

Abstract
We describe a homozygous copy number variant that disrupts the function of Dock2 in a commercially available C57BL/6 mouse strain that is widely used for backcrossing. This Dock2 allele was presumed to have spontaneously arisen in a colony of Irf5 knockout mice. We discovered that this allele has been inadvertently backcrossed into multiple mutant mouse lines, including two lines engineered to be deficient in Siae and Cmah. This particular subline of C57BL/6 mice also exhibits several striking immune phenotypes that have been previously described in the context of Dock2 deficiency. Inadvertent backcrossing of a number of gene-targeted mice into this background has complicated the interpretation of several immunological studies. In light of these findings, published studies involving immune or hematopoietic phenotypes where these mice have been used as controls, as experimental animals, or for backcrossing will need to be reinterpreted.
eTOC Blurb
Gene targeted mice are often backcrossed into the C57BL/6 background. Mahajan et al. find that a homozygous copy number variant disrupts the function of Dock2 in a specific commercially available C57BL/6 mouse strain (C57BL/6NHsd). Published studies using this strain need to be reinterpreted in light of these findings.
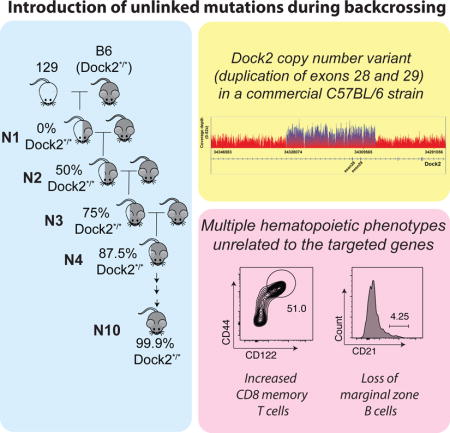
INTRODUCTION
Over the last three decades, gene targeting has emerged as a powerful tool for functional analyses of immune genes in vivo. It has become a common practice to backcross gene-targeted mice for ~10 generations into the C57BL/6 background to facilitate comparisons between gene-targeted mice as well as for adoptive transfer experiments. However, numerous C57BL/6 sublines are now in use across the world(Zurita et al., 2011), and the potential effect of variability among these C57BL/6 sublines on immune phenotypes is often not considered.
We had previously described defects in B cell development in two engineered mice strains with altered sialic acid physiology(Cariappa et al., 2009). Mice with a germline loss of either Siae (sialic acid acetyl esterase) or Cmah (cytidine monophosphate-N-acetylneuraminic acid hydroxylase) were found to lack marginal zone B cells and exhibited hyperactive B cell receptor signaling(Cariappa et al., 2009). Given that these mice generate altered forms of sialic acid that are not recognized by key regulatory Siglecs expressed on B cells (such as CD22/Siglec-2 and Siglec-G), the defects in B cell development observed in these mice were presumed to arise from perturbations in Siglec function(Cariappa et al., 2009; Pillai et al., 2009). In addition, the observed phenotypes were largely compatible with previous studies of Siglec function(Mahajan and Pillai, 2016; Pillai et al., 2009). Notably, both SiaeΔex2/Δex2 and Cmah knockout mice were originally generated at UC San Diego and had been backcrossed into a specific commercially-obtained C57BL/6 background for ten generations(Cariappa et al., 2009; Hedlund et al., 2007). We found that Siae deficient mice unexpectedly lost their aberrant B cell development phenotype upon backcrossing for 13 additional generations into the C57BL/6J (Jackson Laboratories) background. We created an independent knockout line of Siae deficient mice in the C57BL/6N background, and these mice exhibited no defects in B cell development.
Given these discrepant results, we re-examined the genetic basis of aberrant B cell development in SiaeΔex2/Δex2 mice using genetic crosses, SNP arrays and whole genome sequencing. These studies revealed that the defects in B cell development were not linked to Siae, which is present on chromosome 9, but instead to a gene encoding a guanine nucleotide exchange factor, Dock2, on chromosome 11.
Whole genome sequencing revealed a duplication of exons 28 and 29 of Dock2. Surprisingly, an identical mutation in Dock2 had been previously reported in two different colonies of Irf5−/− mice(Purtha et al., 2012). Given that the same mutation in Dock2 was identified in multiple gene-targeted mouse colonies despite different ES cell lines being used to generate these mice, it appeared that it was most likely introduced during backcrossing into the C57BL/6 background. Furthermore, the presence of C57BL/6N SNPs in close linkage with the duplication suggests that it arose in a C57BL/6N subline. Indeed, we were able to find this Dock2 variant (Dock2Hsd) in the homozygous state in the colony of C57BL/6NHsd mice maintained at Harlan Sprague Laboratories (now Envigo Biosciences). We also identified the Dock2Hsd variant in a colony of Cmah knockout mice that had been backcrossed into C57BL/6 mice obtained from Harlan Laboratories. Examination of a range of other commercially available C57BL/6J and C57BL/6N mice revealed only wild type Dock2. It is therefore likely that the Dock2Hsd variant arose within the C57BL/6NHsd mouse colony at Harlan Laboratories. This study raises concerns that many other lines of gene-targeted mice bearing hematopoietic phenotypes may have been inadvertently compromised by backcrosses involving the use of C57BL/6NHsd mice.
