

Abstract
Among other factors, sophistication of immunosuppressive (IS) regimen accounts for the remarkable success attained in the short- and medium-term solid organ transplant (SOT) survival. The use of steroids, mycophenolate mofetil and calcineurin inhibitors (CNI) have led to annual renal graft survival rates exceeding 90% in the last six decades. On the other hand, attrition rates of the allograft beyond the first year have remained unchanged. In addition, there is a persistent high cardiovascular (CV) mortality rate among transplant recipients with functioning grafts. These shortcomings are in part due to the metabolic effects of steroids, CNI and sirolimus (SRL), all of which are implicated in hypertension, new onset diabetes after transplant (NODAT), and dyslipidemia. In a bid to reduce the required amount of harmful maintenance agents, T-cell-depleting antibodies are increasingly used for induction therapy. The downsides to their use are greater incidence of opportunistic viral infections and malignancy. On the other hand, inadequate immunosuppression causes recurrent rejection episodes and therefore early-onset chronic allograft dysfunction. In addition to the adverse metabolic effects of the steroid rescue needed in these settings, the generated proinflammatory milieu may promote accelerated atherosclerotic disorders, thus setting up a vicious cycle. The recent availability of newer agent, belatacept holds a promise in reducing the incidence of metabolic disorders and hopefully its long-term CV consequences. Although therapeutic drug monitoring as applied to CNI may be helpful, pharmacodynamic tools are needed to promote a customized selection of IS agents that offer the most benefit to an individual without jeopardizing the allograft survival.
Keywords: Immunosuppressive therapy, Metabolic adverse effects, Solid organ transplants
Introduction
The remarkable success attained in the short- and medium-term survival rates of solid organ transplant (SOT) is, for the most part, due to the increasing sophistication of immunosuppressive protocols. Prior to the late 1970s, treatment with steroids and azathioprine produced a one-year renal allograft survival rate of 40–50% [Dunea et al. 1965]. The introduction of cyclosporine (CsA) followed in rapid succession by tacrolimus (TAC) and mycophenolate mofetil (MMF) was associated with much lower rate of graft loss. The annual graft survival rate was greater than 90% [Lamb et al. 2011]. However despite the innovative use of immunosuppressive agents (ISAs), the long-term allograft survival has remained unchanged. Whereas there was a drop in the attrition rate in the first year for deceased donor grafts from 20% in 1989 to 7% in 2008, it remained steadily constant at 5–7% over the same period for graft survival beyond 1 year [Lamb et al. 2011]. Unlike the curtailment of the T-cell immunological arm, the lower success attained in the effective suppression of alloreactive antibodies is a major limitation to a long-term graft survival. Of equal importance, and perhaps accounting for the higher mortality rate of transplant recipients with functioning grafts, is the cardiovascular (CV) consequence due to adverse metabolic effects of ISAs [Marcén, 2009; Ghanta et al. 2015]. This review examines the pathophysiologic basis of this metabolic dysregulation with the hope of facilitating smarter applications of these essential therapeutic agents. For the most part, it will highlight the experimental and clinical insight gained in renal transplantation while using this experience as a convenient prototype for other solid organs (including liver, heart and lungs).
Induction therapy and adverse metabolic effects
Both lymphocyte-depleting (e.g. thymoglobulin) and nondepleting antibodies (basiliximab) are used as induction therapies [Chen et al. 2013]. The depletional agents are superior to interleukin (IL)-2 receptor agonists in efficacy but are associated with higher rates of opportunistic viral infection and malignancy [Chen et al. 2013; Pascual et al. 2009; Schold et al. 2015]. Due to the greater proportion of low-immunologic-risk patients, close to 70% of new transplant recipients received IL-2 receptor antagonist in Australasia compared with 27% in the United States [Webster et al. 2010]. Out of the two commonly used depletional agents, alemtuzumab has a 19% higher relative risk for renal allograft loss compared with thymoglobulin [Schold et al. 2015]. There is no direct metabolic benefit with the use of either basiliximab or depletional antibodies. However, the long-lasting immune-suppressive effect of the latter permits the use of lower maintenance doses of steroids or calcineurin inhibitor (CNI) [Chen et al. 2013; Webster et al. 2010]. This therapeutic strategy may result in reduction of drug-associated adverse metabolic effects including hypertension, new onset diabetes after transplant (NODAT) and dyslipidemia [Zsom et al. 2015]. In addition to its immunologic benefits, pulse treatment with methyl prednisolone (5–10 mg/kg) as part of an induction regimen causes a suppression of proinflammatory cytokines (IL-6, IL-8, and tumor necrosis factor [TNF]-a) that are released in response to ischemic reperfusion injury [Schmidt et al. 2007].
Role of combined (maintenance) therapy in adverse metabolic effects
Commonly used maintenance ISAs are steroids, CNI, sirolimus (SRL), MMF, and belatacept. While there is great success with the curtailment of T-cell immunity, these agents are less effective in the prevention of alloreactive antibody response [Chen et al. 2013; Pascual et al. 2009; Schold et al. 2015; Webster et al. 2010]. Furthermore, steroids, SRL, and CNI produce adverse metabolic effects (e.g. hypertension, NODAT, and dyslipidemia) with the potential for negative impacts on allograft and patient survival [Lloberas et al. 2008; Benfield et al. 2010]. The combination of two or more drugs with different mechanisms of action permits the use of a minimum effective dose of each agent while minimizing associated side effects.
Maintenance steroids
The broad spectrum immunosuppressive property of steroids necessitates its use in different clinical settings [Zsom et al. 2015]. However, more than any other maintenance agent, the metabolic impact of steroids has justifiably received the most attention in the literature. There are concerns on the role of steroid-related diabetic nephropathy on the late-onset attrition of renal allografts, and in the promotion of CV morbidity [Zsom et al. 2015]. For these reasons, transplant protocols are designed with the goal of reducing the cumulative dose of steroids [Zsom et al. 2015; Zhang et al. 2013; Knight and Morris, 2010]. Patients placed on such steroid-sparing regimen are characterized by higher rate of acute rejection episodes (AREs) and lower allograft function [Zhang et al. 2013; Knight and Morris, 2010]. Nevertheless despite the associated improvement in metabolic profiles, such protocols often failed to translate into a long-term allograft or patient survival.
Calcineurin inhibitor
With a selective deactivation of T-cell immune function, CNI is the most effective maintenance ISA used for the prevention of allograft rejection [Zsom et al. 2015; Casey and Meier-Kriesche, 2011]. Kidney Disease Improving Global Outcome (KDIGO) recommends TAC, MMF and optional use of steroids as the first line agents. TAC is prescribed to over 96% of patients at the time of hospital discharge for transplant surgery [Rush, 2013]. Although TAC shared a similar mechanism of action with CsA, it has a lower acute rejection (AR) rate and produces a more favorable CV profile [Sandrini et al. 2012; Bakar et al. 2009; Webster et al. 2005a]. In addition to renal injury from prolonged use, other major adverse effects of CNI are hypertension, NODAT and dyslipidemia [Nankivell et al. 2004; Bloom and Reese, 2007]. These events are more likely to occur with the combined treatment of CNI and larger doses of steroids [Lloberas et al. 2008].
Similarly, CNI has been implicated in the promotion of endothelial dysfunction accounting for the impairment in glomerular filtration rate (GFR), cardiac allograft vasculopathy and perhaps contributes to long-term CV morbidity [Yong et al. 2013; Nankivell et al. 2004; Bloom and Reese, 2007]. In a prospective crossover trial, compared with renal recipients switched to SRL, after 5 months of TAC those maintained on the CNI had lower surrogate indices of CV disease that included pulse wave velocity and an aortic augmentation index that was adjusted for heart rate. However as discussed below such short-term benefit of noncalcineurin inhibitor agents has consistently failed to translate into a superior allograft survival in most studies [Guethoff et al. 2015; Zsom et al. 2015].
The concern that drug-related metabolic (and renal) disorders might shorten allograft survival has led to the use of CNI minimization protocols [Zsom et al. 2015; Gallagher et al. 2004; Lebranchu et al. 2009; Holdaas et al. 2011; Budde et al. 2011]. In this regard, the dose of CNI is either reduced or the drug is substituted with an alternative agent (e.g. SRL) at an early or late phase of the transplant. However, such an approach may result in a greater frequency of late-onset AR events [Wang et al. 2013; Burkhalter et al. 2012]. Consequently there may be need for a greater amount of steroid rescue treatments, which in turn may promote metabolic disturbances. In recent times, critical analysis of pre-existing data is refuting the significant role of drug-induced nephrotoxicity in late-onset graft losses while emphasizing the negative impact of de novo generation of donor specific antibodies. Hence rather than the untoward effect of excessive treatment with CNI, inadequate immune suppression may be more deleterious to graft survival [Zsom et al. 2015].
Sirolimus
Because of its antiproliferative effect, there was an initial enthusiasm that SRL might attenuate interstitial fibrosis and therefore prolonged allograft survival. Additional justification for using SRL in place of CNI was rooted in its ability to suppress replication of BK polyomavirus (and therefore prevent BK nephropathy) by interfering with mTOR-S6-Kinase activity [Hirsch et al. 2015]. It is also known for reduction of the more frequent occurrence of nonmelanoma carcinoma in SOT recipients [Tessari and Girolomoni, 2012]. However in the SYMPHONY trial, SRL was associated with the highest rate of AREs. Although there was improvement in transplant function after early substitution of CNI for SRL (3–6 months), there was no allograft survival benefit particularly in the setting of a pre-existing low GFR or proteinuria [Zsom et al. 2015; Ekberg et al. 2007; Chhabra et al. 2012; Lyster et al. 2004]. In addition SRL failed to improve the graft survival when used in combination with CNI [Ekberg et al. 2007; Chhabra et al. 2012]. Rather it increased the propensity of CNI to produce nephrotoxicity, thrombotic microangiopathy, NODAT, and dyslipidemia [Lloberas et al. 2008; Benfield et al. 2010]. Due to the failure of SRL to meet the high expectations of the transplant community and the unexpected high rate of side effects, there has been a downward trend with its use in the last two decades [Lyster et al. 2004; Pape and Ahlenstiel, 2014].
