

Abstract
Sucrose phosphate synthase (SPS) catalyses the transfer of glycosyl group of uridine diphosphate glucose to fructose-6-phosphate to form sucrose-6-phosphate. Plant SPS plays a key role in photosynthetic carbon metabolisms, which activity is modulated by an allosteric activator glucose-6-phosphate (G6P). We produced recombinant sugarcane SPS using Escherichia coli and Sf9 insect cells to investigate its structure-function relationship. When expressed in E. coli, two forms of SPS with different sizes appeared; the larger was comparable in size with the authentic plant enzyme and the shorter was trimmed the N-terminal 20 kDa region off. In the insect cells, only enzyme with the authentic size was produced. We purified the trimmed SPS and the full size enzyme from insect cells and found their enzymatic properties differed significantly; the full size enzyme was activated allosterically by G6P, while the trimmed one showed a high activity even without G6P. We further introduced a series of N-terminal truncations up to 171 residue and found G6P-independent activity was enhanced by the truncation. These combined results indicated that the N-terminal region of sugarcane SPS is crucial for the allosteric regulation by G6P and may function like a suppressor domain for the enzyme activity.
Keywords: allosteric regulation, carbon metabolism, recombinant enzyme, sucrose phosphate synthase, sugarcane
Sucrose is the primary photosynthate in plant leaves and transported to various growing tissues and storage organs for allocation of carbon resources through whole plant body. Sucrose phosphate synthase (SPS) plays a key role in the pathway of sucrose synthesis, which catalyses formation of sucrose-6-phosphate (S6P) by use of fructose-6-phosphate (F6P) and uridine diphosphate-glucose (UDP-G) as substrates (1–3). The activity of SPS is regulated by diurnal cycles of day/night (4) and other environmental factors such as osmotic stress (5) and cold temperature (6). Protein phosphorylation of certain serine residues of SPS polypeptide and allosteric control by metabolites, such as glucose-6-phospate (G6P) and inorganic phosphate, are involved in the regulation of SPS activity (7, 8). Such molecular characteristics of SPS are believed to be crucial for playing the physiological role of regulating photosynthetic carbon flux into sucrose.
Plant SPSs have a molecular mass around 120 kDa and consists of three domains. The central region contains glycosyltransferase domain responsible for the catalytic function of SPS (9). The C-terminal region resembles sucrose phosphate phosphatase and the N-terminal region has no clear similarity with any other proteins. The SPSs from the photosynthetic cyanobacteria (10) and non-photosynthetic bacteria (11) lack the N-terminal domain or both terminal domains of plant SPS, respectively, and unlike plant SPS, their enzyme activity is not allosterically regulated (12, 13). This indicates that the domain(s) characteristic of plant SPS is(are) contributed to the regulatory properties and that, in particular, the unique N-terminal region is likely to be involved in the regulation of enzyme activity.
The crystal structure of SPS from the non-photosynthetic bacterium Halothermothrix orenii, which is composed of glycosyltransferase domain only, was reported and the structure revealed the mechanisms of substrate binding and glycosyl transfer (13). This knowledge could give an important insight into the reaction mechanism of plant SPS as well as the bacterial enzyme. However, the structural basis for the regulatory function of plant SPS still remains totally unknown. With the aim of studying structure-function relationship of the regulatory mechanism of the plant enzyme, we are trying to prepare recombinant plant SPS with quality and amount sufficient for structural analysis. There are several papers reporting expression of plant SPS genes in Escherichia coli (14–16), but resulting recombinant enzymes did not show a clear property of the allosteric regulation. The reason for that is not clearly understood yet.
We have previously cloned cDNAs from sugarcane leaves, SoSPS1 and SoSPS2, encoding two isoforms of SPS (17). SoSPS1 is abundantly expressed in sugarcane leaves and is considered to be a representative of the isoenzyme responsible for photosynthetic carbon allocation with the regulatory function (17). In this study, cDNA of SoSPS1 was expressed in E. coli and insect cells and recombinant enzymes with various lengths of the N-terminal deletion were produced. We report here that the N-terminal region of sugarcane SPS is not essential for the catalytic reaction itself, but plays a role in the allosteric regulation.
Materials and Methods
Construction of various expression plasmids of sugarcane SoSPS1 cDNA in E. coli
To allow the expression of full size enzyme encoded by sugarcane SoSPS1 (gene Accession number AB001337 in GenBank) in E. coli, NcoI/SacI fragment of SoSPS1 including the coding region of the N-terminal part of SPS was ligated into pTrc99A vector (Pharmacia) and the remaining coding region of SoSPS1 starting from XhoI up to SphI (filled in) was inserted into the downstream of the N-terminal part. These steps yielded pTrcSPS(full) for the expression of full-length SPS under control of trc promoter.
To produce SPS fused with a tag, the full coding region of SoSPS1was inserted into pTrcHisA with an N-terminal His6 tag (Life Technologies, Invitrogen). The coding region was amplified by PCR using forward and backward primers generating SbfI and EcoRI sites (Supplementary Table S1). The PCR amplified fragment was digested with SbfI and EcoRI and inserted into PstI and EcoRI sites of the vector.
A series of mutant SPSs with the N-terminal truncation was also constructed in a similar way of the his-tagged full length enzyme. The truncations of the N-terminal region were generated by PCR using six different forward primers with SbfI site and the backward primer containing EcoRI site as used for full-length SPS (Supplementary Table S1). The PCR product of the N-terminal truncated forms designated as HisΔN1, HisΔN2, HisΔN3, HisΔN4, HisΔN5 and HisΔN6 were subcloned into pGEM-T vector (Promega) that contains 3′-T overhangs at the insertion site and then cut with SbfI and EcoRI. The resulting DNA fragments encoding the N-terminal truncated SPSs were inserted into PstI and EcoRI sites of pTrcHisA. All the SPS genes thus constructed were verified by DNA sequencing.