RESULTS
The loss of marginal zone B cells and enhanced CD8+ memory T cell phenotype observed in SiaeΔex2/Δex2 mice is not linked to the loss of Siae
We have previously reported that SiaeΔex2/Δex2 mice, which were backcrossed into the C57BL/6 background for 10 generations (henceforth referred to as N10-Siae mice), show a profound loss of marginal zone (MZ) B cells(Cariappa et al., 2009). In subsequent studies, we noted that N10-Siae mice also exhibit a marked increase in CD8+ CD44+ CD122hi memory-phenotype (MP) cells in the blood and spleen (Figure 1A). Surprisingly, both the defect in MZ B cell development as well as the enhanced CD8+ MP cell phenotype were completely lost upon further backcrossing of N10-Siae mice into the C57BL/6J background for an additional 13 generations (henceforth referred to as N23-Siae mice) (Figure 1B). To further determine whether the loss of Siae per se was responsible for any of the N10 phenotypes, we generated an independent line of Siae deficient mice (Siaetm1a/tm1a) using an ES cell clone bearing a gene-targeted allele of Siae (Siaetm1a(EUCOMM)Wtsi ; MGI ID: 4842607) in the C57BL/6N background (Figure S1). These mice also exhibited normal numbers of MZ B cells and no increase in CD8+ MP cells, confirming that the loss of Siae per se was not responsible for the phenotypes previously observed in N10-Siae mice (Figure 1B).
Figure 1. Siae deficient mice exhibit a background-dependent increase in CD8+ MP cells and a loss of marginal zone B cells.
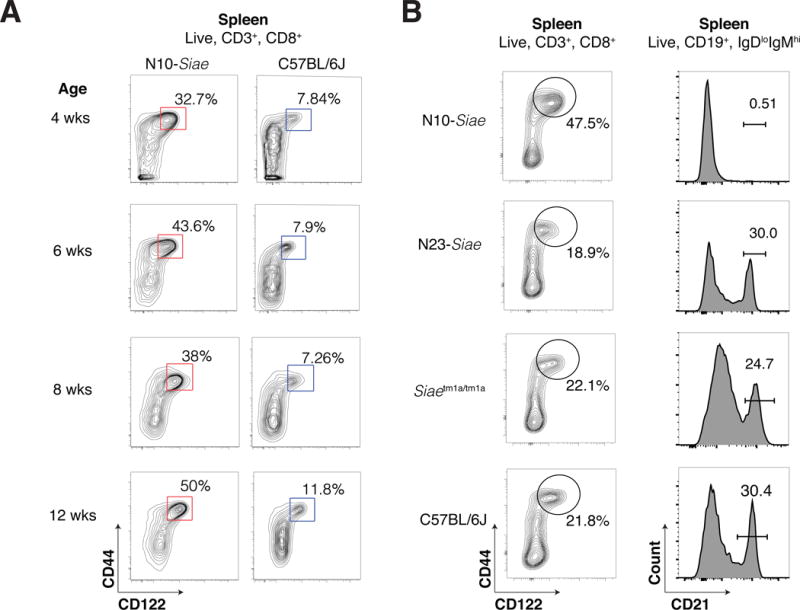
(A) The proportion of CD8+ MP cells in the spleens of one to three month old N10-Siae mice with age-matched C57BL/6J controls are shown. (B) CD8+ MP and marginal zone B cells in N10-Siae, N23-Siae, Siaetm1a/tm1a and C57BL/6J mice. The data shown are representative of 5 mice. See also Figure S1.
The phenotypes observed in N10-Siae mice are inherited in a Mendelian fashion and linked to a locus distinct from Siae
To begin to identify the genetic locus responsible for the anomalous phenotypes observed in N10-Siae mice, we did a test cross to assess the inheritance pattern. N10-Siae × C57BL/6J mice (F1) were generated and backcrossed to N10-Siae mice. We found that the increase in CD8+ MP cells was a Mendelian recessive trait and segregated independently of Siae (Figure 2A). In addition, analysis of these mice showed a 100% linkage between the loss of MZ B cells and an increase CD8+ MP T cells, suggesting that a single genetic locus was likely responsible for both these phenotypes (Figure 2B). Since the proportion of CD8+ MP cells in blood could be easily measured, we used this trait for mapping the pathogenic locus. We also found that 4 of 18 mice that lacked both copies of the SiaeΔex2 allele had an abnormal CD8+ MP population, suggesting that Siae had no contribution to these observed phenotypes (Figure 2C).
Figure 2. The constellation of hematopoietic phenotypes observed in N10-Siae mice are Mendelian, recessive & unlinked to Siae.
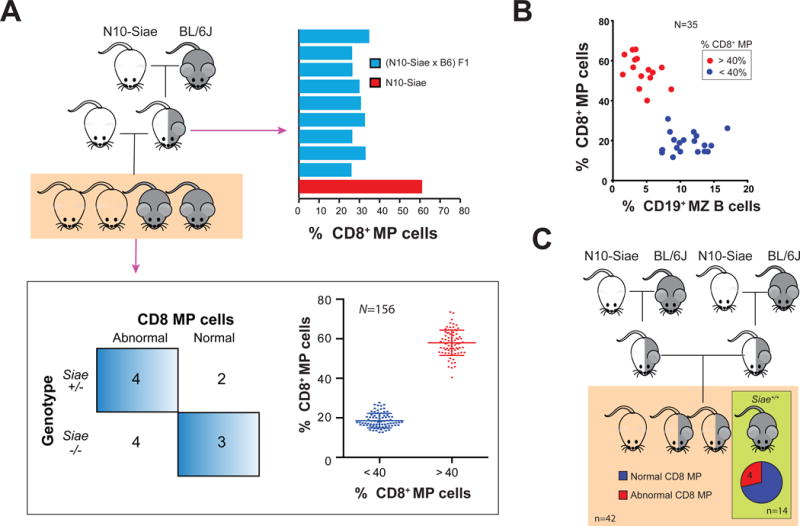
(A) (N10-Siae × C57BL/6J) F1 animals have a normal CD8+ MP cell compartment. Upon subsequent backcrossing to the N10-Siae background, Siae does not segregate with the altered CD8+ MP cells (n=13). Approximately equal number of mice with (n=79) and without (n=77) the phenotype (<40% and >40% CD8 MP cells respectively) are observed. (B) A strong correlation between loss of marginal zone B cells and increase in CD8 MP is observed. (C) ~25% of Siae+/+ (wild-type) mice obtained from an intercross between heterozygous N10-Siae mice exhibit normal CD8+ MP cells.
