20 min) and the pellet resuspended in NaCl (150mM) and Tris.HCl (20 mM, pH6.8).
The mutant isoforms used in this study have been characterised previously; however, A single point ATPase assay (Na2ATP (2mM) and nicardipine (30μM) for 20 min at 37°C) were used to validate each preparation employing a colorimetric assay to detect liberated inorganic phosphate [39]. The nucleotide bound (pre-hydrolytic) conformation of pure, reconstituted ABCB1 was achieved by incubation of the mutant isoforms with the nonhydrolysable ATP analogue AMP-PNP, as previously described [6]. The post-hydrolytic (but pre-ADP or phosphate release) stage was produced using a vanadate trapping procedure [40].
CW EPR Power Saturation Measurements
Room-temperature X-band cw EPR spectra were acquired as described [41] using a Bruker eleXsys E500 spectrometer fitted with a Bruker ER4123D loop-gap resonator and a TPX® capillary setup. The total active sample volume was 3 μl. Experimental parameters for the cw EPR spectra were: Microwave power; 2mW: Field modulation amplitude; 0.2mT: Field modulation frequency; 100 kHz: Sweep width; 14mT: Number of scans; 16 (or 64) scans at ambient temperature. The spectra were quantified by double integration against a known concentration of MTSL to give concentrations between 1.6 to 4 μM MTSL-labelled ABCB1, which were comparable to the concentrations calculated by a standard protein assay. Cw-EPR power saturation measurements [41–44] were performed for each spin-labelled ABCB1 isoform in each of three conformational states (basal, AMP-PNP bound, ADP/Vi bound) after equilibration in N2, air (21 % O2) and N2 in the presence of 10mM NiEDDA. All cw EPR power saturation experiments were repeated at least once with a completely new batch of each ABCB1 isoform. The samples were purged with gas for 20 min prior to, and during, each experiment. Experimental conditions for power saturation experiments were as above with the Microwave power being varied in 2 dB steps (0.1 mW to 40 mW). The accessibility parameter, Π, calculated as previously [45], measures the individual collision frequency of the nitroxide spin label with the paramagnetic reagents. Π is derived from the midpoints of signal saturation at increasing microwave powers according to Equation 1. The line width was normalised to a known reference system: a powder sample of DPPH (α,α’-diphenyl-β-picrylhydrazyl) in KCl.
| Equation (1) |
where ΔP1/2(X) = P1/2(X) – P1/2(N2)
P1/2(X) is the microwave power required to reduce the vertical peak-to-peak amplitude of the first derivative Mi = 0 resonance line of MTSL (A) to half its unsaturated value and is proportional to the longitudinal relaxation rate of the spin label. P is the incident microwave power in milliwatts, I is a scaling factor and ϵ is a measure of the homogeneity of the saturation of the resonance. P1/2(X) is measured in the presence of relaxing agents and P1/2(N2) represents the saturation behaviour in the absence of paramagnetic collisions. Exposure to a paramagnetic agent, such as oxygen or NiEDDA, increases the relaxation rate of the spin label and therefore increases the saturation power, consequently increasing P1/2(X). A mean Π value was calculated for each isoform of ABCB1.
The polarity index, Φ, is calculated as shown previously in [44, 46–48]. Φ has a positive value for spin labels which are embedded in lipid where the environment is hydrophobic and hence collisions with oxygen dominate, whereas if Φ is negative it indicates that a spin label is located in an aqueous phase, either on the surface of the membrane or in transmembrane channels, where collisions with NiEDDA dominate. All data were fitted using Microcal Origin® software.
MD simulations of spin-labelled ABCB1
All MD simulations were performed using the GROMACS (Groningen Machine for Chemical Simulation) package, version 3.3.3, [49] in conjunction with the GROMOS 54A7 force field [50] for proteins. The simple point charge (SPC) water model [51] was used to describe the solvent water. Full details of the simulation parameters used are given in O’Mara and Mark [24]. The pre-equilibrated simulation system containing the central structure of the combined MD trajectory of membrane embedded murine ABCB1 described by O’Mara and Mark [24] was used as the starting conformation for all MD simulations. Point mutations to cysteine covalently bound to the MTSL probe were incorporated into the pre-equilibrated murine ABCB1 system at residues corresponding to 331, 343, 354 and 980. In each case, the appropriate number of counter-ions was included to neutralize the charge on the system. To initiate the simulations, 1000 steps of energy minimization were performed using a steepest descent algorithm, followed by 0.5 ns of unrestrained MD simulation to allow the environment of the cysteine-linked spin label to relax. For each spin-labelled mutation, three independent sets of 30 ns unrestrained MD simulations were performed.