Expression and purification of SoSPS1 in E. coli
The constructed plasmids were introduced into E. coli BL21 (DE3) competent cells (Novagen) to express the full length SPS and various N-terminal truncated forms. The transformed bacterial cells were grown in 50 ml of Luria Broth (LB) media containing 50 μg/ml of ampicillin overnight at 30°C. The seed culture was inoculated into a fermenter containing 8 l of LB media with 50 μg/ml of ampicillin and further grown for 3–5 h until an early log phase. Then, the culture temperature was shifted to 20°C, isopropyl β-d-thiogalactopyranoside (IPTG) was added at a final concentration of 0.05 mM, and the culture was continued overnight. The cells were harvested by centrifugation at 6,000 revolution per minute (rpm) (Sorvall SLC-4000) for 6 min at 4°C and stored at −20°C until use.
The frozen bacterial cells were suspended in extraction buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 1 mM EDTA and 1 mM PMSF], and disrupted by sonication on ice. Cell homogenate was centrifuged at 15,000 rpm (Hitachi T15A37-0029) for 15 min at 4°C and the resulting supernatant was mixed gently with DE52 anion exchange cellulose (Whatman). The mixture was filtered through Miracloth (Calbiochem) and the filtrate was directly loaded onto a column of cOmplete His-Tag Purification Resin (Roche) equilibrated with 50 mM Tris–HCl (pH 7.5)/150 mM NaCl. The column was washed with the same buffer containing 30 mM imidazole and 10% glycerol (w/v) and the resin binding proteins were eluted by increasing concentration of imidazole to 120 mM. The eluted sample from the His-tag resin column was concentrated and applied to size exclusion chromatography on a Superdex 200 column (GE Healthcare Life Sciences) and eluted with 50 mM Tris–HCl (pH 7.5)/150 mM NaCl.
Purification of the full length SPS without His tag was carried out by a combination of ion-exchange, hydrophobic and size exclusion chromatographies as described in the Supplemental section (Supplementary Fig. S2).
Expression of SoSPS1 in Sf9 insect cells and purification of the full-size SPS
SoSPS1 cDNA was PCR-amplified with primers shown in Supplementary Table S2. A 5′-untranslated leader sequence of a lobster tropomyosin cDNA (L21) was included in the forward primer for the enhancement of protein expression (18). The PCR product was ligated into pGEM-T Easy vector (Promega), cut with NotI and EcoRI, and then subcloned into the pFastBac1 (Thermo Fisher Scientific) vector for baculovirus expression in Sf9 insect cells. A tobacco etch virus (TEV) protease cleavage site, His8–myc–His8 tag, and enhanced green fluorescent protein (EGFP) were introduced at the carboxyl terminus of SoSPS1.
The bacmid and baculovirus of SoSPS1 were generated using Bac-to-Bac system (Thermo Fisher Scientific). Sf9 cells were grown to ∼4.0 × 106 cells/ml in an Erlenmeyer flask containing PSFM-J1 medium (Wako) and infected with P2 virus at 28°C. After 24 h post-infection, the cells were further incubated under low temperature at 20°C for 48 h. The cells were harvested by centrifugation at 4,004 rpm (Beckman JLA-8.1000) for 5 min at 4°C, resuspended in extraction buffer [50 mM Tris–HCl (pH 7.5), 1 M NaCl, 20 mM imidazole (pH 7.5), 2 mM DTT, 2 mM benzamidine], and then disrupted by sonication. The insoluble fraction was removed by centrifugation at 18,185 rpm (Beckman Coulter JA-25.50) for 20 min at 4°C and the supernatant was mixed gently with Ni-NTA Agarose (Qiagen) resin for 30 min at 4°C. The resin was washed thoroughly with wash buffer A [50 mM Tris–HCl (pH 7.5), 0.3 M NaCl, 20 mM imidazole (pH 7.5), 2 mM DTT]. SPS–EGFP was eluted with elution buffer [50 mM Tris–HCl (pH 7.5), 0.3 M NaCl, 250 mM imidazole (pH 7.5), 2 mM DTT]. SPS–EGFP fraction was batch incubated with 2 ml of GFP-nanotrap resin for 30 min at 4°C with gentle rotation (19). The resin was washed with wash buffer B [20 mM Tris–HCl (pH7.5), 0.5 M NaCl, 5 mM DTT]. SPS without the tag was eluted from GFP-nanotrap resin by cleaving the linker between SPS and His8–myc–His8–EGFP Tag with TEV protease for 4 h at 4°C, leaving the tag bound to the resin. The eluted SPS was concentrated and further purified by size-exclusion chromatography on a column of Superdex 200 (GE Healthcare Life Sciences) in buffer [20 mM Tris–HCl pH 7.5, 0.5 M NaCl, 5 mM DTT].
Partial purification of SPS from sugarcane and maize leaves
Frozen leaves were pulverized with liquid N2 and the proteins were extracted in three volumes (v/w) of an extraction buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 1 mM EDTA, and 10% PVPP]. The homogenate was centrifuged at 14,000 rpm (Hitachi T15A37-0029) at 4°C for 15 min and the supernatant fluid was filtered through two layers cheesecloth. Crude SPS was recovered by PEG-8000 fractionation between 6% and 12% according to the method described previously (20, 21). The SPS-containing precipitate was resuspended with 50 mM Tris–HCl (pH 7.5) containing 150 mM NaCl and10% glycerol (v/v).
Preparation of SoSPS1 antibody
To detect the expression of SoSPS1 at the protein level, a polyclonal antibody against recombinant SPS protein was prepared. The C-terminal 362 amino acid residues of the coding region of SoSPS1 were ligated into pQE30 vector (Qiagen). The recombinant protein was overexpressed in E. coli and purified using Ni-NTA Agarose (Qiagen). White New Zealand rabbits were immunized against the C-terminal part of SPS and the serum containing polyclonal antibody was prepared.