Mycophenolate mofetil
MMF has no direct metabolic effect but its use may reduce the total amount of steroids, SRL and CNI required for immunosuppression. In a randomized clinical trial (RCT) of steroid minimization strategy, exclusion of MMF caused a higher rate of AREs and a two-fold increase in short-term allograft losses [Knight et al. 2009]. Similarly, patients with single nucleotide polymorphism for uridine diphosphate glucuronosyl-transferase (UGT2B7), the metabolizing enzyme for MMF, experienced higher rate of AREs [Pazik et al. 2013]. In the absence of therapeutic drug monitoring, as it is currently practiced, lower bioavailability of MMF due to such polymorphism may lead to a greater need for steroids or CNI. Studies are needed to validate this hypothesis and to determine if there are metabolic and CV consequences. A proof of significant adverse clinical outcome may justify either therapeutic drug monitoring or avoidance of MMF in this population subset.
Belatacept
Belatacept is a CTLA4Ig fusion protein that blocks costimulatory activation of CD28 receptors on T cells. Compared with CsA, it produces a higher rate of early-onset AREs, greater preservation of 12-month allograft function but a similar rate of 5-year graft survival rate [Rostaing et al. 2013]. In addition, using 7-year risk calculators derived from the ALERT trial, belatacept was projected to reduce major adverse CV events by >20%, and lower the mortality rate by 18–30% [Soveri et al. 2013]. Concern for renal toxicity and metabolic dysregulation has also led to the successful substitution of belatacept for CNI after more than 6 months of treatment [Rostaing et al. 2011; Paz et al. 2014]. In a more recent meta-analysis, belatacept produces a better control of blood pressure, more favorable lipid profiles, and a lower incidence of NODAT in comparison with patients treated with CNI [Masson et al. 2014]. Nevertheless, despite a lower occurrence of chronic allograft fibrosis, there is no difference in either patient or allograft survival rate over a 3-year study period. A longer clinical experience with belatacept will provide an opportunity to evaluate if such metabolic benefits will translate into a long-term CV advantage [Masson et al. 2014; Le Meur et al. 2011].
The potential role of immunologic risk in adverse metabolic effects
Determinants of lower allograft survival in patients with renal transplants include both immunologic and nonimmunologic variables. These include histocompatibility leucocyte antigen (HLA) mis-matches, ABO blood group system incompatibility, panel reactive antibody >30%, donor specific antibody, younger recipients, extended criteria donor, previous transplant rejection, African–American ethnicity, and delayed graft function [KDIGO Transplant Work Group, 2009; Gourishankar et al. 2013]. High immunologic risk individuals frequently require ISAs with considerable therapeutic impact including glucocorticoids (GCs), CNI and T-cell-depleting induction agents [Zsom et al. 2015]. Unfortunately, as previously acknowledged, both steroids and CNI are also notorious for a variety of metabolic disturbances. Therefore a drug regimen that minimizes the use of these agents is more suitable for those at high risk of allograft rejection but at a lower risk of CV morbidity [Zsom et al. 2015; Vincenti et al. 2008; Krämer et al. 2012].
However, despite a greater susceptibility for CV events, compared with Whites, African American patients with high immunologic risk are more likely to sustain AR events in the absence of steroid maintenance [Taber et al. 2013]. Similarly, a population subset at high risk for recurrent glomerulonephritis often requires steroid-based immunosuppression [Padiyar et al. 2010]. In addition, to reduce the likelihood of a second rejection episode, steroid maintenance is warranted in patients who were previously treated for AREs [Matas, 2008; Humar et al. 2007]. Given such diversity with respect to the clinical benefit of ISAs, categorization of the risk profiles is an appropriate strategy for the selection of ISAs in an individual [Zsom et al. 2015; KDIGO Work Group, 2009; Gourishankar et al. 2013; Vincenti et al. 2008; Krämer et al. 2012]. This approach will promote a careful balance in the control of AREs and the rate of drug-induced metabolic complications; and thereby avoid a vicious cycle of persistent proinflammatory milieu with a consequent atherosclerotic CV disease [Zsom et al. 2015].
Use of steroids, cytochrome P450 and drug interactions
Metabolic clearance of prednisolone depends on both hepatic cytochrome P450 and intestinal P-glycoprotein systems [Stratta et al. 2012]. Drug interaction, old age and ethnic differences may account for the random variation in the level of steroid exposure in transplant recipients [Stratta et al. 2012; Tornatore et al. 1995]. Furthermore, due to increased metabolic clearance from large fat mass and higher hepatic blood flow, unadjusted steroid dosing may result in subtherapeutic exposure in obese patients [Tornatore et al. 1995]. Consideration for such pharmacodynamic variability is likely to influence the modality of steroid dosing in the nearest future. For instance, measurement of lymphocyte proliferation in response to a given amount of endogenous cortisol identified those who are likely to fail steroid withdrawal [Takeuchi et al. 2011]. Similarly, the capacity to predict AREs in a given patient may warrant changes in the drug regimen. In this regard, higher expression of RC isoform of CD45 molecule (CD45RC) on the surfaces of CD8 T cells in pretransplant patients was predictive of rejection events [Ordonez et al. 2013].
Furthermore, due to a shared metabolic pathway, there is a pharmacodynamic interaction between steroids, CNI, and SRL, all of which are notable for metabolic side effects [Stratta et al. 2012; Staatz et al. 2010; Ferraris et al. 2011]. By upgrading cytochrome P450 systems, steroid increases the metabolic clearance of both CNI and SRL. However, due to the heterogeneity of single nucleotide polymorphisms, there are inconsistent findings on the relationship between cytochrome P450 genotype and CNI pharmacokinetics [Staatz et al. 2010; Ferraris et al. 2011]. Consequently, metabolic consequences that may arise from a long-term combination of these agents are poorly predicted.
Steroids and adverse metabolic effects
Attempts to eliminate steroids or CNI were motivated by the disproportionate impact of CV disease on the mortality rate among transplant recipients with functioning grafts [Morales et al. 2012]. Adverse effects of prolonged steroid use include hyperlipidemia, growth retardation, obesity, insulin resistance, hypertension, and metabolic bone diseases [Zsom et al. 2015]. Although there were lower metabolic events in clinical trials on steroid-free regimen in patient populations with low immunologic risk, these protocols often resulted in greater occurrence of AREs particularly in the first year of transplantation [Vincenti et al. 2008; Krämer et al. 2012]. Because pulse methyl prednisolone is frequently used in the events of such acute rejection, there are invariably greater cumulative amounts of steroid treatments than otherwise intended in these patients. Nevertheless, compared with those on steroid-based treatment, there was no difference in either patient or allograft survival [Vincenti et al. 2008].
Steroids and oxidative stress
In renal transplantation, oxidative stress often occurs in two clinical settings. As demonstrated in experimental studies, reactive oxygen species are generated in response to reperfusion injury as produced by a frequent event of prolonged cold ischemia time prior to transplant surgery in deceased solid organ donation [Turgay et al. 2012]. Pulse treatment with methyl prednisolone given during transplant surgery may attenuate such proinflammatory oxidative responses. Unlike this beneficial effect, chronic administration of steroids in high dose produces persistent oxidative milieu by inducing carbonylation of skeletal muscle protein [Saidi et al. 2007]. This event may favor the development of hypertension, diabetes mellitus and accelerated atherosclerosis [Calò et al. 2002]. Paradoxically, treatment with low steroid doses on a chronic basis, as currently used in many transplant programs, may downgrade such pro-oxidative tissue inflammation [Saidi et al. 2007].
Steroids, calcineurin inhibitors and hypertension
The prevalence rate of hypertension in adults with renal transplant ranges from 50–80% while that of pediatric recipients varies from 47–82% [Weir et al. 2015]. Poor blood pressure control is not uncommon thereby justifying the need for multiple antihypertensive agents [Kislikova et al. 2015]. This may result in shorter duration of allograft survival and may translate into long-term CV mortality [Weir et al. 2015; Kislikova et al. 2015; Rossi and Vella, 2015]. Treatment with ISAs influences the occurrence of modifiable predictors of hypertension, namely exogenous obesity and AREs [Rossi and Vella, 2015]. Aside from steroid contribution and excessive deposition of fat mass, similar to CNI, it causes proangiotensinogen vasoconstrictive endothelial dysfunction [Calò et al. 2002; Rossi and Vella, 2015; Sander et al. 1996; Pourmand et al. 2015; Bailey et al. 2009; Baum and Moe, 2008]. On the other hand, inadequate treatment with ISAs intensifies recurrent AREs which may result in chronic renal allograft injury and therefore promotes a secondary form of hypertension.
Although steroid suppression of inflammatory cytokines attenuates a hypertensive response to an oxidative state, a dose-dependent vasoreactive effect may override such benefits [Sander et al. 1996]. In addition, due to pre-existing vasculopathy, steroid use, and vasoconstrictive effects of CNI, hypertension is common in the first few days after renal transplant [Sander et al. 1996; Pourmand et al. 2015]. In a few instances, persistent fluid retention due to delayed graft function may contribute to accelerated hypertension. However, the role of fluid retention as a mediator of steroid-induced hypertension may be overstated. An experimental mouse model of chronic steroid use (adrenocorticotrophic hormone [ACTH] infusion) suggests there is only a minimal role for mineralocorticoid activity [Bailey et al. 2009]. Microarray analysis showed no remarkable change in the renal expression of mineralocorticoid target genes including epithelial cell sodium channel (ENaC), SCNN1A, KRAS, AND NEDD4. Indeed there was no evidence for tubular sodium reabsorption, potassium wasting or elevation in plasma renin activity [Bailey et al. 2009]. Rather there was net sodium excretion due to the combined effect of glomerular hyperfiltration and inhibition of proximal tubule reabsorption.