The paramagnetic probe MTSL is a radical compound that carries an unpaired electron on the nitroxide group and has an overall net charge of zero. MTSL was parameterised using the utilities in the Automated Topology Builder (ATB), [52] where the MTSL radical (spin state: doublet for a radical) was optimized at B3LYP/6-31G* level of theory and initial charges were calculated by fitting to the electrostatic potential using the Merz-Kollman fitting scheme. The final optimised topology was generated by the protocols of the ATB and is available together with the starting conformation on the ATB webserver [52].
System set-up
The pre-equilibrated simulation system containing the central structure of the combined MD trajectory of membrane embedded murine ABCB1 described by O’Mara and Mark [24] was used as the starting conformation for all MD simulations. This conformation is available on the ATB webserver repository [52].
Solvent Accessible Surface Area Analysis
The solvent accessible surface area (SASA) of membrane-embedded ABCB1, was calculated for each frame of the MD simulations using the method of Shrake and Rupley [53] and a probe of radius 0.14 nm. The contribution per atom to the solvent accessible surface of the overall system (membrane-embedded ABCB1 covalently modified with MTSL-cysteine) was determined and the total SASA of the covalently modified MTSL-cysteine was calculated from the sum of the individual atomic contributions.
Accessibility of the probe to water, protein and lipids
The number of water molecules and lipid molecules in contact with the cysteine-bound spin label was calculated as the number of molecules found within a 0.4 nm radius of the covalently modified MTSL-cysteine for each frame of the trajectory. This radius was calculated as the minimum distance between the centre of mass of any atom in the group of interest (i.e. amino acids, water or lipids) and the centre of mass of any atom in the covalently modified MTSL-cysteine.
Orientation of the probe
The range of motion of the probe throughout the MD simulations was calculated. Taken with respect to the origin of the coordinate system, the motion of the probe in the z direction was calculated as the angle between the N1 nitrogen MTSL vector and the z-axis was calculated as the arccosine of the vector dot product. To calculate the rotational motion of the probe with respect to the protein backbone, the vector between the MTSL pyrrole nitrogen and the Cα of the covalently bound cysteine, and also the vector between the origin of the coordinate system and the cysteine Cα was calculated for each frame of the MD trajectory. The rotational angle of the probe was calculated as the arccosine of the vector dot product.