SDS–PAGE and western blot analysis
Proteins in the total bacterial extract or in fractions obtained during purification were separated by sodium dodecyl sulphate (SDS) polyacrylamide-gel (10%) electrophoresis (PAGE) as described by Laemmli (22). The gels were directly stained with Coommassie Brilliant Blue or electroblotted onto Immobilon-P transfer membrane (Millipore) for immunodetection with the polyclonal antibodies against SPS or monoclonal antibody against Xpress epitope contained in the tag (Invitrogen). Proteins reacted with the antibodies were visualized with alkaline phosphatase-conjugated goat secondary antibodies (BioRad) using the NBT/BCIP for colour development. The quantification of protein bands stained with Coommassie Brilliant Blue and visualized by the colour development was performed by densitometry using ImageJ software (http://imagej.nih.gov/ij/).
Assay of SPS activity
SPS activity was assayed as described previously (23) with an appropriate modification. The assay mixture (50 μl) contained 50 mM Hepes-NaOH (pH 7.5), 20 mM MgCl2, 20 mM F6P and 20 mM UDP-G. The mixture was incubated at 25–27°C for 10 min and reaction was stopped by an addition of 35 μl of 1 M NaOH, followed by incubation at 95°C for 10 min to decompose unreacted F6P. To determine sucrose formed by the enzyme reaction, 125 μl of 0.1% (w/v) resolcinol in 95% ethanol and 30% (w/v) HCl was added and heated at 85°C for 8 min. The developed color of sucrose derivative was measured at absorbance 520 nm with using a microtiter plate reader (SH-1000, Colona Electric).
Results
Expression, purification and limited-proteolysis of SPS in E. coli
A full length cDNA of sugarcane SPS inserted into pTrc vector was introduced into E. coli cells and its expression was checked by western blot analysis of the total bacterial cell extracts. As shown in Fig. 1A, two major bands with different mobility on the SDS–PAGE gel were detected in the SPS gene transformed cells, but not in the control cells; a larger band with around 120 kDa and a shorter one with 100 kDa were designated as Form A and Form B, respectively. Form A was firstly appeared in the bacterial cells at an early stage of growth, whereas Form B was increased in the cells of prolonged cultivation. Enzyme activity of SPS was measured using the total extracts of bacterial cells which accumulated the two forms with varying ratios (Fig. 1B). The activity level was varied during a time course of the cultivation and the activity profile was similar with the accumulation profile of Form B, suggesting that Form B might be enzymatically more active than Form A.
Fig. 1.
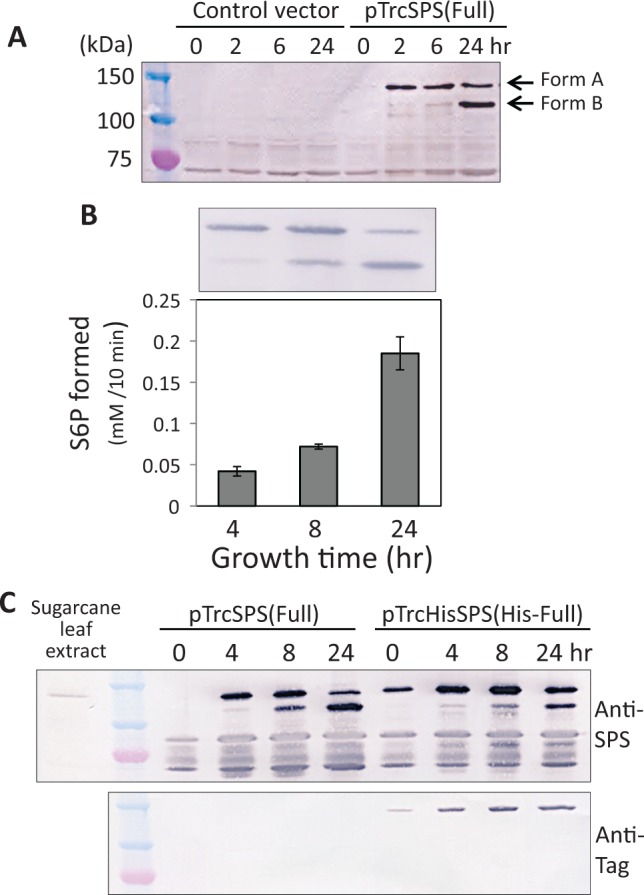
Expression of SPS in E. coli cells. (A) Detection of SPS in E. coli transformed with pTrcSPS(full) or the original vector pTrc99A (control vector). The bacterial cells were grown for 0–24 h and the total bacterial cell proteins were separated by SDS–PAGE, followed by immunodetection with polyclonal antibodies against SPS protein. Two major bands, Form A and B, were differentially accumulated in a culture-time dependent manner. (B) Activity and accumulation levels of SPS. The total extracts prepared from the comparable amounts of bacterial cells cultured for 4, 8 and 24 h were subjected to activity assay and western blotting. (C) Detection of SPS in E. coli transformed with pTrcSPS(full) or pTrcHisSPS(full). The total bacterial cell proteins were analyzed as in (A) by using polyclonal antibodies against SPS protein or monoclonal antibody against the tag region. Sugarcane leaf extracts were also analysed in order to compare molecular sizes of authentic and recombinant SPSs.
The gel mobility of the two forms of recombinant SPS was compared with SPS prepared from sugarcane leaves and Form A was close in size with the authentic enzyme (Fig. 1C). To examine whether Form B was a proteolytic product of Form A by trimming the terminal region of SPS polypeptide, a 6-histidine-epitope-tag (His-tag) was fused to the N-terminus of SPS and its expression profile was monitored by immunoblotting using two different antibodies against SPS polypeptide and the epitope tag. As shown in Fig. 1C, the His-tag SPS (HisFull) was also processed to be the two forms, and Form A of HisFull retained the reactivity with the tag antibody as expected, but Form B did not. This suggested that Form B was most likely a product with the N-terminal region trimmed.