A genetic marker on chromosome 11 segregates with the N10-Siae phenotype
In parallel, we performed a whole genome SNP array (Affymetrix Mouse Diversity array) on two homozygous N10-Siae mice that exhibited a loss of MZ B cells and an increased proportion of CD8+ MP cells. The N10-Siae whole genome SNP arrays yielded an average call rate of 99% with 98.7% homozygosity. Publicly available SNP array data from various 129 substrains and C57BL/6 sublines were used for comparison(Didion et al., 2012). 538,667 SNPs showed a 100% call rate across all the arrays. Given that the SiaeΔex2 allele was generated in an R1 ES cell that is derived from the 129×1/SvJ × 129S1/Sv F1-Kitl<+> background(Nagy et al., 1993), it was not surprising that all the SNPs (276 of 276) in a 6 Mbp region surrounding the Siae locus (chr9:35022155-41040054) that differed from the C57BL/6 reference genome were of 129 origin (Figure 3A and Table S1). Surprisingly, all these 129 origin SNPs were retained in the N23-Siae mice, suggesting that 13 additional generations of backcrossing into C57BL6/J mice had had no further effect on reducing the 129 contribution on chromosome 9 (Figure S3). Considering that ~17.4% of the 538,667 SNPs calls analyzed in the whole genome SNP array were different between the 129 and C57BL/6 backgrounds and the fact that we did not find any genomic region containing two or more contiguous SNPs of 129 origin outside of the chromosome 9 locus containing Siae, we recognized that it was extremely unlikely that a mutation in the gene-targeted ES cell clone of 129 origin had been retained in the N10-Siae mice.
Figure 3. A SNP on chromosome 11 segregates with increase in CD8 memory-phenotype T cells.
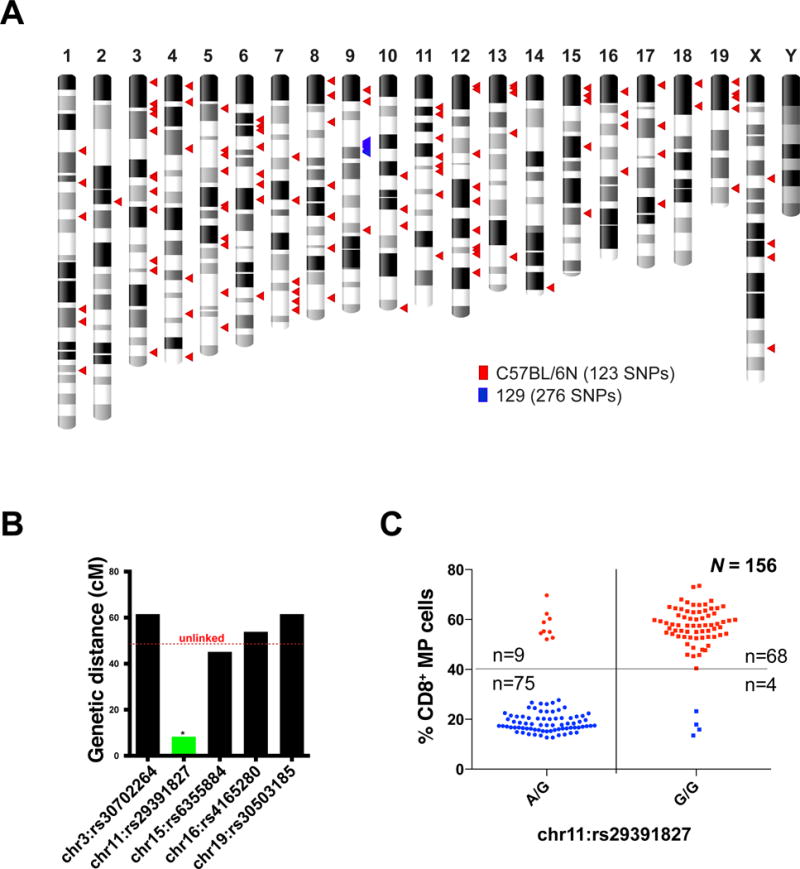
(A) Whole genome SNP array revealed 399 SNPs that differ between N10-Siae and C57BL/6J. 276 SNPs (blue triangles) on chromosome 9 in the Siae locus are shared by 129 strains. 123 SNPs (red triangles) appear to be of C57BL/6N origin. (B) Linkage between candidate SNPs and CD8+ MP expansion in the progeny of test crosses (n=18) described in Figure 2A. A SNP on chromosome 11 (rs29391827) was found to segregate with the CD8 memory phenotype (n=18). (C) This observation was validated in a larger cohort (n=156). See also Figure S2 and Table S1.
All (123 of 123) SNPs on the remaining N10-Siae chromosomes that differed from the C57BL/6J allele, matched the C57BL/6N consensus allele (shared between C57BL/6NTac, C57BL/6Nci and C57BL/6NCrl), suggesting that the pathogenic locus could have arisen from a C57BL/6N strain used previously for backcrossing. We next tested the genetic linkage to a few randomly chosen candidate C57BL/6N SNPs in the (C57BL/6N × C57BL/6J) test crosses (Figure 2A, B). Fortuitously, a SNP of C57BL/6N origin, rs29391827, on chromosome 11 (chr11:50962622) was linked to the increase in CD8+ MP cells in the blood of 18 mice (Figure 3B). The linkage to rs29391827 was validated in a larger cohort of 156 mice and estimated to be at a distance of about 8.3 cM (Figure 3C).