Supplementary Material
The Cα Root mean squared deviation of the ABCB1 TMDs after labelling introduced cysteine residues with MTSL
The MTSL probe is covalently bound to cysteine located at A) 980C, B) 354, C) 343C and D) 331C. The three independent MD simulations performed for each mutation are coloured black, blue and red.
{kind=link}
Accessibility of MTSL labelled residues to quenching agents
a) Representative cw EPR power saturation curves measured for the basal conformation of each isoform of ABCB1 under the conditions shown (O2, N2 and NiEDDA); b) Changes in the O2 power saturation curve of isoform 331C in the three different conformational states (Basal, AMP-PNP and ADP/Vi). This is a representation of the raw cw EPR data before analysis.
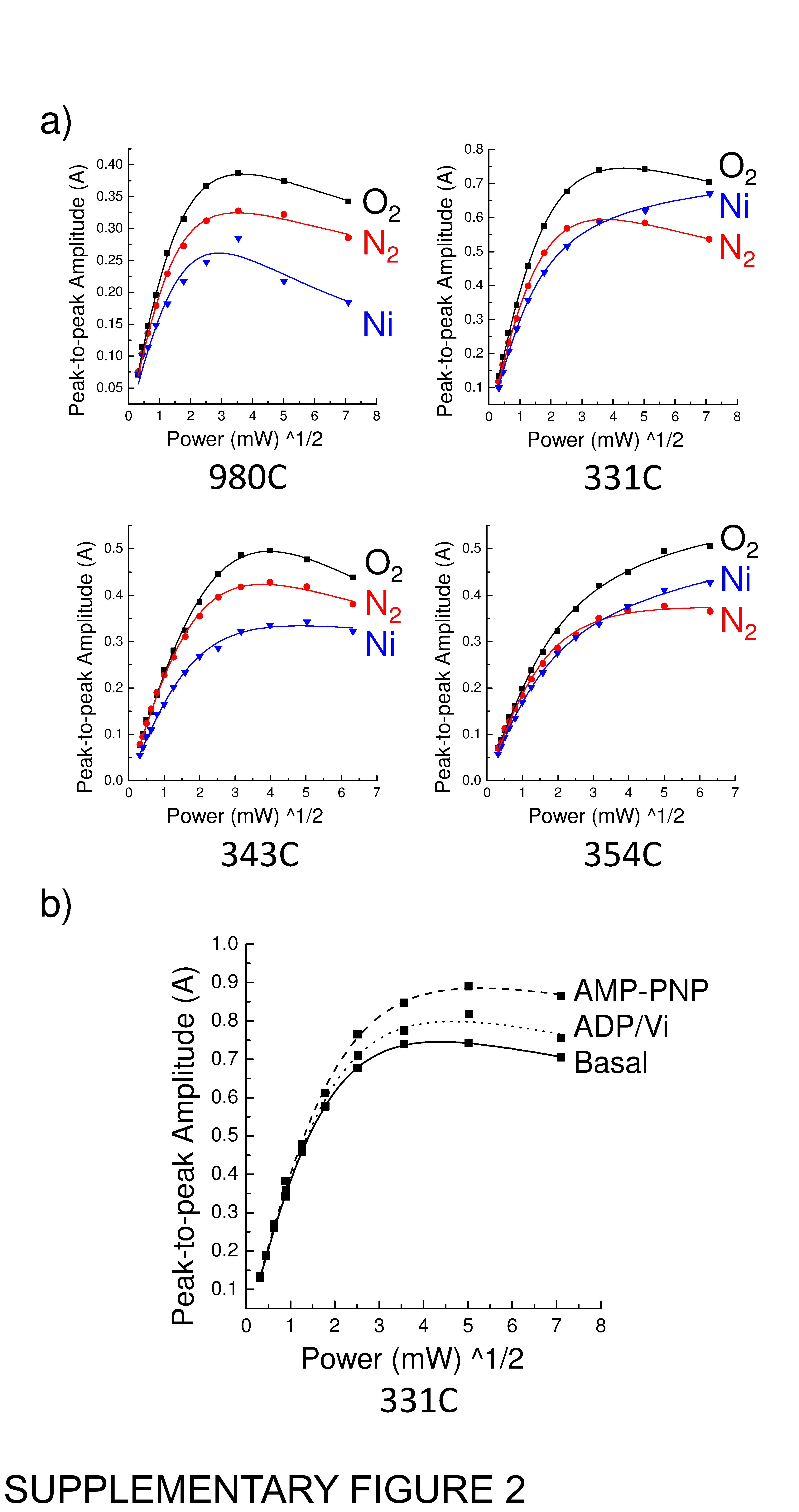{kind=link}
EPR accessibility measurements
a) The accessibility parameter, Π, of nitroxide spin-label to paramagnetic agents oxygen and NiEDDA and the derived polarity index, Φ, for each isoform of ABCB1 in each conformational state: Basal, AMP-PNP and ADP/Vi;
b) schematic to represent each possible state derived from the accessibility results.
{kind=link}
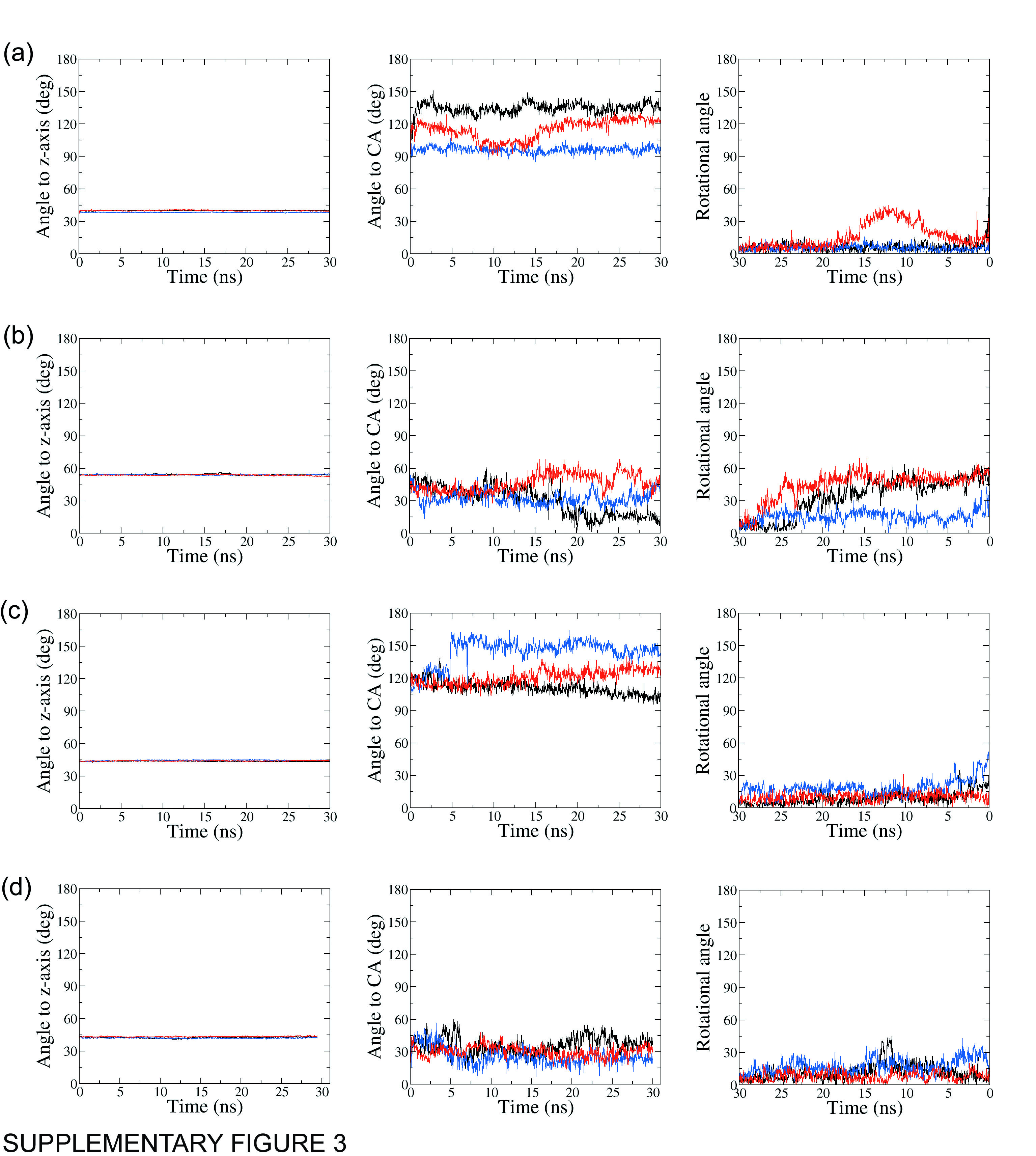
Orientation of the MTSL moiety in ABCB1 mutant isoforms
The angle of the MTSL pyrrol ring N to the Z-axis (left column), to the Cysteine Ca (middle column) and its rotational angle (right column) throughout MD simulations for A) 980C, B) 543C, C) 434C and D) 331C.
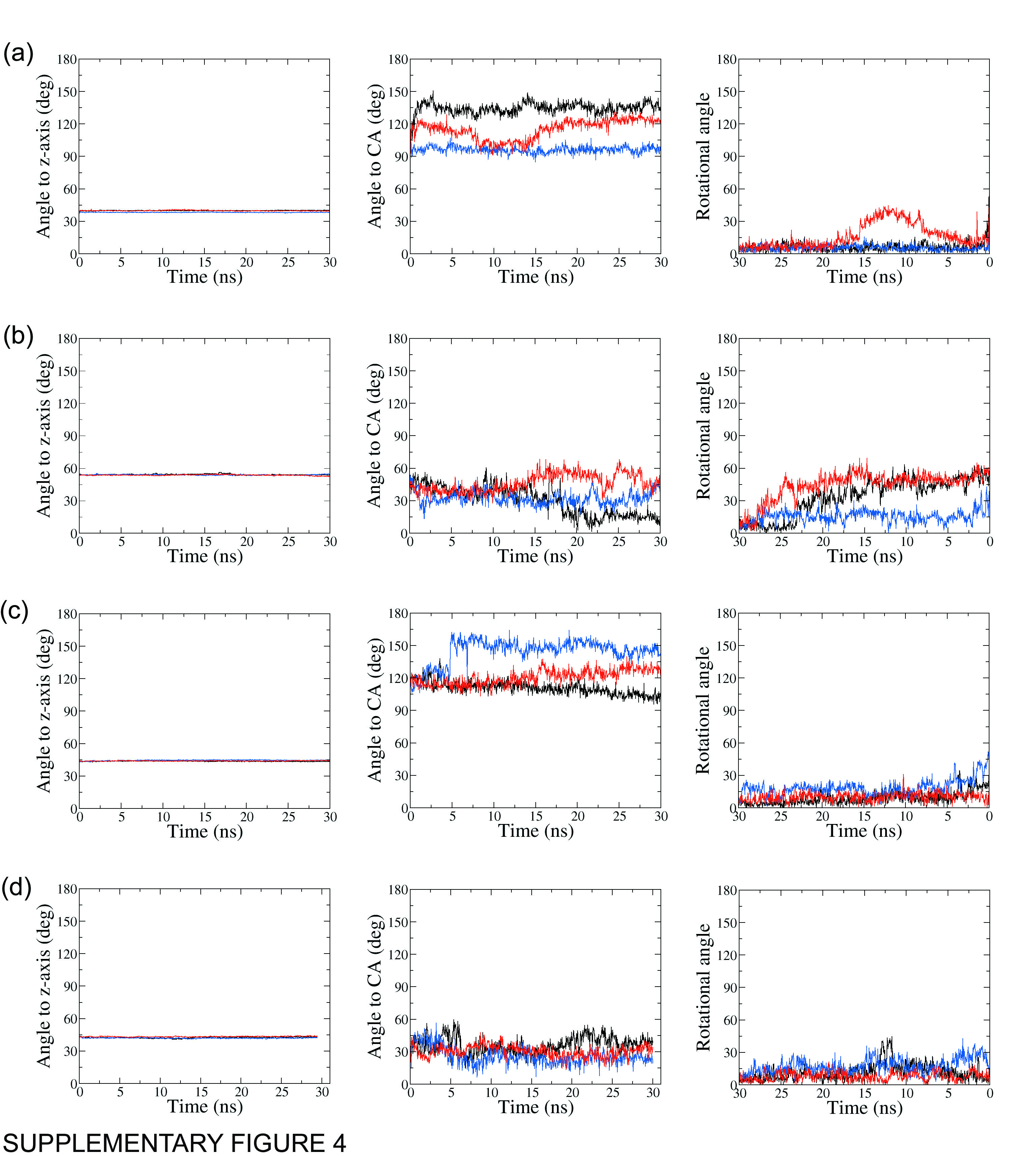{kind=link}
Acknowledgements
The research described in this manuscript was supported by a Wellcome Trust Project Grant (094392/B/10/Z) awarded to RC, IK, AT and FM (who also acknowledges support from a Royal Society Wolfson Research merit Award).
Footnotes
Author Contributions
JvW: performed the EPR spectroscopy experiments, analysed EPR data and contributed to writing the manuscript
RM: performed the molecular biology and protein biochemical experiments and contributed to writing the manuscript
MO’M: performed the molecular modelling experiments, analysed the EPR data and contributed to writing the manuscript
CMcD: contributed to planning experiments, undertook preliminary protein biochemistry and contributed to writing the manuscript
AT: planned experiments and contributed to writing the manuscript
IK: performed molecular biology experiments, planned the study and wrote the manuscript
FM: analysed EPR experiments, planned the study and wrote the manuscript
RC: performed protein biochemical experiments, planned the study and wrote the manuscript
References
- 1.Wu C-P, Hsieh C-H, Wu Y-S. The Emergence of Drug Transporter-Mediated Multidrug Resistance to Cancer Chemotherapy. Molecular Pharmaceutics. 2011;8:1996–2011. doi: 10.1021/mp200261n. [DOI] [PubMed] [Google Scholar]