A more plausible explanation for steroid-induced hypertension is its inhibition of nitric oxide (NO) synthesis. This was experimentally demonstrated by the blockade of steroid-promoted hypertension after an infusion of L-arginine [Baum and Moe, 2008]. Similar to CNI, it is postulated that steroids may increase vasoactive response to angiotensin II by enhancing synthesis of GC receptors on endothelial smooth muscle cell [Pourmand et al. 2015; Baum and Moe, 2008]. Hence knock-out mouse of endothelial GC receptors failed to develop hypertension in response to dexamethasone infusion [Baum and Moe, 2008]. Finally, the modulatory role of oxidative stress in steroid-induced hypertension was suggested by the concurrent attenuation of both oxygen radicals and hypertension after treatment with ramipril [Calò et al. 2002].
Steroids and obesity
In children and adults alike, there is an increasing prevalence of exogenous obesity often with an onset in the first year of solid organ transplantation [Denburg et al. 2010; Hoogeveen et al. 2011; Hanevold et al. 2005]. In a retrospective cohort study, the prevalence rate of obesity increased from 13% at baseline to >30% after 3 months of renal transplantation [Denburg et al. 2010]. Numerous studies support the etiological role of the cumulative dose of steroids in post-transplant obesity [Denburg et al. 2010; Hoogeveen et al. 2011; Hanevold et al. 2005; Foster et al. 2010; Vester et al. 2005]. In one of such studies, there was a significant positive correlation between GC exposure and the Z-score of the body mass index (BMI) [Denburg et al. 2010]. In addition to steroid use, other risk factors for persistent obesity after transplant are younger age group, remote transplant year, maternal obesity, female gender, lower baseline BMI, and ethnic minority [Boschetti et al. 2013; Denburg et al. 2010; Foster et al. 2010; Vester et al. 2005]. Apparently due to its relationship with hepatoportal circulation, truncal adiposity generates proinflammatory milieu by secreting adipokines such as leptin, IL-6 and TNF-a [Hajer et al. 2008; Lambert et al. 2010]. On the other hand, hyperleptinemia promotes insulin resistance; it causes an increase in fasting plasma insulin, post-prandial hyperglycemia and decreased mRNA expression of insulin receptors in skeletal muscle [Wjidan et al. 2015]. As part of metabolic syndrome, there is an increase in the activation of the renal sympathetic system, which in turn causes renin-mediated hypertension [Lambert et al. 2010].
Although the confounding effect of pulse steroid treatment of AREs cannot be excluded, excessive weight gain after SOT is predictive of lower rates of both graft and patient survival [Boschetti et al. 2013; Hoogeveen et al. 2011]. Indeed preadolescent renal patients with post-transplant obesity are at higher risk of death from cardiopulmonary disease and graft loss from thrombosis [Hanevold et al. 2005]. Similarly, there is higher mortality rate in lung transplant recipients with obesity [Upala et al. 2015] and in liver transplant patients with greater amount of abdominal adiposity [Terjimanian et al. 2015]. However, a recent meta-analysis suggests there may be improving survival of allograft recipients in the modern era compared with those transplanted before the year 2000 [Nicoletto et al. 2014].
Immunosuppressive agents and new onset diabetes after transplant
New onset diabetes mellitus is associated with poorer graft survival, higher risk of CV event and greater incidence of patient mortality [Cosio et al. 2005; Israni et al. 2012; Sharif and Baboolai, 2011]. Calcineurin inhibitor, SRL and steroids are estimated to account for 74% incidence of NODAT (Table 1). Other predisposing factors are genetic susceptibility, deceased organ donation, older age (>40 years), male gender, ethnic minority, exogenous obesity, post-transplant weight gain, hyperlipidemia, hypertension, hepatitis C infection, previous transplants, and ⩾3 HLA class-1 mismatches [Bergrem et al. 2010; Gnatta, 2010; Yao et al. 2013]. In a recent meta-analysis, the odds ratio for the development of NODAT in adult patients with cytomegalovirus infection was 1.94 (95% CI = 1.26–2.98) [Einollahi et al. 2014]. In addition, a greater number of metabolic syndrome components increases the likelihood of developing NODAT [Perito et al. 2016]. Hence pretransplant screening for detection of modifiable metabolic events is an appropriate preventive strategy [Juan Khong and Ping Chong, 2014; Langsford and Dwyer, 2015].
Table 1.
Pathogenesis of new onset diabetes mellitus after transplantation as induced by immunosuppressive agents.
| Immunosuppressive agents | Tissue mediating metabolic injury | Mechanism of NODAT |
|---|---|---|
| Steroids | Hepatic cell: gluconeogenesis | Upregulates genes for gluconeogenic enzymes |
| Pancreatic beta cell toxicity | Reduced insulin secretion; followed by hyperinsulinemia | |
| Calcineurin inhibitors: (tacrolimus and cyclosporine) | Pancreatic beta cell toxicity | Hypomagnesemia/ free cytosolic calcium ion: impaired insulin secretion |
| Pancreatic beta cell toxicity | Prevents dephosphorylation of cytoplasmic NFATc | |
| GLUT4 expressing cells: adipocyte and striated muscle | Insulin independent endocytosis of GLUT4 (glucose transporter) | |
| Sirolimus/ everolimus | Hepatic cell: gluconeogenesis | Chronic use inhibits mTORC-2: increase hepatic gluconeogenic enzymes* |
Acute sirolimus treatment inhibits mTORC-1; increases insulin sensitivity.
GLUT4, glucose transporter type 4; mTORC-2, mechanistic target of rapamycin complex-2; NFATc, nuclear factor of activated T cells; NODAT, new onset diabetes after transplantation.
Steroid-induced new onset diabetes after transplant
Most data on the predisposing factors of NODAT support the etiological role of the cumulative dose of steroids [Cole et al. 2013; Schweer et al. 2014]. Indeed the falling incidence of NODAT in recent times may be related to the lower utilization of steroids in many programs [Cole et al. 2013; Schweer et al. 2014; Hjelmesaeth et al. 1997; Luan et al. 2011]. A study showed there is a 5% risk of developing diabetes mellitus for every 0.01 mg/kg/day increase in prednisolone dose [Hjelmesaeth et al. 1997]. Similarly, there is a 42% greater risk of developing NODAT in patients placed on a steroid-based regimen over a period of 3 years [Luan et al. 2011]. However, there have been few studies of high quality that failed to confirm the diabetogenic effect of steroids [Pascual et al. 2012]. In a RCT, although there was no difference in the frequency of NODAT, a greater proportion of patients treated with steroids required insulin (indicating metabolic severity) for the control of diabetes [Woodle et al. 2008].
Perhaps one reason for the disparity in study outcomes is the variation in the diagnostic criteria for post-transplant diabetes [Langsford and Dwyer, 2015]. Furthermore, the benefit derived from a steroid-sparing regimen may be offset by the harmful metabolic effect of the greater need of CNI to prevent AREs in the control arms [Schweer et al. 2014; Webster et al. 2005b]. As for the pathogenesis, GC produces hyperglycemia by inducing the transcription genes for hepatic gluconeogenic enzymes [Vadlamudi et al. 1993] (Table 1). Hence, the initial functional inhibition of beta cells by steroids is overcome by glucose-induced hyperinsulinemia. However, an excessive amount of insulin is invariably cytotoxic; it inhibits the replication of the pancreatic islet cells. Similarly synergistic cytotoxic injury may be produced by the larger doses of a concurrent treatment with CNI [Choi et al. 2006]. Ultimately, persistent stimulation of a fewer number of beta cells causes pancreatic cell hypertrophy (and established diabetes mellitus).
Calcineurin inhibitor and new onset diabetes after transplant
Compared with CsA, there is a 25% greater incidence of NODAT in patients treated with maintenance TAC [Luan et al. 2011]. There is a dose–effect relationship such that greater exposure of CNI results in higher incidence of NODAT [Chan et al. 2012]. However, compared with lower doses of CsA and SRL, low-dose TAC has the greatest potential for NODAT [Ekberg et al. 2007]. In addition, transplant recipients with prevailing hyperlipidemia, a traditional CV risk factor, are more likely to sustain CNI-induced diabetes mellitus [Porrini et al. 2008]. On the other hand, early discontinuation or reduction in the dose of CNI may reverse the diabetic complication [Veroux et al. 2013]. The mechanism by which CNI induces diabetes mellitus involves multiple concurrent variables (Table 1 and Figure 1). Hypomagnesemia, a common side effect of CNI, impairs pancreatic insulin secretion by increasing calcium concentration within the beta cells [Rodríguez-Morán and Guerrero-Romero, 2011]. Similarly, free cytosolic calcium ions in other cells may promote insulin resistance by the activation of protein kinase C, a constitutive regulator of insulin receptor substrate [Rodríguez-Morán and Guerrero-Romero, 2011].
Figure 1.
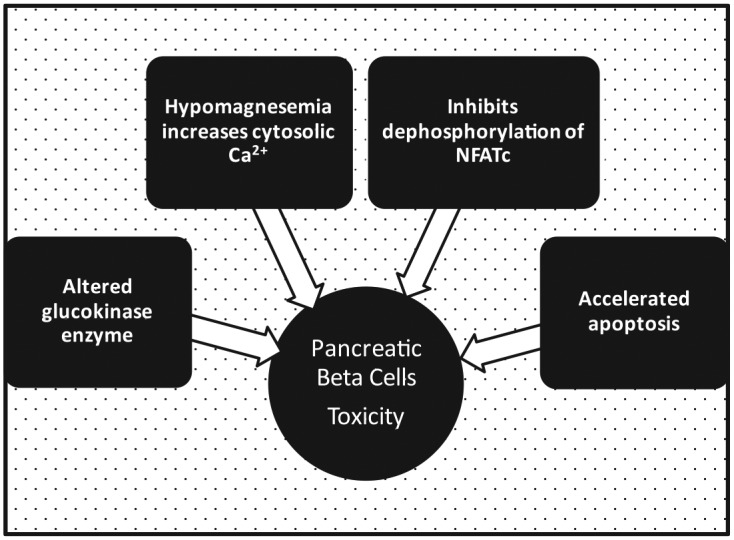
Mechanisms of calcineurin-inhibitor induction of pancreatic beta cell toxicity in new onset diabetes mellitus after transplant.