A region on chr11:30552213-35421130 is tightly associated with the N10-Siae phenotype
We next performed whole genome sequencing of an N10-Siae mouse that exhibited the phenotypic changes described. Single end sequencing was performed for 85 cycles on a NextSeq500 instrument, yielding a total of ~40Gbp of sequence data. 93.9% of the reads mapped to the reference mouse genome (65.42% of the reads mapped to unique sites on the genome). A mean coverage depth of 15.33 fold was obtained. The GATK variant calling pipeline was used to identify positions on chromosome 11 that differed from the reference genome (Figure S3)(McKenna et al., 2010). This analysis confirmed the results of the whole genome SNP array (for SNPs with adequate sequencing coverage) and also revealed a few additional SNPs that were not covered by the SNP array that differed between N10-Siae and C57BL/6J. All the protein coding variants (3 of 3) that we observed within 25 Mbp of rs29391827 on chromosome 11 were previously reported SNPs present in one or more inbred mouse strains (rs240617401, rs8261521 and rs6268547). As structural variants could not be ruled out on the basis of this analysis, we used a panel of SNPs that differed between N10-Siae and C57BL/6J to analyze mice shown in Figure 3 that showed discrepant phenotypes with respect to rs29391827. This allowed us to progressively narrow the pathogenic genetic locus to a 5 Mbp region on chromosome 11 (chr11:30552213-35421130) (Figure S4).
A Dock2 duplication on chromosome 11 accounts for the observed N10-Siae phenotype
A focused examination of the whole genome sequence in the chr11:30,552,213–35,421,130 region revealed a ~2-fold increase in coverage in a 23.5 Kbp region encompassing exons 28 and 29 of the Dock2 gene (Figure 4A). This was reminiscent of a previously published report identifying a duplication involving exons 28 and 29 of Dock2 in Irf5−/− mice(Purtha et al., 2012). Indeed, the presence of an identical duplication in Dock2 was confirmed in N10-Siae mice using a previously reported PCR designed to identify this duplication (Figure 4B)(Yasuda et al., 2013). Sequencing of the PCR product revealed that the breakpoint in the Dock2 duplication (chr11:34329863-34306424; GRCm38/mm10 assembly) was identical in Irf5−/− and N10-Siae mice (Figure S4). As Cmah−/− mice also lack MZ B cells(Cariappa et al., 2009), we looked for the presence of the Dock2Hsd allele in 3 Cmah−/− mice. The Dock2Hsd allele was present in 3 of 3 Cmah−/− mice analyzed (Figure 4B). This duplication has been demonstrated to be a loss-of-function allele as it results in a frameshift mutation and nonsense-mediated decay of Dock2 mRNA(Purtha et al., 2012).
Figure 4. A genomic segment encoding exons 28 and 29 of Dock2 is duplicated in N10-Siae and Cmah−/− mice.
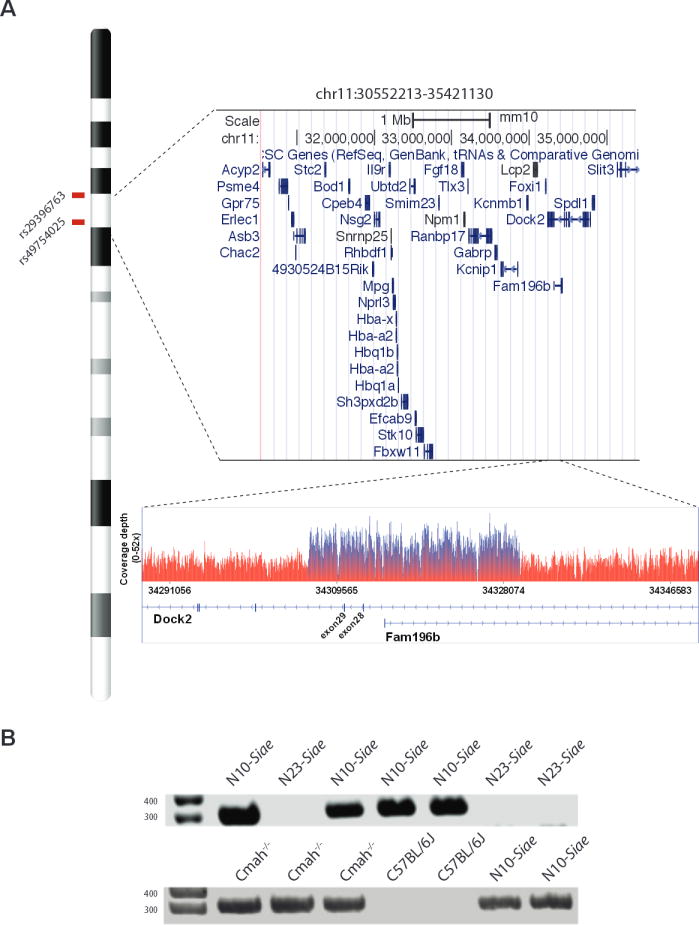
(A) An abrupt increase in coverage depth within the putative pathogenic locus on chromosome 11 mapped by SNPs ecompasses exons 28 and 29 of Dock2 in the N10-Siae genome. (B) PCR detection of Dock2 duplication in N10-Siae, Cmah−/−, N23-Siae and C57BL6/J mice. See also Figures S3, S4, and S5.
The mutant Dock2 allele was introduced by backcrossing into C57BL/6NHsd mice
Given that the same duplication was seen in three independently gene-targeted mice, it confirmed our suspicion that the mutant Dock2 allele was inadvertently introduced during backcrossing. As previously noted, the SNPs flanking the duplication were of C57BL6/N origin (rs29473246 at chr11:33548367, rs29414108 at chr11:43358462), suggesting that this duplication arose in a C57BL/6N substrain. Indeed, a survey of several commercially available C57BL6/N substrains showed the presence of a duplication in exons 28 and 29 of Dock2 with an identical breakpoint in 44 of 44 C57BL/6NHsd mice analyzed (Figure 5A and Figure S5). We henceforth refer to this allele as Dock2Hsd. The Dock2Hsd allele was not detected in any of the other C57BL/6N strains analyzed viz. C57BL/6NTac, C57BL/6NCrl, C57BL/6NJ (Figure S5). This Dock2 duplication was also not observed in the C57/BL6NJ genome published by the Mouse Genomes Project (Wellcome Trust Sanger Institute)(Yalcin et al., 2012). Thus, the Dock2Hsd allele appears to be a copy number variant that has been fixed in the C57BL/6NHsd strain and may have been introduced into numerous other gene-targeted mouse lines during backcrossing. Consistent with this, both SiaeΔex2/Δex2 and Cmah−/− mice were generated from ES cells of 129 origin and backcrossed into the C57BL/6 background at UCSD using mice obtained from Harlan Laboratories.