- 2.Darby RA, Callaghan R, McMahon RM. P-glycoprotein inhibition: the past, the present and the future. Curr Drug Metab. 2011;12:722–731. doi: 10.2174/138920011798357006. [DOI] [PubMed] [Google Scholar]
- 3.Crowley E, Callaghan R. Multidrug efflux pumps: drug binding--gates or cavity? FEBS J. 2010;277:530–539. doi: 10.1111/j.1742-4658.2009.07484.x. [DOI] [PubMed] [Google Scholar]
- 4.Jones PM, O’Mara ML, George AM. ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem Sci. 2009;34:520–531. doi: 10.1016/j.tibs.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Loo TW, Clarke DM. Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate. J Biol Chem. 1997;272:31945–31948. doi: 10.1074/jbc.272.51.31945. [DOI] [PubMed] [Google Scholar]
- 6.Martin C, Higgins CF, Callaghan R. The vinblastine binding site adopts high- and low- affinity conformations during a transport cycle of P-glycoprotein. Biochemistry. 2001;40:15733–15742. doi: 10.1021/bi011211z. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg MF, Velarde G, Ford RC, Martin C, Berridge G, Kerr ID, Callaghan R, Schmidlin A, Wooding C, Linton KJ, et al. Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 2001;20:5615–5625. doi: 10.1093/emboj/20.20.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell PL, Sharom FJ. Conformational and functional characterization of trapped complexes of the P-glycoprotein multidrug transporter. Biochem J. 2006;399:315–323. doi: 10.1042/BJ20060015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonveaux N, Shapiro AB, Goormaghtigh E, Ling V, Ruysschaert JM. Secondary and tertiary structure changes of reconstituted P-glycoprotein A Fourier transform attenuated total reflection infrared spectroscopy analysis. J Biol Chem. 1996;271:24617–24624. doi: 10.1074/jbc.271.40.24617. [DOI] [PubMed] [Google Scholar]
- 10.Maki N, Moitra K, Ghosh P, Dey S. Allosteric modulation bypasses requirement for ATP hydrolysis in regnerating low-affinity transition state conformation of human P-glycoprotein. J Biol Chem. 2006 doi: 10.1074/jbc.M512579200. [DOI] [PubMed] [Google Scholar]
- 11.McDevitt CA, Crowley E, Hobbs G, Starr KJ, Kerr ID, Callaghan R. Is ATP binding responsible for initiating drug translocation by the multidrug transporter ABCG2? FEBS J. 2008;275:4354–4362. doi: 10.1111/j.1742-4658.2008.06578.x. [DOI] [PubMed] [Google Scholar]
- 12.Rothnie A, Callaghan R, Deeley RG, Cole SP. Role of GSH in estrone sulfate binding and translocation by the multidrug resistance protein 1 (MRP1/ABCC1) J Biol Chem. 2006;281:13906–13914. doi: 10.1074/jbc.M600869200. [DOI] [PubMed] [Google Scholar]