The two forms showed distinctive affinity with DEAE cellulose; Form A and Form B were recovered in the unbound and bound fractions, respectively, and the fraction containing Form B showed an activity higher than that containing Form A (Supplementary Fig. S1). As shown in Supplementary Fig. S2, Form B was successfully purified to a certain level of homogeneity by a combination of conventional chromatographic procedures. No clear data on the N-terminal amino acid sequence of Form B were obtained by automatic Edman degradation procedure, suggesting that its N-terminus might be blocked. Form A is hardly purified due to its non-binding nature to ion exchange resin (Supplementary Fig. S1).
Production of the N-terminal truncated forms of SPS
As the N-terminal truncation would give a significant effect on its enzymatic nature as described above, a series of the N-terminal truncated forms of SPS with different lengths deleted was considered to be useful for further investigation of an effect of the N-terminal region, if any, on the enzyme activity. Using the expression vector of the His-tag SPS as described above, the N-terminal truncated derivatives designated as HisΔN1 to HisΔN6 were constructed (Fig. 2A). Their expression profiles were immunologically monitored using two antibodies against SPS polypeptide and the tag region (Fig. 2B). All genes encoding for the N-terminal truncated forms were expressed in the bacterial cells and polypeptides with shorter sizes corresponding to the length of truncation were found as a major band in bacterial cells grown for 4 h, which was reacted with both antibodies. After overnight cultivation, additional polypeptide with the size equivalent with Form B appeared in all the truncated SPSs except for HisΔN6. These shorter polypeptides were not reacted with the tag-specific antibody, indicating that the limited proteolysis occurred irrespective of the length of the N-terminal region. HisΔN6 did not produce such shorter band and this was most probably due to the lack of the proteolytic site, indicating that the cleavage site should be within the regions between HisΔN5 and HisΔN6. It is noted that HisΔN6 lost Ser162, which was proposed to be one of phosphorylated serine residues involved in the modulation of SPS activity (17) Accumulation level of HisΔN1 became lower than a limit of our immunological detection during the overnight growth. We are unable to explain reason for this phenomenon at moment, although this was reproducible.
Fig. 2.
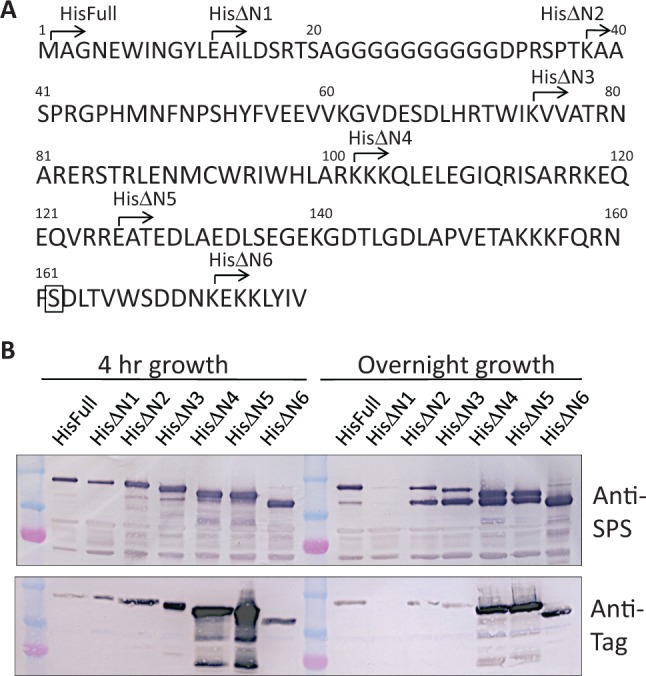
Expression of the N-terminal truncated forms of SPS in E. coli. (A) Sites of the N-terminal truncation of SPS. SoSPS1 genes with deletion of the increasing length of the N-terminal coding region were inserted into pTrcHis vector to construct fusion with his-tag. The resulting truncated forms of SPS were designated as HisΔN1 to HisΔN6. Ser162 marked with box is proposed to be a phosphorylation site (17). (B) E. coli cells transformed with the N-terminal truncated constructs were grown for 4 h or overnight. The total cell proteins from each transformant were analysed by Western blotting using antibodies against SPS polypeptide and the tag region as in Fig. 1C. Major SPS bands with decreasing size corresponding to the length of truncation were detected with both antibodies. Additional polypeptides with the size equivalent to Form B (Fig. 1A) were detected with anti-SPS antibody, but not with anti-tag antibody.
The N-terminal truncated forms of SPS were subjected to chromatography on a Ni-resin column and representative data of HisΔN5 is shown in Fig. 3A. This purification step gave us an advantage to remove efficiently the limited proteolysis product. Fractions containing HisΔN5 were combined and further subjected to a size exclusion chromatography. HisΔN5 was eluted as a single major peak at a retention volume corresponding to a molecular size of 370–380 kDa (Fig. 3C), indicative of an oligomeric structure (probably tetramer) in a native state. HisΔN4 and HisΔN6 were also eluted in the same profile with HisΔN5 (data not shown). All truncated forms except for HisΔN1 were thus successfully prepared by using the Ni-resin column as shown in Fig. 4A. Purification of HisFull and HisΔN1 was not successful, since they were hardly bound to the Ni-resin column. The reason for this phenomenon was not clear, but we presume that the His-tag region might be masked within the whole structure of SPS molecule in a manner not accessible with the resin. The presence of His-tag had been confirmed in an SDS-denatured state by immunoblotting with the tag-specific antibody (Fig. 2B).
Fig. 3.