Figure 5. The Dock2 duplication is present in the C57BL/6NHsd strain, which exhibits multiple characteristics of Dock2 deficiency.
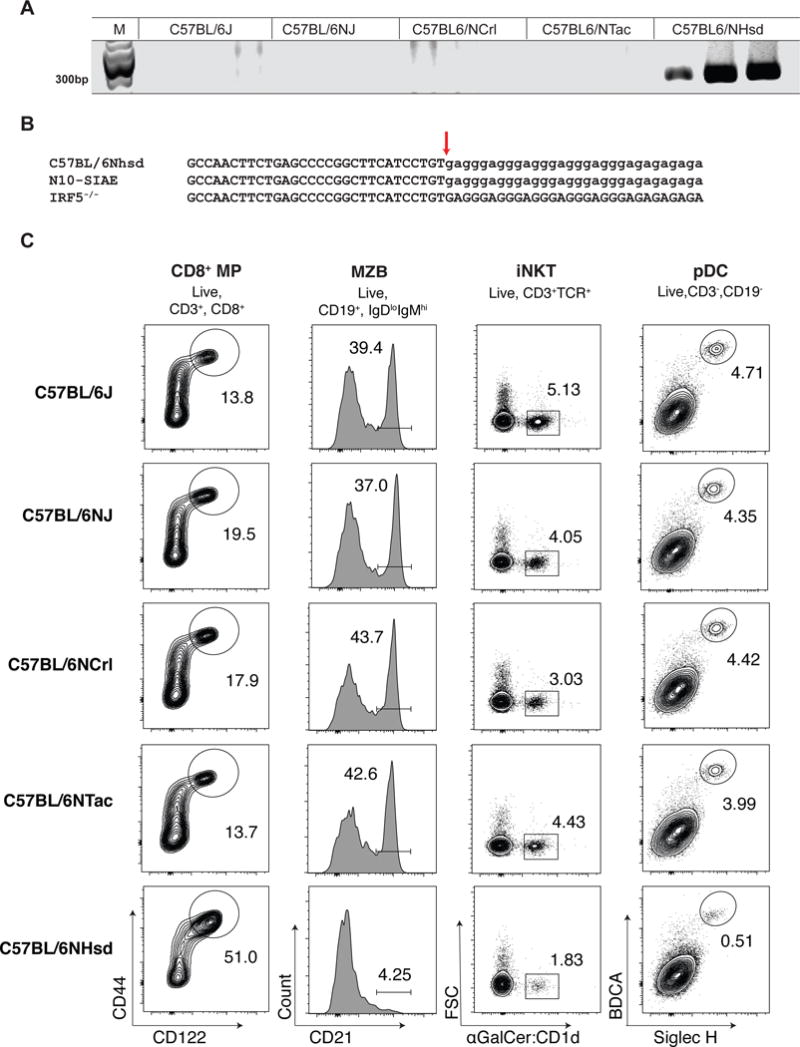
(A) PCR detection of Dock2 duplication in C57BL/6NHsd mice and other C57BL6/N sublines (C57BL6/NTac and C57BL6/NJ). (B) Sequences of the Dock2 duplication breakpoint (arrow) from C57BL6/NHsd, N10-Siae and IRF5−/− mice. A low complexity sequence at the site of the breakpoint is shown in lower case. (C) Proportion of CD8+ MP cells, marginal zone B cells, invariant NK T cells and plamsmacytoid dendritic cells C57BL/6NHsd, C57BL6/NTac and C57BL6/J mice. The data shown are representative of 5 mice each. See also Figure S5.
The Irf5−/− mice were generated from a E14K ES cell (129OlaHsd background) and backcrossed into the C57BL/6 background before being distributed to various investigators from a laboratory in Japan (Takaoka et al., 2005). However, the Dock2 duplication was found only in two of three colonies derived from the original Irf5−/− colony, which led the investigators to believe that the Dock2 duplication arose spontaneously within a subline of Irf5−/− mice(Purtha et al., 2012). Like the N10-Siae mice, C57BL/6NHsd mice exhibit an expansion of CD8+ MP T cells (Figure 5C and Figure S5). C57BL/6NHsd mice also exhibit other phenotypes that have been previously observed in the Dock2−/− mice(Fukui et al., 2001). Both Dock2−/− and C57BL/6NHsd mice exhibit splenic cytopenia as well as a profound loss of marginal zone B cells, invariant NKT cells and plasmacytoid dendritic cells (Figure 5C)(Fukui et al., 2001; Gotoh et al., 2008; Kunisaki et al., 2006). The similarities between Dock2−/− and C57BL/6NHsd mice support the notion that the Dock2Hsd allele inactivates the function of Dock2 by creating a premature stop codon that contributes to nonsense mediated decay of the resultant mRNA.
DISCUSSION
Striking phenotypes have been discovered in a number of gene-targeted mice that were previously assumed to be the result of the homozygous loss of a specific gene. In order to explain the loss of phenotypes in SiaeΔex2/Δex2 upon further backcrossing into the C57B6/J background, we generated an additional mutant allele of Siae. This mutant mouse also lacked the phenotypes that we had previously reported.