- 13.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo RP, Harrell PM, Trinh YT, Zhang QH, Urbatsch IL, et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Callaghan R, George AM, Kerr ID. 8.8 Molecular Aspects of the Translocation Process by ABC Proteins. In: Edward HE, editor. Comprehensive Biophysics. Elsevier; Amsterdam: 2012. pp. 145–173. [Google Scholar]
- 15.Choi K, Chen C-J, Kriegler M, Roninson IB. An altered pattern of cross-resistance in multidrug resistant human cells results from spontaneous mutations in the mdr1 (P-glycoprotein) gene. Cell. 1988;53:519–529. doi: 10.1016/0092-8674(88)90568-5. [DOI] [PubMed] [Google Scholar]
- 16.Jin MS, Oldham ML, Zhang Q, Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 2012 doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crowley E, O’Mara ML, Kerr ID, Callaghan R. Transmembrane helix 12 plays a pivotal role in coupling energy provision and drug binding in ABCB1. FEBS J. 2010;277:3974–3985. doi: 10.1111/j.1742-4658.2010.07789.x. [DOI] [PubMed] [Google Scholar]
- 18.Rothnie A, Storm J, Campbell J, Linton KJ, Kerr ID, Callaghan R. The topography of transmembrane segment six is altered during the catalytic cycle of P-glycoprotein. J Biol Chem. 2004;279:34913–34921. doi: 10.1074/jbc.M405336200. [DOI] [PubMed] [Google Scholar]
- 19.Storm J, Modok S, O’Mara ML, Tieleman DP, Kerr ID, Callaghan R. Cytosolic Region of TM6 in P-Glycoprotein: Topographical Analysis and Functional Perturbation by Site Directed Labeling. Biochemistry. 2008;47:3615–3624. doi: 10.1021/bi7023089. [DOI] [PubMed] [Google Scholar]
- 20.Lee JY, Urbatsch IL, Senior AE, Wilkens S. Projection structure of P-glycoprotein by electron microscopy - Evidence for a closed conformation of the nucleotide binding domains. J Biol Chem. 2002;277:40125–40131. doi: 10.1074/jbc.M206871200. [DOI] [PubMed] [Google Scholar]
- 21.Lee JY, Urbatsch IL, Senior AE, Wilkens S. Nucleotide-induced structural changes in P-glycoprotein observed by electron microscopy. J Biol Chem. 2008;283:5769–5779. doi: 10.1074/jbc.M707028200. [DOI] [PubMed] [Google Scholar]
- 22.Rosenberg MF, Callaghan R, Ford RC, Higgins CF. Structure of the multidrug resistance P-glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. J Biol Chem. 1997;272:10685–10694. doi: 10.1074/jbc.272.16.10685. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg MF, Kamis AB, Callaghan R, Higgins CF, Ford RC. Three-dimensional structures of the mammalian multidrug resistance P-glycoprotein demonstrate major conformational changes in the transmembrane domains upon nucleotide binding. J Biol Chem. 2003;278:8294–8299. doi: 10.1074/jbc.M211758200. [DOI] [PubMed] [Google Scholar]