Ca2+, calcium ions; NFATc, cytoplasmic nuclear factor of activated T cells
Calcineurin-inhibitor (CNI) causes new onset diabetes after transplant (NODAT) by producing pancreatic beta cells toxicity with reduction in insulin secretion and a cellular resistance to the effect of insulin. It causes hypomagnesemia which impairs beta cell insulin secretion by increasing cytosolic calcium ion concentration. CNI prevents dephosphorylation of NFATc, a transcription factor for genes that promotes islet cell proliferation. It also reduces efficient insulin secretion by causing functional alteration of glucokinase enzyme. Finally, a high dose of CNI causes apoptosis of pancreatic beta cells.
Furthermore, CNI prevents dephosphorylation of cytoplasmic nuclear factor of activated T cells (NFATc), a transcription factor for genes that promotes islet cell proliferation [Rostambeigi et al. 2011]. By a similar mechanism it reduces the efficiency of insulin secretion by causing functional alteration of glucokinase enzyme [Rostambeigi et al. 2011; Penfornis and Kury-Paulin, 2006]. Albeit in high doses, in vitro treatment with CsA has also been shown to produce direct pancreatic toxicity by inducing apoptosis of beta cells [Penfornis and Kury-Paulin, 2006]. Apart from the deficient pancreatic secretion of insulin, end-organ resistance is depicted by the inhibition of glucose uptake by human adipocytes exposed to therapeutic doses of both CsA and TAC [Pereira et al. 2014]. This metabolic effect is mediated by the endocytic removal of glucose transporter type 4 (GLUT4) from the cell surfaces, and it is independent of insulin signal transduction.
Sirolimus and new onset diabetes after transplant
Perhaps due to the variation in study quality, data on the role of SRL in the etiology of NODAT showed inconsistent result [Veroux et al. 2013; Johnston et al. 2008; Gyurus et al. 2011; Teutonico et al. 2005]. In an analysis of a US registry, compared with its combination with MMF, addition of SRL to CNI increases the risk of developing NODAT [Johnston et al. 2008]. Older recipients of renal transplants are more susceptible to SRL-induced diabetes mellitus [Gyurus et al. 2011]. However, not all studies affirmed the diabetogenic effect of SRL. In one study, after switching patients from a CNI-based protocol to SRL, there was no difference in the outcome of an oral glucose tolerance test [Teutonico et al. 2005]. Furthermore, compared with CNI, the diabetogenic response to SRL may be less intense. Hence its substitution for CNI has led to the resolution of NODAT in some instances [Veroux et al. 2013].
Demonstrating a biphasic biologic effect, acute SRL treatment enhances insulin sensitivity by inhibition of the mTOR complex 1 (mTORC-1) [Table 1, Figure 2]. Paradoxically, its chronic use produces insulin resistance by the disruption of mTORC-2. Activation of mTORC-1 by cellular exposure to excessive nutrients promotes insulin resistance by enhancing (serine) phosphorylation of IRS-1. The latter in turn decreases the activation of phosphatidyl inositol 3-kinase, and thereby creates a negative feedback loop for insulin action [Houde et al. 2010]. By blocking mTORC-1, SRL produces a starvation-like signal despite nutrient abundance and was therefore considered a potential therapeutic agent for attenuating insulin resistance. However, a paradoxical effect of glucose intolerance and hyperlipidemia was observed with its use in transplant recipients [Houde et al. 2010]. This is because chronic treatment with SRL produces hyperglycemia by overriding the inhibitory effect of insulin on the transcription of hepatic gluconeogenic enzymes.
Figure 2.

Biphasic effect of sirolimus: acute treatment enhances insulin sensitivity while chronic dosing causes insulin resistance.
Acute sirolimus treatment enhances insulin sensitivity by inhibition of mechanistic target of rapamycin complex 1 (mTORC1). It decreases serine phosphorylation of insulin receptor substrate-1, which in turn activates phosphatidyl inositol 3-kinase, and thereby sensitizes the cell to insulin action. Paradoxically, chronic administration of sirolimus produces hyperglycemia by overriding the inhibitory effect of insulin on the transcription of hepatic gluconeogenic enzymes.
Immunosuppressive agents calcineurin inhibitors and dyslipidemia
Partly due to intense immunosuppression, dyslipidemia occurs within the first year in more than 80% of adult renal transplant recipients [Spinelli et al. 2011]. Persistent lipid disorders are more likely to occur in those patients with pretransplant hyperlipidemia [Razeghi et al. 2011]. The most significant risk factor is the cumulative dose of steroids particularly when used in combination with CsA or SRL [Claes et al. 2012].
Steroids and dyslipidemia
Chronic steroid use increases free fatty acid synthetase and upregulates hepatic synthesis of very low-density lipoprotein (VLDL) (Table 2). It also reduces the synthesis of low-density lipoprotein (LDL) receptor and inhibits the activity of lipoprotein lipase [Razeghi et al. 2011]. The sum effect is an increase in serum total cholesterol, high serum triglyceride, elevated level of VLDL but a reduction in plasma high-density lipoprotein (HDL) cholesterol [Ferraris et al. 2007].
Table 2.
Pathogenesis of dyslipidemia due to immunosuppressive agents.
| Immunosuppressive agents | Pathogenesis of dyslipidemia | Pattern of dyslipidemia |
|---|---|---|
| Steroids | Increases FFA synthetase |
|
| Reduces LDL receptor | ||
| Reduces lipoprotein lipase activity | ||
| Calcineurin inhibitors: TAC/ CsA | CsA inhibits mitochondrial 27-hydroxylase and increases HMGR activity |
|
| CsA binds LDL receptor | ||
| Increases hepatic lipase and reduces lipoprotein lipase | ||
| Sirolimus/ everolimus | Reduces hepatic lipase but increases lipase enzyme in adipocytesDownregulates peroxisome proliferator-activated receptor-γ 2 (PPAR-γ 2) |
|
CsA, cyclosporine; FFA, free fatty acid; HDL, high-density lipoprotein; HMGR, 3 hydroxy - 3-methyl-glutaryl-CoA reductase; LDL, low-density lipoprotein; TAC, tacrolimus; TC, total cholesterol; TG, triglyceride; VLDL, very low-density lipoprotein.
and dyslipidemia
CsA causes dose-dependent inhibition of mitochondrial 27-hydroxylase (CYP27A1) and therefore increases the expression of 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGR), a key regulatory enzyme in cholesterol synthesis [Gueguen et al. 2007] (Table 2). CsA impairs the clearance of VLDL and LDL cholesterol by binding to the LDL receptor. In addition, it increases the activity of hepatic lipase, which in turn converts intermediate-density lipoprotein to LDL. It also downregulates lipoprotein lipase enzyme activity [Derfler et al. 1991; Sugioka et al. 2006]. Furthermore, CsA is transported in the circulation by LDL cholesterol particles, which facilitates its intracellular uptake by LDL receptors. CsA effect on lipid metabolism is facilitated by the concurrent use of high-dose steroids. Elevated cytoplasmic concentration may promote endothelial inflammation by increasing the susceptibility of LDL cholesterol to oxidative milieu [Gueguen et al. 2007]. On the other hand, compared with CsA, TAC produces less lipid disturbances. Indeed a switch to TAC from a CsA-based regimen is associated with resolution of hyperlipidemia [Bakar et al. 2009].
Depicting its role in the regulation of lipid homeostasis, mice lacking calcineurin Aβ (CnAβ) developed hyperlipidemia [Suk et al. 2013]. Calcineurin is associated with greater degree of lipolysis in adipose tissues, a process mediated by the β-adrenergic G-protein-coupled receptor signaling pathway and sustained by intracellular activation of cyclic AMP and protein kinase A [Suk et al. 2013]. Knock-out mice (CnAβ) developed hyperlipidemia by enhancing the signal transduction and by upregulation of the metabolic pathway. CsA toxicity may be aggravated by a higher serum level of saturated fatty acid, which may be seen in patients with steroid-induced obesity and drug-induced hyperlipidemia [Luo et al. 2012]. This was demonstrated in an experimental study in which cotreatment of hepatic cells with palmitic acid and CsA caused dose-dependent cytotoxicity and apoptosis. The cellular injury was associated with mitochondrial dysfunction and may be mediated by activation of c-Jun N-terminal kinase (JNK).
Sirolimus and dyslipidemia
Sirolimus increases lipase enzyme activity in adipose tissue but causes a reduction in the synthesis of hepatic lipoprotein lipase (Table 2). At 2 weeks after addition of SRL to the regimen of CsA and prednisone in transplant recipients, there is a dose-dependent increase in total plasma cholesterol, plasma LDL, serum triglyceride, and ApoB-100 [Morrisett et al. 2003]. Normal lipid levels are restored by about 4 weeks after discontinuation of the drug [Spinelli et al. 2011; Morrisett et al. 2003]. In animal models, SRL impairs circulating triglyceride hydrolysis (lipoprotein lipase), cellular fatty acid uptake, and lipid synthesis (lipin 1) by downregulation of peroxisome proliferator-activated receptor-γ 2 (PPAR-γ 2) [Houde et al. 2010] (Table 2). It blocks the modulatory effects of mTORC-1 on adipogenesis through the AKT-mediated phosphorylation of tuberous sclerosis complex 2. Consequently, the reduction in the capacity of adipose tissue clearance of fat results in hyperlipidemia [Houde et al. 2010]. Paradoxically, prolonged SRL treatment in experimental animals causes a reduction in food intake and weight loss while there is an increase in energy expenditure.
Immunosuppressive agents and bone metabolism
Steroids and linear growth
The initial wave of clinical trials on steroid minimization was conducted in pediatric populations out of concern for its negative impact on longitudinal skeletal growth in children. A 2-year RCT of late steroid withdrawal (>3 months) using CsA and MMF as a maintenance therapy showed a positive catch-up growth and a favorable metabolic impact [Höcker et al. 2010]. In a 2-year follow up of a similar study, the skeletal growth advantage was sustained, and it was observed as more pronounced in prepubertal children [Webb, 2015] However, compared with the control, there was no difference in AREs, nor in patient or allograft survival.