In order to explain this loss of immune phenotypes we undertook a series of studies involving conventional genetics, whole genome genotyping and whole genome sequencing. This allowed us to identify a causal mutation on chromosome 11 that was responsible for the previously observed immune phenotypes such as the loss of marginal zone B cells and the increase in CD8+ memory phenotype T cells in SiaeΔex2/Δex2 mice. We then also found this same mutation, a homozygous duplication of two exons of Dock2 that creates a non-functional product, in Cmah mutant mice. Interestingly, an identical mutation had been previously found in Irf5−/− mice, originally generated in Japan, and had been assumed to have arisen as a spontaneous mutation(Purtha et al., 2012; Takaoka et al., 2005). The most parsimonious explanation for a specific mutation being present in three different gene-targeted mice, all derived from different ES cells, two generated originally in California and a third in Japan, is that they had all been acquired by backcrossing into an unidentified mutant C57BL/6 strain. It should be noted that while the loss of MZ B cells in Cmah mutant mice may be ascribed to the acquired Dock2 mutation, there are other phenotypes that have been reported in these mice that may relate to the absence of the N-glycolyl form of sialic acid observed in the absence of the Cmah protein(Naito et al., 2007). Thus it remains possible that some functional phenotypes in such mice are related to altered interactions between sialic acids and Siglecs.
Because our SNP arrays showed the presence of specific C57BL/6N SNPs we had a high degree of suspicion that this mutation had been acquired from a commercial C57BL/6N strain. We surveyed commercial C57BL/6N sublines and discovered that this Dock2 gene duplication that was present only in C57BL/6N mice obtained from Harlan Laboratories (C57BL/6NHsd). Intriguingly, the set of 123 SNPs outside the Siae locus that differed between N10-Siae and C57BL/6J also comprised 97% of the SNP alleles (n = 128) from a pool of 538,667 SNPs analyzed in the whole genome arrays that differ between C57BL/6J and C57BL/6N sublines but are identical to the consensus 129 alleles. Given that C57BL/6J and C57BL/6N strains were derived from a common stock of C57BL/6 mice, the differences between C57BL/6J and C57BL/6N substrains have been so far attributed to genetic drift. In contrast, our analysis indicates that C57BL/6N mice may have derived some genetic contribution from the 129 strain at a remote time during their history, prior to the divergence of various C57BL/6N sublines.
SiaeΔex2/Δex2 and Cmah deficient mice were backcrossed at UC San Diego into the C57BL/6 background using mice obtained from Harlan Laboratories Inc. between 2005–2008. Initial reports of Irf5−/− mice backcrossed into the C57BL/6 background that bear phenotypes (e.g. lack of type I interferon) attributable to Dock2 deficiency were published in 2007(Gotoh et al., 2010, 2008; Yasuda et al., 2007). Thus, we believe that the Dock2 copy number variant has been present in the C57BL/6NHsd colony at Harlan Laboratories for at least a decade. The C57BL/6NHsd mice were derived by Harlan Laboratories from the breeding nucleus of the C57BL/6N colony maintained at the NIH in 1988. Harlan Laboratories Inc. was acquired by Envigo Biosciences in 2015. The C57BL/6N subline originated from the C57BL/6J colony at Jackson Laboratory in 1951(Mekada et al., 2009). Given that other sublines derived from C57BL/6N and C57BL/6J strains lack the Dock2Hsd variant, it is formally possible that a spontaneous mutation present in the C57BL/6N founders was expanded at Harlan Laboratories. However, it is more likely that the Dock2 duplication arose later in the C57BL/6NHsd colony after it diverged from the NIH breeding stock and was fixed by the breeding strategies based on continued brother-sister mating that are designed to minimize genetic drift in mouse sublines. Analysis of archived tissues from C57BL/6NHsd mice will help estimate when the Dock2Hsd variant arose, and help assess how many studies have been affected. Despite the relatively large size of the Dock2 gene, coding variants of Dock2 have not been reported in other inbred mouse strains(Sherry et al., 2001).
The Dock2Hsd variant is particularly significant as it causes wide-ranging hematopoietic phenotypes in a strain of mice that is widely used for immunological studies. Since 2003, the Jackson Laboratory has implemented a Genetic Stability Program in selected mouse strains to limit cumulative genetic drift, including that caused by copy number variation, by regularly rebuilding foundation stocks from cryopreserved, pedigreed embryos every few generations(Taft et al., 2006). Commercial breeders of inbred mouse strains as well as individual research groups maintaining knockout lines over long periods should consider the implications of the choice of their breeding strategies on genetic drift within the colony.
The recent availability of ES cells from a C57BL/6 background obviates the need for backcrossing but ES cells derived from the C57BL/6NHsd background should be tested for the presence of the Dock2Hsd allele(Skarnes et al., 2011). Given that Dock2 is expressed primarily in the hematopoietic lineage, immune phenotypes that have been studied in the context of B6 mice from Harlan Laboratories should be reviewed carefully. The Dock2Hsd allele has likely affected the interpretation of a large number of studies in which C57BL/6NHsd mice obtained from Harlan Sprague were utilized as controls. We have identified a large number of studies in the literature that suggest to us that a plethora of assumed phenotypes should be revisited. A PCR screening protocol for the Dock2Hsd allele is included under Supplemental Experimental Procedures.
Loss-of-function variants of other genes such as Nnt, Mnrn1, Rd8 and Cyfip2 have been described in one or more sublines of C57BL/6 and have been linked to glucose intolerance, impaired platelet function, retinal degeneration and altered cocaine response respectively(Freeman et al., 2006; Kumar et al., 2013; Mattapallil et al., 2012; Reheman et al., 2010). Loss of function variants of Scna are seen in some sublines of C57BL/6N as well as in ES cells of C57BL/6N origin but result in no obvious phenotype(Specht and Schoepfer, 2001). A copy number variant that alters the expression of insulin-degrading enzyme (Ide) and fibroblast growth factor binding protein 3 (Fgfbp3) genes has been reported to have achieved a high allele frequency in C57BL6/J mice(Watkins-Chow and Pavan, 2008). While it is difficult to estimate the precise degree of genetic drift that results in deleterious or loss-of-function mutations, subline-specific variants are not uncommon and should be considered as a possible cause of phenotypic discrepancies in mouse sublines. Furthermore, the specific substrain of mice used for experiments or for backcrossing should be clearly documented.