- 24.O’Mara ML, Mark AE. Effect of environment on membrane protein structure: P-glycoprotein under physiological conditions. J Chem Theory Comput. 2012 doi: 10.1021/ct300254y. [DOI] [PubMed] [Google Scholar]
- 25.Crowley E, O’Mara ML, Reynolds C, Tieleman DP, Storm J, Kerr ID, Callaghan R. Transmembrane helix 12 modulates progression of the ATP catalytic cycle in ABCB1. Biochemistry. 2009;48:6249–6258. doi: 10.1021/bi900373x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rothnie A, Storm J, Campbell J, Linton KJ, Kerr ID, Callaghan R. The topography of transmembrane segment six is altered during the catalytic cycle of P-glycoprotein. J Biol Chem. 2004;279:34913–34921. doi: 10.1074/jbc.M405336200. [DOI] [PubMed] [Google Scholar]
- 27.Storm J, O’Mara M, Crowley E, Peall J, Tieleman PD, Kerr ID, Callaghan R. Residue G346 In Transmembrane Segment Six Is Involved In Inter-Domain Communication In P-Glycoprotein. Biochemistry. 2007;46:9899–9910. doi: 10.1021/bi700447p. [DOI] [PubMed] [Google Scholar]
- 28.Crowley E, O’Mara ML, Kerr ID, Callaghan R. Transmembrane Helix 12 Plays a Pivotal Role in Coupling Energy Provision and Drug Binding in ABCB1. FEBS Journal. 2010 doi: 10.1111/j.1742-4658.2010.07789.x. In Press. [DOI] [PubMed] [Google Scholar]
- 29.Rothnie A, Storm J, McMahon R, Taylor A, Kerr ID, Callaghan R. The coupling mechanism of P-glycoprotein involves residue L339 in the sixth membrane spanning segment. FEBS Lett. 2005;579:3984–3990. doi: 10.1016/j.febslet.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 30.Loo TW, Clarke DM. Determining the structure and mechanism of the human multidrug resistance P-glycoprotein using cysteine-scanning mutagenesis and thiol- modification techniques. Biochim Biophys Acta. 1999;1461:315–325. doi: 10.1016/s0005-2736(99)00165-0. [DOI] [PubMed] [Google Scholar]
- 31.Loo TW, Bartlett MC, Clarke DM. Disulfide cross-linking analysis shows that transmembrane segments 5 and 8 of human P-glycoprotein are close together on the cytoplasmic side of the membrane. J Biol Chem. 2004;279:7692–7697. doi: 10.1074/jbc.M311825200. [DOI] [PubMed] [Google Scholar]
- 32.Loo TW, Clarke DM. Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J Biol Chem. 2000;275:39272–39278. doi: 10.1074/jbc.M007741200. [DOI] [PubMed] [Google Scholar]
- 33.Loo TW, Clarke DM. Determining the dimensions of the drug-binding domain of human P-glycoprotein using thiol cross-linking compounds as molecular rulers. J Biol Chem. 2001;276:36877–36880. doi: 10.1074/jbc.C100467200. [DOI] [PubMed] [Google Scholar]
- 34.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 35.Oh KJ, Altenbach C, Collier RJ, Hubbell WL. Site-directed spin labeling of proteins Applications to diphtheria toxin. Methods Mol Biol. 2000;145:147–169. doi: 10.1385/1-59259-052-7:147. [DOI] [PubMed] [Google Scholar]