Steroids and bone disease
Despite normal serum calcium, phosphorous and intact parathyroid hormone (PTH) in transplant recipients, osteitis fibrosa, osteomalacia, and adynamic bone disease are commonly observed on evaluation by bone biopsy [Evenepoel, 2013]. Observational study showed there was a positive correlation between cumulative steroid doses, accelerated cancellous bone remodeling, and loss of bone mineral density [Pichett et al. 1996; Julian et al. 1991]. Steroids produce a negative calcium balance by reducing intestinal absorption while increasing its urinary excretion [Canalis et al. 2007]. This metabolic aberration may impair the restoration of pretransplant hyperparathyroidism. In addition, onset of chronic allograft dysfunction may lead to a persistent metabolic acidosis, 1, 25 vitamin D deficiency, and a more rapid progression of bone disease [Evenepoel, 2013]. Perhaps for these reasons, vertebral fracture occurs at a higher rate than in the general population as observed in a cross sectional study [Jiménez et al. 2015]. Its higher frequency of occurrence also correlates with greater cumulative dose of steroids and a longer pretransplant duration of dialysis [Patel et al. 2001]. However, there is a failure to confirm the predictive relationship of bone fracture with steroid use in studies with long-term follow up [Malluche et al. 2010; O’Shaughnessy et al. 2002].
Steroid decreases bone-forming osteoblastic cells, increases osteocyte apoptosis, and attenuates both insulin-like growth factor-1 (IGF-1) and transforming growth factor-β (TGF-β). It causes bone resorption by stimulating osteoprotegerin ligand (OPG-L) and promotes inactivation of osteoprotegerin (OPG), a soluble neutralizing receptor [Hofbauer et al. 1999]. Lower doses of steroids, its use as alternate-day regimen, and correction of hypogonadism may ameliorate the severity of bone disease [Weisinger et al. 2006]. In addition, patients on a GC-withdrawal protocol had a net gain in bone mineral density; a benefit that was more pronounced in patients with severe kidney disease [Ing et al. 2011]. There are limited data on the safety and effectiveness of bisphosphonates in transplant settings. In a prospective study of liver transplant recipients, trabecular bone mineralization was preserved while there was no beneficial effect on the cortical bone mass [Pennisi et al. 2007].
Calcineurin inhibitors, sirolimus and bone metabolism
Due to concurrent use of CNI with steroids, studies on their solitary effect on bone metabolism are limited [Malluche et al. 2010; Marcén et al. 2006; Briner et al. 1995]. Confounding effects of steroid use may account for the conflicting outcome of the clinical studies. Both TAC and CsA stimulate osteoclastic activity in excess of deficit in osteoblastogenesis, and therefore enhance cancellous bone loss [Kirino et al. 2004]. Elevation in serum PTH, increased serum alkaline phosphatase, a rise in serum osteocalcin, and hyperparathyroid bone changes in the face of normal value of 1, 25-dihydroxyl vitamin D level suggest there is a skeletal end-organ resistance [Briner et al. 1995; Kirino et al. 2004]. Despite the proof of higher bone turnover rate, there is no evidence for a greater risk of skeletal fracture with the use of these agents [Patel et al. 2001]. On the other hand, in vitro studies showed mTOR inhibitor (SRL and EVR) enhances osteoblast differentiation by inactivation of notch pathway and upregulation of Runx2 [Huang et al. 2015]. It also decreases the formation of osteocytes with a resultant preservation of cancellous bone mass [Kneissel et al. 2004].
Summary
We have achieved a remarkable stride in the provision of targeted immunosuppression particularly against alloreactive T cells. The limitation of modern ISAs in curtailing chronic antibody-mediated allograft injury has necessitated the continuous beneficial role of steroids in the past few decades. Unfortunately, high-impact therapeutic agents such as steroids, CNI and SRL are associated with multiple adverse metabolic effects (e.g. hypertension, NODAT, and dyslipidemia), and a potential for long-term CV mortality. Improved awareness of the pathophysiologic bases for such metabolic complications will promote appropriate selection of ISAs that offers the most benefit in an individual patient while minimizing the potential loss of allograft function.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Bailey M., Mullins J., Kenyon C. (2009) Mineralocorticoid and glucocorticoid receptors stimulate epithelial sodium channel activity in a mouse model of Cushing syndrome. Hypertension 54: 890–896. [DOI] [PubMed] [Google Scholar]
- Bakar F., Keven K., Dogru B., Aktan F., Erturk S., Tuzuner A., et al. (2009) Low-density lipoprotein oxidizability and the alteration of its fatty acid content in renal transplant recipients treated with cyclosporine/tacrolimus. Transplant Proc 41:1630–1633. [DOI] [PubMed] [Google Scholar]
- Baum M., Moe O. (2008) Glucocorticoid-mediated hypertension: does the vascular smooth muscle hold all the answers? J Am Soc Nephrol 19: 1251–1253. [DOI] [PubMed] [Google Scholar]
- Benfield M., Bartosh S., Ikle D., Warshaw B., Bridges N., Morrison Y., et al. (2010) Randomized double-blind, placebo controlled trial of steroid withdrawal after pediatric renal transplantation. Am J Transplant 10: 81–88. [DOI] [PubMed] [Google Scholar]
- Bergrem H., Valderhaug T., Hartmann A., Bergrem H., Hjelmesaeth J., Jenssen T. (2010) Glucose tolerance before and after renal transplantation. Nephrol Dial Transplant 25: 985–992. [DOI] [PubMed] [Google Scholar]
- Bloom R., Reese P. (2007) Chronic kidney disease after nonrenal solid-organ transplantation. J Am Soc Nephrol 18: 3031–3041. [DOI] [PubMed] [Google Scholar]
- Boschetti S., Nogueira P., Pereira A., Fisberg M., Pestana J. (2013) Prevalence, risk factors, and consequences of overweight in children and adolescents who underwent renal transplantation – Short- and medium-term analysis. Pediatr Transplant 17: 41–47. [DOI] [PubMed] [Google Scholar]
- Briner V., Thiel G., Monier-Faugere M., Bognar B., Landmann J., Kamber V., et al. (1995) Prevention of cancellous bone loss but persistence of renal bone disease despite normal 1,25 vitamin D levels two years after kidney transplantation. Transplantation 59: 1393–1400. [DOI] [PubMed] [Google Scholar]
- Budde K., Becker T., Arns W., Sommerer C., Reinke P., Eisenberger U., et al. (2011) Everolimus- based, calcineurin-inhibitor-free regimen in recipients of de-novo kidney transplants: an open-label, randomised, controlled trial. Lancet 377: 837–847. [DOI] [PubMed] [Google Scholar]
- Burkhalter F., Oettl T., Descoeudres B., Bachmann A., Guerke L., Mihatsch M., et al. (2012) High incidence of rejection episodes and poor tolerance of sirolimus in a protocol with early steroid withdrawal and calcineurin inhibitor-free maintenance therapy in renal transplantation: experiences of a randomized prospective single-center study. Transplant Proc 44: 2961–2965. [DOI] [PubMed] [Google Scholar]
- Calò L., Davis P., Giacon B., Pagnin E., Sartori M., Riegler P., et al. (2002) Oxidative stress in kidney transplant patients with calcineurin inhibitor-induced hypertension: effect of ramipril. J Cardiovasc Pharmacol 40: 625–631. [DOI] [PubMed] [Google Scholar]
- Canalis E., Mazziotti G., Giustina A., Bilezikian J. (2007) Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int 18: 1319–1328. [DOI] [PubMed] [Google Scholar]
- Casey M., Meier-Kriesche H. (2011) Calcineurin inhibitors in kidney transplantation: friend or foe? Curr Opin Nephrol Hypertens 20: 610–615. [DOI] [PubMed] [Google Scholar]
- Chan L., Andres A., Bunnapradist S., Gugliuzza K., Parasuraman R., Peddi V., et al. (2012) Renal function and NODM in de novo renal transplant recipients treated with standard and reduced levels of tacrolimus in combination with ECMPS. J Transplant 2012: 941640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra D., Skaro A., Leventhal J., Dalal P., Shah G., Wang E., et al. (2012) Long-term kidney allograft function and survival in prednisone-free regimens: tacrolimus/mycophenolate mofetil versus tacrolimus/sirolimus. Clin J Am Soc Nephrol 7: 504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S., Jin Sun Jang J., Hong S., Jun D., Park S. (2006) Exercise and dexamethasone oppositely modulate B-cell function and survival via independent pathways in 90% pancreatectomized rats. J Endocrinol 190: 471–482. [DOI] [PubMed] [Google Scholar]
- Chen G., Gu J., Qiu J., Wang C., Fei J., Deng S., et al. (2013) Efficacy and safety of thymoglobulin and basiliximab in kidney transplant patients at high risk for acute rejection and delayed graft function. Exp Clin Transplant 11: 310–314. [DOI] [PubMed] [Google Scholar]
- Claes K., Meier-Kriesche H., Schold J., Vanrenterghem Y., Halloran P., Ekberg H. (2012) Effect of different immunosuppressive regimens on the evolution of distinct metabolic parameters: evidence from the Symphony study. Nephrol Dial Transplant 27: 850–857. [DOI] [PubMed] [Google Scholar]
- Cole E., Prasad G., Cardella C., Kim J., Tinckam K., Cattran D., et al. (2013) A pilot study of reduced dose cyclosporine and corticosteroids to reduce new onset diabetes mellitus and acute rejection in kidney transplant recipients. Transplant Res 2: 1 PMID: 23369458. DOI: 10.1186/2047144021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosio F., Kudva Y., van der Velde M., Larson T., Textor S., Griffin M., et al. (2005) New onset hyperglycemia and diabetes are associated with increased cardiovascular risk after kidney transplantation. Kidney Int 67: 2415–2421. [DOI] [PubMed] [Google Scholar]