Purification of the N-terminal truncated and full-length forms of SPS expressed in E. coli and insect cells, respectively. (A) Chromatography of HisΔN5 on a column of Ni-NTA resin. The crude extracts of E. coli cells expressing HisΔN5 was applied on the column and eluted successively with three buffers containing increasing concentrations of imidazole, 0, 30 and 120 mM (see the Materials and Methods for details). The eluates were analyzed by SDS–PAGE, followed by staining with Coomassie Brilliant Blue. (B) Sequential chromatography of Full (Sf9) on columns of Ni-NTA resin and GFP nanotrap resin. The crude extracts of insect cells expressing full-length SPS with its C-terminus fused with his-tag and EGFP were chromatographed on the Ni-NTA column by increasing imidazole concentrations from 0, 20 and 250 mM. Fractions eluted with the buffer containing 250 mM imidazole were applied to the GFP-nanotrap column and further purified by TEV protease treatment (see the Materials and Methods for details). Fractions obtained by the chromatography were analyzed as in (A). (C) Size exclusion chromatography of HisΔN5 and Full (Sf9). HisΔN5 and Full (Sf9) obtained as described above were further purified by size exclusion chromatography on a column of Superdex S-200. HisΔN5 and Full (Sf9) were eluted at comparable elution volumes of 10.4 and 10.5 ml, respectively. Calibration of molecular size was carried out with marker proteins; thyrogloburin (669 kDa) at 9.1 ml, ferritin (440 kDa) at 10.2 ml, aldolase (158 kDa) at 12.1 ml, ovalbumin (44 kDa) at 14.8 ml, carbonic anhydrase (29 kDa) at 16.2 ml and ribonuclease A (13.7 kDa) at 17.8 ml.
Fig. 4.
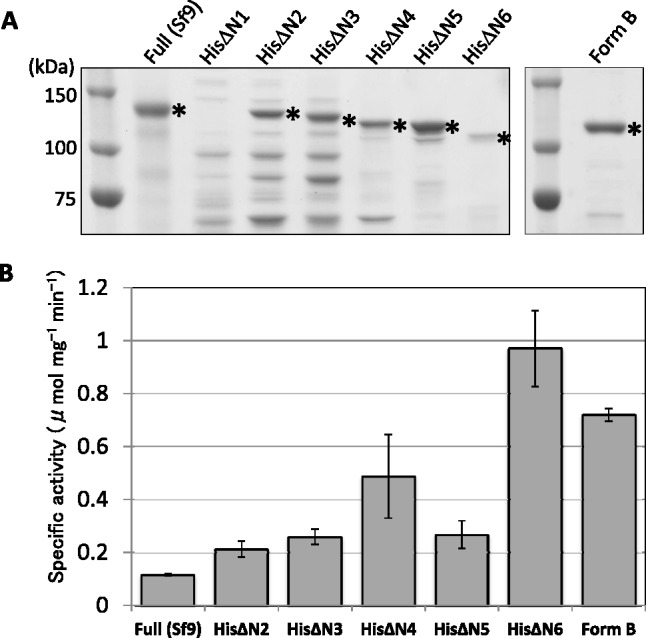
Comparison of activity of the N-terminal truncated and full-length forms of SPS. (A) SDS-PAGE analysis of various forms of SPS used for activity measurement. All the N-terminal truncated forms of SPS were prepared by His-tag affinity chromatography as described in Fig. 3A. HisΔN1 was not sufficiently obtained due to a low expression in the bacterial cells (Fig. 2B). HisΔN4, HisΔN5, and HisΔN6 were further purified by size exclusion chromatography on a Superdex S-200 column. Full (Sf9) was prepared as described in Fig. 3B. Amounts of sample loaded on a gel were based on the activity unit to give an assay condition suitable for colorimetric quantification of enzyme reaction as described in Materials and Methods. Protein bands were visualized by Coomassie Brilliant Blue staining and quantification of various forms of SPS (protein bands marked by star) were carried out by densitometry using imageJ software with an appropriate standard protein. (B) Specific activity of the N-terminal truncated and full-length forms of SPS. The enzyme activity was measured in the presence of saturated levels of two substrates, 10–20 mM UDP-G and 20 mM F6P as described in Materials and Methods. Specific activity was calculated with the concentration of each enzyme determined as in (A).
Expression of SPS in insect cells
Preparation of the full length SPS was not successful in an E. coli recombinant expression system. Then, baculovirus expression system using the Sf9 insect cell line was applied. The C-terminus of SPS fused with His tag and EGFP was expressed in Sf9 cells and resulting expressed SPS was purified by successive chromatographies on columns of Ni resin and GFP-nanotrap resin (Fig. 3B). The EGFP tag region was removed by digestion with TEV protease and the full length SPS thus obtained, Full (Sf9), was confirmed to have a molecular size equivalent with authentic SPS from sugarcane leaves (data not shown). Full (Sf9) was subjected to a size exclusion chromatography and eluted with an elution time comparable with that of HisΔN5 (Fig. 3C).
Effect of the N-terminal truncation on enzyme activity
Activity of Form B and the truncation forms of HisΔN2 to HisΔN6, using at least partially purified preparation (Fig. 4A), were measured in the presence of the saturating concentrations of F6P and UDP-G, together with that of the full length SPS. As shown in Fig. 4B, the truncated forms showed an increasing tendency of the specific activity and this was the most remarkable for HisΔN6 that showed an increase in the activity higher by 10-fold than that of the full-length SPS. These results were in good agreement with the above-mentioned observation that activity of SPS in the E. coli cells became higher when accumulation level of Form B was increased over that of Form A (Fig. 1B).
As the activity of plant SPS is known to be affected by G6P, assay were carried out under increasing concentrations of two substrates in the presence or absence of G6P (Fig. 5A and B). The activity of the full length SPS was significantly enhanced in the presence of G6P and affinities for the two substrates were increased. The activity of the N-terminal truncated forms was not significantly influenced by the addition of G6P under all conditions tested in this experiment and this was a sharp contrast with the results of full-length enzyme. It is noteworthy that affinities for UDP-G and F6P of the N-terminal truncated forms were high even in the absence of G6P. These combine data suggested that the N-terminal region of SPS, which is not proximal with the catalytic domain in the primary structure, would give a significant generative influence on the catalytic domain to lower enzyme activity and that thus, the deletions of this N-terminal region was resulted in a disappearance of the suppressive effect.