MATERIALS AND METHODS
Mice
An ES cell clone (EPD0679_2_E06) bearing a targeted disruption of the Siae locus (Siaetm1a(EUCOMM)Wtsi ; MGI ID: 4842607) was obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM)(Collins et al., 2007). The Siaetm1a(EUCOMM)Wtsi allele (Siaetm1a) contains a beta-galactosidase reporter and a polyadenylation signal in intron 3 of Siae. Gene targeting had been initially confirmed using long-range PCR by EUCOMM, and additionally validated by us with a Southern Blot using the non-isotopic BrightStar Biodetect kit (Ambion Inc.) (Figure S1). The ES cells were then injected into C57BL/6 blastocysts at the Transgenic Core Facility at Brigham and Women’s Hospital. The resulting chimeric mice were crossed into C57BL6/NTac. An Siaetm1a founder was backcrossed for one generation into C57BL6/NTac and intercrossed to obtain Siaetm1a/tm1a homozygous mice. C57BL6/NHsd mice were purchased from Harlan Sprague Dawley, Frederick, MD (acquired by Envigo Biosciences in 2015).
Genetic mapping
Whole genome SNP arrays were performed at the Microarray Core Facility at the Dana Farber Cancer Institute using the Affymetrix Mouse Diversity Array platform. Analysis was performed using the Affymetrix Genotyping Console version 4.2.0.26. Publicly available SNP array data from various 129 substrains and C57BL/6 sublines were used for comparison(Didion et al., 2012). We focused on 538,667 SNPs that were concordant among all the C57BL/6N substrains, all the 129 strains and both the N10-Siae mice analyzed respectively. These were intended to represent the ancestral alleles of the 129 and C57BL6/N strains. Candidate SNPs were evaluated using PCR and Sanger sequencing. Sequencing chromatograms were analyzed using Mutation Surveyor v3.24 (Softgenetics).
Whole genome sequencing
A whole genome library was constructed using the Kapa HyperPlus kit following manufacturer’s recommendations. Briefly, 1 μg of genomic DNA was enzymatically fragmented, end repaired, A-tailed and ligated to Illumina Truseq adapters. No library amplification was performed to avoid introducing any coverage bias. The library was sequenced on the Illumina NextSeq 500 for 85 cycles using the NextSeq 500/550 High Output v2 kit. A total of 567,104,840 single-end reads were aligned to the Mus musculus genome (GRCm38/mm10, December 2011 build) using Bowtie2(Langmead and Salzberg, 2012). For a total 93.96% of reads mapping to the reference genome, 65.42% of reads mapped to unique sites on the genome, and 28.54% of reads mapped to multiple regions. The GATK pipeline was used to identify variants(McKenna et al., 2010). Variants with quality scores < 30 or allele frequency <100% were excluded, yielding 35,340 variants. Variant and read densities were calculated using BEDtools on Galaxy and visualized using the R statistical programming language(Quinlan and Hall, 2010).
Flow cytometry
Mouse splenocytes or peripheral blood were hemolyzed with ACK buffer, stained with fluorescently conjugated antibodies analyzed on a BD LSR II flow cytometer. The following antibody clones were used in this study: anti-CD122 (TMB1), anti-CD44 (IM7), anti-CD3 (17A2), anti-CD8 (53-6.7) anti-CD19 (6D5) anti-B220 (RA3-6B2), anti-CD21(7E9), anti-IgM (RMM-1), anti-CD1d (1B1), and anti-IgD (11-26c.2a) from Biolegend.
Statistical Analysis
Statistical analysis was performed using Graphpad Prism 6. The significance of differences between groups was evaluated using the Student’s t-test.
Supplementary Material
Highlights.
-
-
A spontaneous Dock2 mutation was found in a widely used C57BL/6 mouse strain
-
-
The Dock2Hsd allele has been inadvertently introduced into several gene-targeted mice
-
-
The Dock2Hsd allele may confound the interpretation of several gene-targeting studies
-
-
Published studies using C57BL/6NHsd mice may need to be revisited
Acknowledgments
We thank Patrick Secrest for the examination of mice currently at UCSD. This work was supported by grants from the NIH (AI064930 and GM32373).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: Conceptualization, V.S.M., E.D. and S.P.; Methodology, V.S.M., E.D., H.M.; Software, V.V., V.S.M., R.M.; Investigation, V.S.M., E.D., H.M.; Resources, A.V.; Writing – Original Draft, V.S.M., E.D., S.P.; Writing – Review & Editing, V.S.M., E.D., H.M., S.P.; Visualization, V.S.M., E.D., V.V.; Supervision, S.P., V.S.M.
Accession numbers: NCBI BioProjects Accession Number: PRJNA318274.