- Denburg M., Pradhan M., Shults J., Jones A., Palmer J., Baluarte H., et al. (2010) Longitudinal relations between obesity and hypertension following pediatric renal transplantation. Pediatr Nephrol 25: 2129–2139. [DOI] [PubMed] [Google Scholar]
- Derfler K., Hayde M., Heinz G., Hirschl M., Steger G., Hauser A., et al. (1991) Decreased postheparin lipolytic activity in renal transplant recipients with cyclosporin A. Kidney Int 40: 720–727. [DOI] [PubMed] [Google Scholar]
- Dunea G., Nakamoto S., Straffon R., Figueroa J., Versaci A., Shibagaki M., et al. (1965) Renal homotransplantation in 24 Patients. Brit Med J 1: 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einollahi B., Motalebi M., Salesi M., Ebrahimi M., Taghipour M. (2014) The impact of cytomegalovirus infection on new-onset diabetes mellitus after kidney transplantation: a review on current findings. J Nephropathol 3: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekberg H., Tedesco-Silva H., Demirbas A., Vítko S., Nashan B., Gürkan A., et al. (2007) ELITE-Symphony Study. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med 357: 2562–2575. [DOI] [PubMed] [Google Scholar]
- Evenepoel P. (2013) Recovery versus persistence of disordered mineral metabolism in kidney transplant recipients. Semin Nephrol 33: 191–203. [DOI] [PubMed] [Google Scholar]
- Ferraris J., Argibay P., Costa L., Jimenez G., Coccia P., Ghezzi L., et al. (2011) Influence of CYP3A5 polymorphism on tacrolimus maintenance doses and serum levels after renal transplantation: age dependency and pharmacological interaction with steroids. Pediatr Transplant 15: 525–532. [DOI] [PubMed] [Google Scholar]
- Ferraris J., Pasqualini T., Alonso G., Legal S., Sorroche P., Galich A., et al. (2007) Deflazacort Study Group. Effects of deflazacort vs. methylprednisone: a randomized study in kidney transplant patients. Pediatr Nephrol 22: 734–741. [DOI] [PubMed] [Google Scholar]
- Foster B., Martz K., Gowrishankar M., Stablein D., Al-Uzri A. (2010) Weight and height changes and factors associated with greater weight and height gains after pediatric renal transplantation: A NAPRTCS study. Transplantation 89: 1103–1112. [DOI] [PubMed] [Google Scholar]
- Gallagher M., Hall B., Craig J., Berry G., Tiller D., Eris J. (2004) A randomized controlled trial of cyclosporine withdrawal in renal-transplant recipients: 15-year results. Transplantation 78: 1653–1660. [DOI] [PubMed] [Google Scholar]
- Ghanta M., Kozicky M., Jim B. (2015) Pathophysiologic and treatment strategies for cardiovascular disease in end-stage renal disease and kidney transplantations. Cardiol Rev 23: 109–118. [DOI] [PubMed] [Google Scholar]
- Gnatta D., Keitel E., Heineck I., Cardoso B., Rodrigues A., Michel K., et al. (2010) Use of tacrolimus and the development of posttransplant diabetes mellitus: a Brazilian single-center, observational study. Transplant Proc 42: 475–478. [DOI] [PubMed] [Google Scholar]
- Gourishankar S., Grebe S., Mueller T. (2013) Prediction of kidney graft failure using clinical scoring tools. Clin Transplant 27: 517–522. [DOI] [PubMed] [Google Scholar]
- Gueguen Y., Ferrari L., Souidi M., Batt A., Lutton C., Siest G., et al. (2007) Compared effect of immunosuppressive drugs cyclosporine A and rapamycin on cholesterol homeostasis key enzymes CYP27A1 and HMG-CoA reductase. Basic Clin Pharmacol Toxicol 100: 392–397. [DOI] [PubMed] [Google Scholar]
- Guethoff S., Stroeh K., Grinninger C., Koenig M., Kleinert E., Rieger A., et al. (2015) De novo sirolimus with low-dose tacrolimus versus full-dose tacrolimus with mycophenolate mofetil after heart transplantation – 8-year results. J Heart Lung Transplant 34: 634–642. [DOI] [PubMed] [Google Scholar]
- Gyurus E., Kaposztas Z., Kahan B. (2011) Sirolimus therapy predisposes to newonset diabetes mellitus after renal transplantation: a long term analysis of various treatment regimens. Transplant Proc 43: 15831592. [DOI] [PubMed] [Google Scholar]
- Hajer G., van Haeften T., Visseren F. (2008) Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur Heart J 29: 2959–2971. [DOI] [PubMed] [Google Scholar]
- Hanevold C., Ho P., Talley L., Mitsnefes M. (2005) Obesity and renal transplant outcome: a report of the North American Pediatric Renal Transplant Cooperative Study. Pediatrics 115: 352–356. [DOI] [PubMed] [Google Scholar]
- Hirsch H., Yakhontova K., Lu M., Manzetti J. (2015) BK polyomavirus replication in renal tubular epithelial cells is inhibited by sirolimus, but activated by tacrolimus through a pathway involving FKBP-12. Am J Transplant doi: 10.1111/ajt.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmesaeth J., Hartmann A., Kofstad J., Stenstrøm J., Leivestad T., Egeland T., et al. (1997) Glucose intolerance after renal transplantation depends upon prednisolone dose and recipient age. Transplantation 64: 979–983. [DOI] [PubMed] [Google Scholar]
- Höcker B., Weber L., Feneberg R., Drube J., John U., Fehrenbach H., et al. (2010) Improved growth and cardiovascular risk after late steroid withdrawal: 2-year results of a prospective, randomised trial in paediatric renal transplantation. Nephrol Dial Transplant 25: 617–624. [DOI] [PubMed] [Google Scholar]
- Hofbauer L., Gori F., Riggs B., Lacey D., Dunstan C., Spelsberg T., et al. (1999) Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology 140: 4382–4389. [DOI] [PubMed] [Google Scholar]
- Holdaas H., Rostaing L., Serón D., Cole E., Chapman J., Fellstrøm B., et al. (2011) Conversion of long-term kidney transplant recipients from calcineurin inhibitor therapy to everolimus: a randomized, multicenter, 24-month study. Transplantation 92: 410–418. [DOI] [PubMed] [Google Scholar]
- Hoogeveen E., Aalten J., Rothman K., Roodnat J., Mallat M., Borm G., et al. (2011) Effect of obesity on the outcome of kidney transplantation: a 20-year follow-up. Transplantation 91: 869–874. [DOI] [PubMed] [Google Scholar]
- Houde V., Brûlé S., Festuccia W., Blanchard P., Bellmann K., Deshaies Y., et al. (2010) Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 59: 1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B., Wang Y., Wang W., Chen J., Lai P., Liu Z., et al. (2015) mTORC1 prevents preosteoblast differentiation through the notch signaling pathway. PLoS Genet 11: e1005426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humar A., Gillingham K., Kandaswamy R, Payne W., Matas A. (2007) Steroid avoidance regimens: a comparison of outcomes with maintenance steroids versus continued steroid avoidance in recipients having an acute rejection episode. Am J Transplant 7: 1948–1953. [DOI] [PubMed] [Google Scholar]
- Ing S., Sinnott L., Donepudi S., Davies E., Pelletier R., Lane N. (2011) Change in bone mineral density at one year following glucocorticoid withdrawal in kidney transplant recipients. Clin Transplant 25: E113–E123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israni A., Snyder J., Skeans M., Kasiske B. (2012) Clinical diagnosis of metabolic syndrome: predicting new-onset diabetes, coronary heart disease, and allograft failure late after kidney transplantation, Transplant Int 25: 748–757. [DOI] [PubMed] [Google Scholar]
- Jiménez S., Marcén R., Vaamonde C., Caballero C., Fernández-Rodríguez A., Villafruela J., et al. (2015) Bone fractures and lumbar mineral density after renal transplantation. A long-term cross-sectional study. Clin Transplant. DOI: 10.1111/ctr.12666. [DOI] [PubMed] [Google Scholar]
- Johnston O., Rose C., Webster A., Gill J. (2008) Sirolimus is associated with newonset diabetes in kidney transplant recipients. J Am Soc Nephrol 19: 14111418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan Khong M., Ping Chong C. (2014) Prevention and management of new-onset diabetes mellitus in kidney transplantation. Neth J Med 72: 127–134. [PubMed] [Google Scholar]
- Julian B., Laskow D., Dubovsky J., Dubovsky E., Curtis J., Quarles L. (1991) Rapid loss of vertebral mineral density after renal transplantation. N Engl J Med 325: 544–550. [DOI] [PubMed] [Google Scholar]
- KDIGO (Kidney Disease: Improving Global Outcomes) Transplant Work Group (2009) KDIGO clinical practice guidelines for the care of kidney transplant recipients. Am J Transplant 9: S1–S157. [Google Scholar]
- Kirino S., Fukunaga J., Ikegami S., Tsuboi H., Kimata M., Nakata N., et al. (2004) Regulation of bone metabolism in immunosuppressant (FK506)-treated rats. J Bone Miner Metab 22: 554–560. [DOI] [PubMed] [Google Scholar]
- Kislikova M., Seras M., Monfa E., Rodrigo E., Fernandez-Fresnedo G., Ruiz J., et al. (2015) Number of antihypertensive drugs at 1 year after kidney transplantation. Transplant Proc 47: 76–77. [DOI] [PubMed] [Google Scholar]
- Kneissel M., Luong-Nguyen N., Baptist M., Cortesi R., Zumstein-Mecker S., Kossida S., et al. (2004) Everolimus suppresses cancellous bone loss, bone resorption, and cathepsin K expression by osteoclasts. Bone 35: 1144–1156. [DOI] [PubMed] [Google Scholar]
- Knight S., Morris P. (2010) Steroid avoidance or withdrawal after renal transplantation increases the risk of acute rejection but decreases cardiovascular risk. A meta-analysis. Transplantation 89: 1–14. [DOI] [PubMed] [Google Scholar]
- Knight S., Russell N., Barcena L., Morris P. (2009) Mycophenolate mofetil decreases acute rejection and may improve graft survival in renal transplant recipients when compared with azathioprine: a systematic review. Transplantation 87: 785–794. [DOI] [PubMed] [Google Scholar]