Fig. 5.

Kinetic properties of the N-terminal truncated and full-length forms of SPS. (A) Activity of various forms of SPS was measured in the assay mixture containing 20 mM UDP-G and increasing concentrations of F6P from 0 to 10 mM in the presence or absence of 6 mM G6P. The amount of S6P formed for 10 min after an addition of each enzyme was determined calorimetrically as described in Materials and Methods. (B) Activity of various forms of SPS was measured in the assay mixture containing 10 mM F6P and increasing concentrations of UDP-G from 0 to 20 mM in the presence or absence of 6 mM G6P. Reaction rate was determined as in (A).
G6P is known to be an allosteric effector in many plant SPSs. SPSs from sugarcane and maize leaves are members of such allosteric enzymes. Maize SPS has a high sequence homology with sugarcane SPS (17). By using SPS partially purified from maize leaves as a control of authentic enzyme, it was further examined how allosteric characteristics were retained in the full length and N-terminal truncated SPSs (HisΔN3 and HisΔN4). Under three different concentrations of F6P, G6P-dependent activation of SPS activity was measured. As shown in Fig. 6, the full length recombinant SPS was found to be activated by G6P in a concentration dependent manner up to around 5 mM, and this tendency was comparable with that of maize leaf SPS. Such clear allosteric property was not observed in the N-terminal truncated enzymes.
Fig. 6.
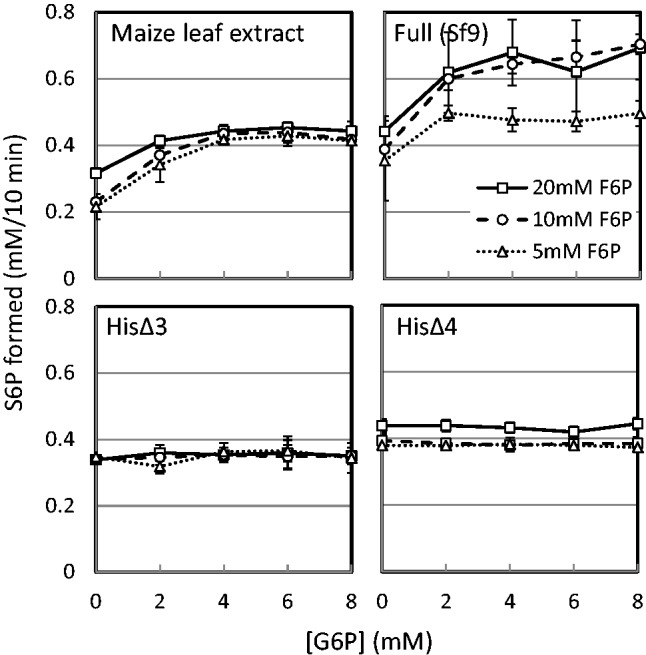
An allosteric effect of G6P on the activity of recombinant SPSs and authentic SPS from plant leaves. Activity of Full (Sf9), HisΔN3, and HisΔN4 was measured in the assay mixture containing 20 mM UDP-G and 5, 10 or 20mM F6P. Effect of allosteric activator G6P was examined by the addition of increasing concentrations from 0 to 8 mM. As a control for the allosteric activation of SPS by G6P, a crude SPS fraction prepared from maize leaves was also assayed side by side. Reaction rate was determined as in Fig. 5A.
Discussion
It is well-known that the activity of plant SPS, a key enzyme in sucrose biosynthesis, is modulated by metabolic effectors, such as G6P and inorganic phosphate, and by reversible phosphorylation of certain serine residues. To elucidate the structural basis for the regulation of SPS activity, the enzyme needs to be prepared in an amount suitable for structural and kinetic analyses. There were several reports for preparation of SPS from plant tissues or from recombinant bacterial cells; spinach and maize SPSs from leaves of each plant (2, 7, 20), transgenic tobacco leaves from spinach mutant SPS (24), maize and spinach leaf SPSs from recombinant E. coli cells (14, 15), and potato tuber SPS from recombinant E. coli cells (16). SPSs from plant leaves were reported to be proteolytically cleaved in the course of purification to produce two fragments around 90–100 and 30 kDa. Recombinant SPS expressed in the bacterial cells was also processed to a cleaved form shorter by 20–30 kDa than the original size. All recombinant enzymes were shown to be active, but it was not clearly described whether their activity was modulated by the metabolic effectors or not. This is most probably because the full length and cleaved enzymes could not be separately characterized. More extensive and detailed studies were apparently necessary for understanding of structure-function relationship of plant SPSs.
In this study, we expressed sugarcane SPS in E. coli and observed that the original SPS with a size of 120 kDa was trimmed down to be a shorter form with 100 kDa during bacterial cultivation. When His-tag was fused at the N-terminus of SPS, the trimmed form had no tag-region (Fig. 1), indicating that the proteolytic cleavage occurs at the N-terminal side. Then, we have obtained a series of mutants of SPS with the N-terminal truncation of increasing lengths from 37 to 171 residues (Fig. 2A). Purification of SPS with His-tag resin allowed us to separate the original and its trimmed forms of SPS. As the full-length SPS was not successfully prepared in E. coli cells, we utilized insect cells for preparation of recombinant SPS. The full-length enzyme was successfully purified as a fusion with the C-terminal His-tag and EGFP, which could be cleaved off during purification (Fig. 3B).