- 36.Crowley E, O’Mara ML, Kerr ID, Callaghan R. Transmembrane helix 12 plays a pivotal role in coupling energy provision and drug binding in ABCB1. FEBS J. 2010;277:3974–3985. doi: 10.1111/j.1742-4658.2010.07789.x. [DOI] [PubMed] [Google Scholar]
- 37.Callaghan R, Berridge G, Ferry DR, Higgins CF. The functional purification of P-glycoprotein is dependent on maintenance of a lipid-protein interface. Biochim Biophys Acta. 1997;1328:109–124. doi: 10.1016/s0005-2736(97)00079-5. [DOI] [PubMed] [Google Scholar]
- 38.Modok S, Heyward C, Callaghan R. P-glycoprotein retains function when reconstituted into a sphingolipid- and cholesterol-rich environment. J Lipid Res. 2004;45:1910–1918. doi: 10.1194/jlr.M400220-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Taylor AM, Storm J, Soceneantu L, Linton KJ, Gabriel M, Martin C, Woodhouse J, Blott E, Higgins CF, Callaghan R. Detailed characterization of cysteine-less P-glycoprotein reveals subtle pharmacological differences in function from wild-type protein. Brit J Pharmacol. 2001;134:1609–1618. doi: 10.1038/sj.bjp.0704400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urbatsch IL, Sankaran B, Weber J, Senior AE. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J Biol Chem. 1995;270:19383–19390. doi: 10.1074/jbc.270.33.19383. [DOI] [PubMed] [Google Scholar]
- 41.Altenbach C, Flitsch SL, Khorana HG, Hubbell WL. Structural studies on transmembrane proteins 2 Spin labeling of bacteriorhodopsin mutants at unique cysteines. Biochemistry. 1989;28:7806–7812. doi: 10.1021/bi00445a042. [DOI] [PubMed] [Google Scholar]
- 42.Altenbach C, Froncisz W, Hyde JS, Hubbell WL. Conformation of spin-labeled melittin at membrane surfaces investigated by pulse saturation recovery and continuous wave power saturation electron paramagnetic resonance. Biophys J. 1989;56:1183–1191. doi: 10.1016/S0006-3495(89)82765-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klug CS, Feix JB. Methods and applications of site-directed spin labeling EPR spectroscopy. Method Cell Biol. 2008;84:617–658. doi: 10.1016/S0091-679X(07)84020-9. [DOI] [PubMed] [Google Scholar]
- 44.Altenbach C, Greenhalgh DA, Khorana HG, Hubbell WL. A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc Natl Acad Sci USA. 1994;91:1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farahbakhsh ZT, Altenbach C, Hubbell WL. Spin Labeled Cysteines as Sensors for Protein Lipid Interaction and Conformation in Rhodopsin. Photochem Photobiol. 1992;56:1019-&. doi: 10.1111/j.1751-1097.1992.tb09725.x. [DOI] [PubMed] [Google Scholar]
- 46.Altenbach C, Marti T, Khorana HG, Hubbell WL. Transmembrane protein structure: spin labeling of bacteriorhodopsin mutants. Science. 1990;248:1088–1092. doi: 10.1126/science.2160734. [DOI] [PubMed] [Google Scholar]