- Krämer B., Klinger M., Vítko Š., Glyda M., Midtvedt K., Stefoni S., et al. (2012) Tacrolimus-based, steroid-free regimens in renal transplantation: 3-year follow-up of the ATLAS trial. Transplantation 94: 492–498. [DOI] [PubMed] [Google Scholar]
- Lamb K., Lodhi S., Meier-Kriesche H. (2011) Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transpl 11: 450–462. [DOI] [PubMed] [Google Scholar]
- Lambert G., Straznicky N., Lambert E., Dixon J., Schlaich M. (2010) Sympathetic nervous activation in obesity and the metabolic syndrome–causes, consequences and therapeutic implications. Pharmacol Ther 126: 159–172. [DOI] [PubMed] [Google Scholar]
- Langsford D., Dwyer K. (2015) Dysglycemia after renal transplantation: Definition, pathogenesis, outcomes and implications for management. World J Diabetes 6: 1132–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Meur Y., Thierry A., Glowacki F., Rerolle J., Garrigue V., Ouali N., et al. (2011) Early steroid withdrawal and optimization of mycophenolic acid exposure in kidney transplant recipients receiving mycophenolate mofetil. Transplantation 92: 1244–1251. [DOI] [PubMed] [Google Scholar]
- Lebranchu Y., Thierry A., Toupance O., Westeel P., Etienne I., Thervet E., et al. (2009) Efficacy on renal function of early conversion from cyclosporine to sirolimus 3 months after renal transplantation: concept study. Am J Transplant 9: 1115–1123. [DOI] [PubMed] [Google Scholar]
- Lloberas N., Torras J., Alperovich G., Cruzado J., Giménez-Bonafé P., Herrero-Fresneda I., et al. (2008) Different renal toxicity profiles in the association of cyclosporine and tacrolimus with sirolimus in rats. Nephrol Dial Transplant 23: 3111–3119. [DOI] [PubMed] [Google Scholar]
- Luan F., Steffick D., Akinlolu O. (2011) New-onset diabetes mellitus in kidney transplant recipients discharged on steroid-free immunosuppression. Transplantation 91: 334–341. [DOI] [PubMed] [Google Scholar]
- Luo Y., Rana P., Will Y. (2012) Cyclosporine A and palmitic acid treatment synergistically induce cytotoxicity in HepG2 cells. Toxicol Appl Pharmacol 261: 172–180. [DOI] [PubMed] [Google Scholar]
- Lyster H., Panicker G., Leaver N., Banner N. (2004) Initial experience with sirolimus and mycophenolate mofetil for renal rescue from cyclosporine nephrotoxicity after heart transplantation. Transplant Proc 36: 3167–3170. [DOI] [PubMed] [Google Scholar]
- Malluche H., Monier-Faugere M., Herberth J. (2010) Bone disease after renal transplantation. Nat Rev Nephrol 6: 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcén R. (2009) Immunosuppressive drugs in kidney transplantation: impact on patient survival, and incidence of cardiovascular disease, malignancy and infection. Drugs 69: 2227–2243. [DOI] [PubMed] [Google Scholar]
- Marcén R., Caballero C., Pascual J., Teruel J., Tenorio M., Ocaña J., et al. (2006) Lumbar bone mineral density in renal transplant patients on neoral and tacrolimus: a four-year prospective study. Transplantation 81: 826–831. [DOI] [PubMed] [Google Scholar]
- Masson P., Henderson L., Chapman J., Craig J., Webster A. (2014) Belatacept for kidney transplant recipients. Cochrane Database Syst Rev 11: CD010699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matas A. (2008) Steroid elimination-who, when, how? Transplant Proc 40: S52–S56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales J., Marcén R., del Castillo D., Andres A., Gonzalez-Molina M., Oppenheimer F., et al. (2012) Risk factors for graft loss and mortality after renal transplantation according to recipient age: a prospective multicentre study. Nephrol Dial Transplant 27: iv39–iv46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisett J., Abdel-Fattah G., Kahan B. (2003) Sirolimus changes lipid concentrations and lipoprotein metabolism in kidney transplant recipients. Transplant Proc 35: 143S–150S. [DOI] [PubMed] [Google Scholar]
- Nankivell B., Borrows R., Fung C., O’Connell P., Chapman J., Allen R. (2004) Calcineurin inhibitor nephrotoxicity: longitudinal assessment by protocol histology. Transplantation 78: 557–565. [DOI] [PubMed] [Google Scholar]
- Nicoletto B., Fonseca N., Manfro R., Gonçalves L., Leitão C., Souza G. (2014) Effects of obesity on kidney transplantation outcomes: a systematic review and meta-analysis. Transplantation 98: 167–176. [DOI] [PubMed] [Google Scholar]
- Ordonez L., Bernard I., Chabod M., Augusto J., Lauwers-Cances V., Cristini C., et al. (2013) A higher risk of acute rejection of human kidney allografts can be predicted from the level of CD45RC expressed by the recipients’ CD8 T cells. PLoS One 8: e69791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shaughnessy E., Dahl D., Smith C., Kasiske B. (2002) Risk factors for fractures in kidney transplantation. Transplantation 74: 362–366. [DOI] [PubMed] [Google Scholar]
- Padiyar A., Augustine J., Bodziak K., Aeder M., Schulak J., Hricik D. (2010) Influence of African-American ethnicity on acute rejection after early steroid withdrawal in primary kidney transplant recipients. Transplant Proc 42: 1643–1647. [DOI] [PubMed] [Google Scholar]
- Pape L., Ahlenstiel T. (2014) mTOR inhibitors in pediatric kidney transplantation. Pediatr Nephrol 29: 1119–1129. [DOI] [PubMed] [Google Scholar]
- Pascual J., Royuela A., Galeano C., Crespo M., Zamora J. (2012) Very early steroid withdrawal or complete avoidance for kidney transplant recipients: a systematic review. Nephrol Dial Transplant 27: 825832. [DOI] [PubMed] [Google Scholar]
- Pascual J., Zamora J., Galeano C., Royuela A., Querela C. (2009) Steroid avoidance or withdrawals for kidney transplant recipients. Cochrane Database Syst Rev 4: CD007669. [DOI] [PubMed] [Google Scholar]
- Patel S., Kwan J., McCloskey E., McGee G., Thomas G., Johnson D., et al. (2001) Prevalence and causes of low bone density and fractures in kidney transplant patients. J Bone Miner Res 16: 1863–1870. [DOI] [PubMed] [Google Scholar]
- Paz M., Roberti J., Mos F., Cicora F. (2014) Conversion to belatacept-based immunosuppression therapy in renal transplant patients. Transplant Proc 46: 2987–2990. [DOI] [PubMed] [Google Scholar]
- Pazik J., Ołdak M., Lewandowski Z., Podgórska M., Sitarek E., Płoski R., et al. (2013) Uridine diphosphate glucuronosyl transferase 2B7 variant p.His268Tyr as a predictor of kidney allograft early acute rejection. Transplant Proc 45: 1516–1519. [DOI] [PubMed] [Google Scholar]
- Penfornis A., Kury-Paulin S. (2006) Immunosuppressive drug-induced diabetes. Diabetes Metab 32: 539–546. [DOI] [PubMed] [Google Scholar]
- Pennisi P., Trombetti A., Giostra E., Mentha G., Rizzoli R., Fiore C. (2007) Pamidronate and osteoporosis prevention in liver transplant recipients. Rheumatol Int 27: 251–256. [DOI] [PubMed] [Google Scholar]
- Pereira M., Palming J., Rizell M., Aureliano M., Carvalho E., Svensson M., Eriksson J. (2014) Cyclosporine A and tacrolimus reduce the amount of GLUT4 at the cell surface in human adipocytes: increased endocytosis as a potential mechanism for the diabetogenic effects of immunosuppressive agents. J Clin Endocrinol Metab 99: E1885–E1894. [DOI] [PubMed] [Google Scholar]
- Perito E., Lustig R., Rosenthal P. (2016) Metabolic syndrome components after pediatric liver transplantation: prevalence and the impact of obesity and immunosuppression. Am J Transplant. DOI: 10.1111/ajt.13714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichette V., Bonnardeaux A., Prudhomme L., Gagné M., Cardinal J., Ouimet D. (1996) Long-term bone loss in kidney transplant recipients: a cross-sectional and longitudinal study. Am J Kidney Dis 28: 105–114. [DOI] [PubMed] [Google Scholar]
- Porrini E., Delgado P., Alvarez A., Cobo M., Pérez L., González-Posada J., et al. (2008) The combined effect of pre-transplant triglyceride levels and the type of calcineurin inhibitor in predicting the risk of new onset diabetes after renal transplantation. Nephrol Dial Transplant 23: 1436–1441. [DOI] [PubMed] [Google Scholar]
- Pourmand G., Dehghani S., Rahmati M., Mehrsai A., Gooran S., Alizadeh F., et al. (2015) Does hypertension remain after kidney transplantation? Acta Med Iran 53: 297–300. [PubMed] [Google Scholar]
- Razeghi E., Shafipour M., Ashraf H., Pourmand G. (2011) Lipid disturbances before and after renal transplant. Exp Clin Transplant 9: 230–235. [PubMed] [Google Scholar]
- Rodríguez-Morán M., Guerrero-Romero F. (2011) Insulin secretion is decreased in non-diabetic individuals with hypomagnesaemia. Diabetes Metab Res Rev 27: 590–596. [DOI] [PubMed] [Google Scholar]
- Rossi A., Vella J. (2015) Hypertension, living kidney donors, and transplantation: where are we today? Adv Chronic Kidney Dis 22: 154–164. [DOI] [PubMed] [Google Scholar]
- Rostaing L., Massari P., Garcia V., Mancilla-Urrea E., Nainan G., del Carmen Rial M., et al. (2011) Switching from calcineurin inhibitor-based regimens to a belatacept-based regimen in renal transplant recipients: a randomized phase II study. Clin J Am Soc Nephrol 6: 430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostaing L., Vincenti F., Grinyó J., Rice K., Bresnahan B., Steinberg S., et al. (2013) Long-term belatacept exposure maintains efficacy and safety at 5 years: results from the long-term extension of the BENEFIT study. Am J Transplant 13: 2875–2883. [DOI] [PubMed] [Google Scholar]
- Rostambeigi N., Lanza I., Dzeja P., Deeds M., Irving B., Reddi H., et al. (2011) Unique cellular and mitochondrial defects mediate FK506-induced islet β-cell dysfunction. Transplantation 91: 615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush D. (2013) The impact of calcineurin inhibitors on graft survival. Transplant Rev (Orlando) 27: 93–99. [DOI] [PubMed] [Google Scholar]