By kinetic analysis of the full length and truncated forms of sugarcane SPS thus prepared, three important results were obtained: (i) the full length enzyme showed a remarkable allosteric activation by G6P, while none of the truncated enzymes tested here had such characteristics (Fig. 6), (ii) specific activity of the full length and truncated SPSs showed a tendency that longer truncation has higher activity (Fig. 4) and (iii) whether G6P was added or not, the truncated forms showed similar substrate affinity with F6P and UDP-G, while the full length enzyme showed a higher substrate affinity when G6P was present (Fig. 5A and B).
Plant SPSs consist of three domains, glucosyltransferase domain (GTD) (55 kDa), the C-terminal phosphohydrolase domain (30 kDa) and the N-terminal domain (20 kDa). The N-terminal domain has no apparent homology with known motifs (25). Notably, cyanobacteria SPSs (10) and H. orenii SPS (HoSPS) (13), which lack the N-terminal domain of plant SPSs, are not allosterically regulated, implying an involvement of the N-terminal domain in the allosteric regulation. Our present data give a clear piece of evidence that the N-terminal region composed of at least 171 residues of sugarcane SPS is crucial for its allosteric sensitivity to G6P and that the N-terminal truncation as short as 37 residues leads the mutant enzyme to be constitutively active with the allosteric sensitivity lost. The N-terminal region of plant SPS was postulated to be intrinsic disordered by bioinformatics analysis (13). It has been proposed that polypeptide regions with intrinsic disorders play a role for modulation of allostery (26), such as optimizing high or low affinity binding and controlling the ability to interact the protein surface (27). Therefore, plant SPS might be one of examples for such allosteric modulation.
SPS is classified as a member of glycosyltransferases-B (GT-B) family, known as glycogen phosphorylase glycosyltransferase superfamily (13). It was previously reported that plant SPSs are tetramer or dimer depending on experimental conditions (9, 15) and that cyanobacterial and bacterial SPSs are monomeric enzymes (25). Our results showed that both full-length (Full-Sf9) and N-terminal truncation (HisΔN5) enzymes were indicated to be an oligomer (Fig. 3). Therefore, N-terminal region of plant SPS itself is not directly involved in the oligomerization.
The modular architecture of sucrose biosynthesis enzymes has recently been suggested to represent oligomeric proteins, including SPS and sucrose synthase (SUS) (28). All SPSs and SUSs share 20–33% homology for the sequences of GTD (25) and the 3-D structures of GTDs from Arabidopsis thaliana SUS (AtSUS) and bacterial SPS (HoSPS) were well superimposed (29). Furthermore, AtSUS appears as a tetramer to generate the most compact oligomer, which revealed the N-terminal half and C-terminal extensions compromise the subunit interface (29). The similarity between GTDs of SPSs and yeast glycogen synthase (Gsy2p) gave a possible mechanism for plant SPS regulation (13). In eukaryotes, glycogen synthase is oligomeric and the modulation of activity involves phosphorylation and G6P allostery (30). The G6P binding to Gsy2p, leading a substantial conformation reorganization at the subunit interface, increases the catalytic efficiency (31). By analogy, it could be hypothesized that the N-terminal domain of plant SPS plays a direct role in allostery by inducing a structural perturbation upon binding to G6P, which might include three-model states as an unphosphorylated state with intermediate activity, phosphorylated state with reduced activity, and G6P-bound state with high activity. Our present work has not given any information on phosphorylation status of recombinant SPS except that loss of Ser162 in HisΔN6, which is one of phosphorylated serine residues (17), has no significant effect on SPS activity (Fig. 5) and further works will be necessary to draw more extensive story of the allostery of plant SPS.
Eventually, present biochemical studies has clarified the functional roles of the N-terminal region of sugarcane SPS. It would be advantageous to analysis both the full length and the N-terminal truncated forms for elucidation of the precise model of the allostery. In addition, dynamic simulations analysis may useful for mapping the energy landscape of allosteric process (32). These combined studies would provide an insight into molecular mechanism of SPS for controlling carbon partitioning in plant sucrose biosynthesis.
Supplementary Data
Supplementary Data are available at JB Online.
Funding
This work was supported by the International Collaborative Research Program of Institute for Protein Research, Osaka University, ICR-14-03 and –05, Japan and partly by Ministry of Research, Technology and Higher Education of the Republic of Indonesia (PUPT).
Conflict of Interest
None declared.