References
- Cariappa A, Takematsu H, Liu H, Diaz S, Haider K, Boboila C, Kalloo G, Connole M, Shi HN, Varki N, Varki A, Pillai S. B cell antigen receptor signal strength and peripheral B cell development are regulated by a 9-O-acetyl sialic acid esterase. J Exp Med. 2009;206:125–138. doi: 10.1084/jem.20081399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Rossant J, Wurst W. A mouse for all reasons. Cell. 2007;128:9–13. doi: 10.1016/j.cell.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Didion JP, Yang H, Sheppard K, Fu CP, McMillan L, de Villena FPM, Churchill GA. Discovery of novel variants in genotyping arrays improves genotype retention and reduces ascertainment bias. BMC Genomics. 2012;13:34. doi: 10.1186/1471-2164-13-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman HC, Hugill A, Dear NT, Ashcroft FM, Cox RD. Deletion of nicotinamide nucleotide transhydrogenase: a new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes. 2006;55:2153–2156. doi: 10.2337/db06-0358. [DOI] [PubMed] [Google Scholar]
- Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, Inayoshi A, Noda M, Oike M, Shirai T, Sasazuki T. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature. 2001 doi: 10.1038/35090591. [DOI] [PubMed] [Google Scholar]
- Gotoh K, Tanaka Y, Nishikimi A, Inayoshi A, Enjoji M, Takayanagi R, Sasazuki T, Fukui Y. Differential requirement for DOCK2 in migration of plasmacytoid dendritic cells versus myeloid dendritic cells. Blood. 2008;111:2973–2976. doi: 10.1182/blood-2007-09-112169. [DOI] [PubMed] [Google Scholar]
- Gotoh K, Tanaka Y, Nishikimi A, Nakamura R, Yamada H, Maeda N, Ishikawa T, Hoshino K, Uruno T, Cao Q, Higashi S, Kawaguchi Y, Enjoji M, Takayanagi R, Kaisho T, Yoshikai Y, Fukui Y. Selective control of type I IFN induction by the Rac activator DOCK2 during TLR-mediated plasmacytoid dendritic cell activation. J Exp Med. 2010 doi: 10.1084/jem.20091776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund M, Tangvoranuntakul P, Takematsu H, Long JM, Housley GD, Kozutsumi Y, Suzuki A, Wynshaw-Boris A, Ryan AF, Gallo RL, et al. N-glycolylneuraminic acid deficiency in mice: implications for human biology and evolution. Mol Cell Biol. 2007;27:4340–4346. doi: 10.1128/MCB.00379-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Kim K, Joseph C, Kourrich S, Yoo SH, Huang HC, Vitaterna MH, de Villena FPM, Churchill G, Bonci A, Takahashi JS. C57BL/6N mutation in cytoplasmic FMRP interacting protein 2 regulates cocaine response. Science. 2013;342:1508–1512. doi: 10.1126/science.1245503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Tanaka Y, Sanui T, Inayoshi A, Noda M, Nakayama T, Harada M, Taniguchi M, Sasazuki T, Fukui Y. DOCK2 is required in T cell precursors for development of Valpha14 NK T cells. J Immunol. 2006;176:4640–4645. doi: 10.4049/jimmunol.176.8.4640. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan VS, Pillai S. Sialic acids and autoimmune disease. Immunol Rev. 2016;269:145–161. doi: 10.1111/imr.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, Caspi RR. The Rd8 Mutation of the Crb1 Gene Is Present in Vendor Lines of C57BL/6N Mice and Embryonic Stem Cells, and Confounds Ocular Induced Mutant Phenotypes. Investigative Opthalmology & Visual Science. 2012;53:2921. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekada K, Abe K, Murakami A, Nakamura S, Nakata H, Moriwaki K, Obata Y, Yoshiki A. Genetic differences among C57BL/6 substrains. Exp Anim. 2009;58:141–149. doi: 10.1538/expanim.58.141. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci U S A. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y, Takematsu H, Koyama S, Miyake S, Yamamoto H, Fujinawa R, Sugai M, Okuno Y, Tsujimoto G, Yamaji T, Hashimoto Y, Itohara S, Kawasaki T, Suzuki A, Kozutsumi Y. Germinal center marker GL7 probes activation-dependent repression of N-glycolylneuraminic acid, a sialic acid species involved in the negative modulation of B-cell activation. Mol Cell Biol. 2007;27:3008–3022. doi: 10.1128/MCB.02047-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S, Cariappa A, Pirnie SP. Esterases and autoimmunity: the sialic acid acetylesterase pathway and the regulation of peripheral B cell tolerance. Trends Immunol. 2009;30:488–493. doi: 10.1016/j.it.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purtha WE, Swiecki M, Colonna M, Diamond MS, Bhattacharya D. Spontaneous mutation of the Dock2 gene in Irf5−/− mice complicates interpretation of type I interferon production and antibody responses. Proceedings of National Academy of Sciences. 2012;109 doi: 10.1073/pnas.1118155109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reheman A, Tasneem S, Ni H, Hayward CPM. Mice with deleted multimerin 1 and alpha-synuclein genes have impaired platelet adhesion and impaired thrombus formation that is corrected by multimerin 1. Thromb Res. 2010;125:e177–83. doi: 10.1016/j.thromres.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht CG, Schoepfer R. Deletion of the alpha-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci. 2001;2:11. doi: 10.1186/1471-2202-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft RA, Davisson M, Wiles MV. Know thy mouse. Trends Genet. 2006;22:649–653. doi: 10.1016/j.tig.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano SI, Honda K, Ohba Y, Mak TW, Taniguchi T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- Watkins-Chow DE, Pavan WJ. Genomic copy number and expression variation within the C57BL/6J inbred mouse strain. Genome Res. 2008;18:60–66. doi: 10.1101/gr.6927808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin B, Adams DJ, Flint J, Keane TM. Next-generation sequencing of experimental mouse strains. Mamm Genome. 2012;23:490–498. doi: 10.1007/s00335-012-9402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Nündel K, Watkins AA, Dhawan T, Bonegio RG, Ubellacker JM, Marshak-Rothstein A, Rifkin IR. Phenotype and function of B cells and dendritic cells from interferon regulatory factor 5-deficient mice with and without a mutation in DOCK2. Int Immunol. 2013;25:295–306. doi: 10.1093/intimm/dxs114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Richez C, Maciaszek JW, Agrawal N, Akira S, Marshak-Rothstein A, Rifkin IR. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol. 2007;178:6876–6885. doi: 10.4049/jimmunol.178.11.6876. [DOI] [PubMed] [Google Scholar]
- Zurita E, Chagoyen M, Cantero M, Alonso R, González-Neira A, López-Jiménez A, López-Moreno JA, Landel CP, Benítez J, Pazos F, Montoliu L. Genetic polymorphisms among C57BL/6 mouse inbred strains. Transgenic Res. 2011;20:481–489. doi: 10.1007/s11248-010-9403-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.