- 47.Malmberg NJ, Falke JJ. Use of EPR power saturation to analyze the membrane-docking geometries of peripheral proteins: applications to C2 domains. Annu Rev Bioph Biom. 2005;34:71–90. doi: 10.1146/annurev.biophys.34.040204.144534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nielsen RD, Che K, Gelb MH, Robinson BH. A ruler for determining the position of proteins in membranes. J Am Chem Soc. 2005;127:6430–6442. doi: 10.1021/ja042782s. [DOI] [PubMed] [Google Scholar]
- 49.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- 50.Schmid N, Eichenberger A, Choutko A, Riniker S, Winger M, Mark A, van Gunsteren W. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. European Biophysics Journal. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- 51.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. Intermol Forces. 1981;11:331–338. [Google Scholar]
- 52.Malde AK, Zuo L, Breeze M, Stroet M, Poger D, Nair PC, Oostenbrink C, Mark AE. An Automated force field Topology Builder (ATB) and repository: version 1.0. J Theor Comput Chem. 2011;12:4026–4037. doi: 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
- 53.Shrake A, Rupley JA. Environment and exposure to solvent of protein atoms - lysozyme and insulin. J Mol Biol. 1973;79:351–371. doi: 10.1016/0022-2836(73)90011-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Cα Root mean squared deviation of the ABCB1 TMDs after labelling introduced cysteine residues with MTSL
The MTSL probe is covalently bound to cysteine located at A) 980C, B) 354, C) 343C and D) 331C. The three independent MD simulations performed for each mutation are coloured black, blue and red.
Accessibility of MTSL labelled residues to quenching agents
a) Representative cw EPR power saturation curves measured for the basal conformation of each isoform of ABCB1 under the conditions shown (O2, N2 and NiEDDA); b) Changes in the O2 power saturation curve of isoform 331C in the three different conformational states (Basal, AMP-PNP and ADP/Vi). This is a representation of the raw cw EPR data before analysis.
EPR accessibility measurements
a) The accessibility parameter, Π, of nitroxide spin-label to paramagnetic agents oxygen and NiEDDA and the derived polarity index, Φ, for each isoform of ABCB1 in each conformational state: Basal, AMP-PNP and ADP/Vi;
b) schematic to represent each possible state derived from the accessibility results.
Orientation of the MTSL moiety in ABCB1 mutant isoforms
The angle of the MTSL pyrrol ring N to the Z-axis (left column), to the Cysteine Ca (middle column) and its rotational angle (right column) throughout MD simulations for A) 980C, B) 543C, C) 434C and D) 331C.