Supplementary Material
Glossary
Abbreviations
- F6P
fructose-6-phophate
- G6P
glucose-6-phosphate SPS, sucrose phosphate synthase
- UDP-G
uridine diphosphate glucose
References
- 1.Leloir L.F., Cardini C.E. (1954) The biosynthesis of sucrose phosphate. J. Biol. Chem. 214, 157–165 [PubMed] [Google Scholar]
- 2.Amir J., Preiss J. (1982) Kinetic characterization of spinach leaf sucrose-phosphate synthase. Plant Physiol. 69, 1027–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huber S.C., and Huber J.L. (1996) Role and regulation of sucrose-phosphate synthase in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 431–444 [DOI] [PubMed] [Google Scholar]
- 4.Ohsugi R., Huber S.C. (1987) Light modulation and localization of sucrose phosphate synthase activity between mesophyll cell and bundle sheath cells in C4 species. Plant Physiol. 84, 1096–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quick P., Siegl G., Neuhaus E., Feil R., Stitt M. (1989) Short-term water stress leads to a stimulation of sucrose synthesis by activating sucrose-phosphate synthase. Planta. 177, 535–546 [DOI] [PubMed] [Google Scholar]
- 6.Hurry V.M., Malmberg G., Gardeström P., Öquist G. (1994) Effects of a short-term shift to low temperature and of long-term cold hardening on photosynthesis and ribulose-1,5-bisphoshate carboxylase/oxygenase and sucrose phosphate synthase activity in leaves of winter rye (Secale cereal L.). Plant Physiol. 106, 983–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doehlert D.C., Huber S.C. (1983) Regulation of spinach leaf sucrose phosphate synthase by glucose-6-phosphate, inorganic phosphate, and pH. Plant Physiol. 73, 989–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salerno G.L., Pontis H.G. (1978) Sucrose phosphate synthase: separation from sucrose synthetase and a study of its properties. Planta. 142, 41–48 [DOI] [PubMed] [Google Scholar]
- 9.Castleden C.K., Aoki N., Gillespie V.J., MacRae E.A., Quick W.P., Buchner P., Foyer C.H., Furbank R.T., Lunn J.E. (2004) Evolution and function of the sucrose-phosphate synthase gene families in wheat and other grasses. Plant Physiol. 135, 1753–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lunn J.E., Price G.D., Furbank R.T. (1999) Cloning and expression of a prokaryotic sucrose-phosphate synthase gene from the cyanobacterium Synechocystis sp. PCC 6803. Plant Mol. Biol. 40, 297–305 [DOI] [PubMed] [Google Scholar]
- 11.Huynh F., Tan T.C., Swaminathan K., Patel B.K.C. (2004) Expression, purification, and preliminary crystallographic analysis of sucrose phosphate synthase (SPS) from Halothermothrix orenii. Acta Crystallogr. Sect F. Struct. Biol. Cryst. Commun. 61, 116–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lunn J.E., Gillespie V.J., Furbank R.T. (2003) Expression of a cyanobacterial sucrose-phosphate synthase from Synechocystis sp. PCC 6803 in transgenic plants. J. Exp. Bot. 381, 223–237 [DOI] [PubMed] [Google Scholar]
- 13.Chua T.K., Bujnicki J.M., Tan T.C., Huynh F., Patel B.K. (2008) The structure of sucrose phosphate synthase from Halothermothrix orenii reveals its mechanism of action and binding mode. Plant Cell. 20, 1059–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Worrell A.C., Bruneau J.M., Summerfelt K., Boersig M., Voelker T.A. (1991) Expression of maize sucrose phosphate synthase in tomato alters leaf carbohydrate partitioning. Plant Cell. 3, 1121–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sonnewald U., Quick W.P., MacRae E., Krause K.-P., Stitt M. (1993) Purification, cloning and expression of spinach leaf sucrose-phosphate synthase in Escherichia coli. Planta. 189, 174–181 [DOI] [PubMed] [Google Scholar]
- 16.Chen W.L., Yang C.C., Lee P.D. (2007) Cloning and expression of sweet potato tuber sucrose phosphate synthase gene in Escherichia coli. Taiwanese J. Agric. Chem. Food Sci. 45(2, 91–100 [Google Scholar]
- 17.Sugiharto B., Sakakibara H., Sumadi, Sugiyama T. (1997) Differential expression of two genes for sucrose-phosphate synthase in sugarcane: molecular cloning of the cDNAs and comparative analysis of gene expression. Plant Cell Physiol. 38(8, 961–965 [DOI] [PubMed] [Google Scholar]
- 18.Sano K.I., Maeda K., Oki M., Maéda Y. (2002) Enhancement of protein expression in insect cells by a lobster tropomyosin cDNA leader sequence. FEBS Lett. 532, 143–146 [DOI] [PubMed] [Google Scholar]
- 19.Rothbauer U., Zolghadr K., Muyldermans S., Schepers A., Cardoso M.C., Leonhard H. (2008) A versatile nanotrap for biochemical and functional studies with fluorescent fusion protein. Mol. Cell. Proteomics 7, 282–289 [DOI] [PubMed] [Google Scholar]
- 20.Torres W.-K., Kerr P.S., Huber S.C. (1987) Isolation and characterization of multiple form of maize leaf sucrose-phosphate synthase. Physiol. Plantarium 70, 653–658 [Google Scholar]
- 21.Walker J.L., Huber S.C. (1989) Purification and preliminary characterization of sucrose-phosphate synthase using monoclonal antibodies. Plant Physiol. 89, 518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 23.Huber S.C., Kerr P.S., Rufty T.W. (1985) Diurnal changes in sucrose phosphate synthase activity in leaves. Physiol. Plant. 64, 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toroser D., McMichael R., Jr., Krause K.P., Kurreck J., Sonnewald U., Stitt M., Huber S.C. (1999) Site-directed mutagenesis of serine 158 demonstrates its role in spinach leaf sucrose-phosphate synthase modulation. Plant J. 17(4), 407–413 [DOI] [PubMed] [Google Scholar]
- 25.Cumino A., Curatti L., Giarrocco L., Salerno G.L. (2002) Sucrose metabolism: Anabaena sucrose-phosphate synthase and sucrose-phosphate phosphatase define minimal functional domains shuffled during evolution. FEBS Lett. 517, 19–23 [DOI] [PubMed] [Google Scholar]
- 26.Ferreon A.C.M., Ferreom J.C., Wright P.E., Deniz A.A. (2013) Modulation of allostery by protein intrinsic disorder. Nature 498, 308–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hilser V.J., Thompson E.B. (2007) Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. USA 104, 8311–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salerno G.L., Curatti L. (2003) Origin of sucrose metabolism in higher plants: when, how and why? Trends Plant Sci. 8, 63–69 [DOI] [PubMed] [Google Scholar]
- 29.Zheng Y., Anderson S., Zhang Y., Garavito R.M. (2011) The structure of sucrose synthase-1 from Arabidopsis thaliana and its functional implications. J. Biol. Chem. 286, 36108–36118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pederson B.A., Cheng C., Wilson W.A., Roach P.J. (2000) Regulation of glycogen synthase. J. Biol. Chem. 275, 27753–27761 [DOI] [PubMed] [Google Scholar]
- 31.Baskaran S., Roach P.J., Roach D., Hurley T.D. (2010) Structural basis for glucose-6-phosphate activation of glycogen synthase. Proc. Natl. Acad. USA 107, 17563–17568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motlagh H.N., Wrabl J.O., Li J., Hilser V.J. (2014) The ensemble nature of allostery. Nature, 508, 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.