- Saidi R., Chang J., Verb S., Brooks S., Nalbantoglu I., Adsay V., et al. (2007) The effect of methyl prednisolone on warm ischemia-reperfusion injury in the liver. Am J Surg 193: 345–347. [DOI] [PubMed] [Google Scholar]
- Sander M., Lyson T., Thomas G., Victor R. (1996) Sympathetic neural mechanisms of cyclosporine-induced hypertension. Am J Hypertens 9: 121S–138S. [DOI] [PubMed] [Google Scholar]
- Sandrini S., Aslam N., Tardanico R., Setti G., Bossini N., Valerio F., et al. (2012) Tacrolimus versus cyclosporine for early steroid withdrawal after renal transplantation. J Nephrol 25: 43–49. [DOI] [PubMed] [Google Scholar]
- Schmidt S., Hamann S., Langrehr J., Höflich C., Mittler J., Jacob D., et al. (2007) Preoperative high-dose steroid administration attenuates the surgical stress response following liver resection: results of a prospective randomized study. J Hepatobiliary Pancreat Surg 14: 484–492. [DOI] [PubMed] [Google Scholar]
- Schold J., Poggio E., Goldfarb D., Kayler L., Flechner S. (2015) Clinical outcomes associated with induction regimens among retransplant kidney recipients in the United States. Transplantation 99: 1165–1171. [DOI] [PubMed] [Google Scholar]
- Schweer T., Gwinner W., Scheffner I., Schwarz A., Haller H., Blume C. (2014) High impact of rejection therapy on the incidence of posttransplant diabetes mellitus after kidney transplantation. Clin Transplant 28: 512519. [DOI] [PubMed] [Google Scholar]
- Sharif A., Baboolai K. (2011) Complications associated with new-onset diabetes after kidney transplantation. Nat Rev Nephrol 8: 34–42. [DOI] [PubMed] [Google Scholar]
- Soveri I., Snyder J., Holdaas H., Holme I., Jardine A., L’Italien G., et al. (2013) The external validation of the cardiovascular risk equation for renal transplant recipients: applications to BENEFIT and BENEFIT-EXT trials. Transplantation 95: 142–147. [DOI] [PubMed] [Google Scholar]
- Spinelli G., Felipe C., Park S., Mandia-Sampaio E., Tedesco-Silva H., Medina-Pestana J. (2011) Lipid profile changes during the first year after kidney transplantation: risk factors and influence of the immunosuppressive drug regimen. Transplant Proc 43: 3730–3737. [DOI] [PubMed] [Google Scholar]
- Staatz C., Goodman L., Tett S. (2010) Effect of CYP3A and ABCB1 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of calcineurin inhibitors: Part I. Clin Pharmacokinet 49: 141–175. [DOI] [PubMed] [Google Scholar]
- Stratta P., Quaglia M., Cena T., Antoniotti R., Fenoglio R., Menegotto A., et al. (2012) The interactions of age, sex, body mass index, genetics, and steroid weight-based doses on tacrolimus dosing requirement after adult kidney transplantation. Eur J Clin Pharmacol 68: 671–680. [DOI] [PubMed] [Google Scholar]
- Sugioka N., Kokuhu T., Okamoto M., Yoshimura N., Ito Y., Shibata N., et al. (2006) Effect of plasma lipid on pharmacokinetics of ciclosporin and its relationship with plasma prednisolone level in renal transplant patients. J Pharm Pharmacol 58: 1193–1200. [DOI] [PubMed] [Google Scholar]
- Suk H., Zhou C., Yang T., Zhu H., Yu R., Olabisi O., et al. (2013) Ablation of calcineurin Aβ reveals hyperlipidemia and signaling cross-talks with phosphodiesterases. J Biol Chem 288: 3477–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taber D., Meadows H., Pilch N., Chavin K., Baliga P., Egede L. (2013) The impact of diabetes on ethnic disparities seen in kidney transplantation. Ethn Dis 23: 238–244. [PubMed] [Google Scholar]
- Takeuchi H., Matsuno N., Hirano T., Gulimire M., Hama K., Nakamura Y., et al. (2011) Steroid withdrawal based on lymphocyte sensitivity to endogenous steroid in renal transplant recipients. Biol Pharm Bull 34: 1578–8153. [DOI] [PubMed] [Google Scholar]
- Terjimanian M., Harbaugh C., Hussain A., Olugbade K., Waits S., Wang S., et al. (2015) Abdominal adiposity, body composition and survival after liver transplantation. Clin Transplant doi: 10.1111/ctr.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessari G., Girolomoni G. (2012) Nonmelanoma skin cancer in solid organ transplant recipients: update on epidemiology, risk factors, and management. Dermatol Surg 38: 1622–1630. [DOI] [PubMed] [Google Scholar]
- Teutonico A., Schena P., Di Paolo S. (2005) Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol 16: 31283135. [DOI] [PubMed] [Google Scholar]
- Tornatore K., Biocevich D., Reed K., Tousley K., Singh J., Venuto R. (1995) Methylprednisolone pharmacokinetics, cortisol response, and adverse effects in black and white renal transplant recipients. Transplantation 59: 649–795. [DOI] [PubMed] [Google Scholar]
- Turgay M., Turgay F., Devrim E., Kucuksahin O., Caydere M., Durak I. (2012) The effects of dexamethasone on oxidant/antioxidant status in kidneys of rats administered mercuric chloride. Bratisl Lek Listy 113: 10–13. [DOI] [PubMed] [Google Scholar]
- Upala S., Panichsillapakit T., Wijarnpreecha K., Jaruvongvanich V., Sanguankeo A. (2015) Underweight and obesity increase the risk of mortality after lung transplantation: a systematic review and meta-analysis. Transpl Int. DOI: 10.1111/tri.12721 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Vadlamudi S., Hiremagalur B., Tao L., Kalhan S., Kalaria R., Kaung H., et al. (1993) Long-term effects on pancreatic function of feeding a HC formula to rats during the preweaning period. Am J Physiol 265: E565–E571. [DOI] [PubMed] [Google Scholar]
- Veroux M., Tallarita T., Corona D., Sinagra N., Giaquinta A., Zerbo D., et al. (2013) Conversion to sirolimus therapy in kidney transplant recipients with new onset diabetes mellitus after transplantation. Clin Dev Immunol 2013: 496974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester U., Schaefer A., Kranz B., Wingen A., Nadalin S., Paul A., et al. (2005) Development of growth and body mass index after pediatric renal transplantation. Pediatr Transplant 9: 445–449. [DOI] [PubMed] [Google Scholar]
- Vincenti F., Schena F., Paraskevas S., Hauser I., Walker R., Grinyo J. (2008) A randomized, multicenter study of steroid avoidance, early steroid withdrawal or standard steroid therapy in kidney transplant recipients. Am J Transplant 8: 307–316. [DOI] [PubMed] [Google Scholar]
- Wang R., Xu Y., Wu J., Wang Y., He Q., Chen J. (2013) Reduced-dose cyclosporine with mycophenolate mofetil and prednisone significantly improves the long-term glomerular filtration rate and graft survival. Intern Med 52: 947–953. [DOI] [PubMed] [Google Scholar]
- Webb N., Douglas S., Rajai A., Roberts S., Grenda R., Marks S., et al. (2015) Corticosteroid-free kidney transplantation improves growth: 2-year follow-up of the TWIST randomized controlled trial. Transplantation 99: 1178–1185. [DOI] [PubMed] [Google Scholar]
- Webster A., Ruster L., McGee R., Matheson S., Higgins G., Willis N., et al. (2010) Interleukin 2 receptor antagonists for kidney transplant recipients. Cochrane Database Syst Rev 1: CD003897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster A., Woodroffe R., Taylor R., Chapman J., Craig J. (2005a) Tacrolimus versus ciclosporin as primary immunosuppression for kidney transplant recipients: Meta-analysis and meta-regression of randomised trial data. BMJ 331: 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster A., Woodroffe R., Taylor R., Chapman J., Craig J. (2005b) Tacrolimus versus cyclosporin as primary immunosuppression for kidney transplant recipients. Cochrane Database Syst Rev 4: CD003961. [DOI] [PubMed] [Google Scholar]
- Weir M., Burgess E., Cooper J., Fenves A., Goldsmith D., McKay D., et al. (2015) Assessment and management of hypertension in transplant patients. J Am Soc Nephrol 26: 1248–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisinger J., Carlini R., Rojas E., Bellorin-Font E. (2006) Bone disease after renal transplantation. CJASN 1: 1300–1313. [DOI] [PubMed] [Google Scholar]
- Wjidan K., Ibrahim E., Caszo B., Gnanou J., Singh H. (2015) Dysregulation of glucose homeostasis following chronic exogenous administration of leptin in healthy Sprague-Dawley rats. J Clin Diagn Res 9: OF06–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodle E., First M., Pirsch J., Shihab F., Gaber A., Van Veldhuisen P. (2008) A prospective, randomized, doubleblind, placebocontrolled multicenter trial comparing early (7 day) corticosteroid cessation versus longterm, lowdose corticosteroid therapy. Ann Surg 248: 564577. [DOI] [PubMed] [Google Scholar]
- Yao B., Chen X., Shen F., Xu W., Dong T., Chen L., et al. (2013) The incidence of posttransplantation diabetes mellitus during follow-up in kidney transplant recipients and relationship to Fok1 vitamin D receptor polymorphism. Transplant Proc 45: 194–196. [DOI] [PubMed] [Google Scholar]
- Yong K., Nguyen H., Hii L., Chan D., Boudville N., Messineo A., et al. (2013) Association of a change in immunosuppressive regimen with hemodynamic and inflammatory markers of cardiovascular disease after kidney transplantation. Am J Hypertens 26: 843–849. [DOI] [PubMed] [Google Scholar]
- Zhang X., Huang H., Han S., Fu S., Wang L. (2013) Is it safe to withdraw steroids within seven days of renal transplantation? Clin Transplant 27: 1–8. [DOI] [PubMed] [Google Scholar]
- Zsom L., Wagner L., Fülöp T. (2015) Minimization vs tailoring: Where do we stand with personalized immunosuppression during renal transplantation in 2015? World J Transplant 5: 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]