

Abstract
Objectives:
To report characteristics, treatment, and outcomes for an international cohort of patients with extraskeletal osteosarcoma (ESOS).
Materials and Methods:
Through the Rare Cancer Network, retrospective data on patients with ESOS were collected. Patient characteristics, multimodality treatment information, and survival status were analyzed.
Results:
Thirty-seven patients in 4 health care institutions were identified. Thirty-one (86%) patients had grade 3 or 4 tumors. Most patients (27 [73%]) had stage III disease. Fourteen (38%) received neoadjuvant chemotherapy or chemoradiation. Of 28 (85%) who underwent surgery, 21 (75%) had free margins achieved and 15 (41%) subsequently received adjuvant chemotherapy. At median follow-up of 45 months, 20 (55%) patients were alive, 13 (43%) of whom were disease free. Univariate analysis showed that poor overall survival was related to stage IV (P<0.001), no surgery (P<0.001), primary size >10 cm (P=0.002), and age (P=0.002). In multivariate analysis, primary size >10 cm (P=0.005) was prognostic for overall survival. For patients without metastases, univariate analysis showed disease-free survival (DFS) related to primary size >10 cm (P=0.003), surgery (P=0.004), local recurrence (P=0.003), and age (P<0.001). In multivariate analysis for DFS, primary size >10 cm (P=0.01) and older age (P<0.001) were significant for worse outcome.
Conclusions:
Multimodality treatment remains standard for localized ESOS, with indications for neoadjuvant therapy less clear. Larger tumor size and older age were prognostic of poorer DFS.
Key Words: extraskeletal osteosarcoma, multimodality therapy, outcome, soft-tissue sarcoma, Rare Cancer Network
Osteosarcoma is a common primary bone cancer which is invasive and arises from mesenchymal cells. It produces osteoid, which is capable of invading local tissue structures and causing metastasis.1 Extraskeletal osteosarcomas (ESOS) are a rare variant of osteosarcoma located in soft tissue and organs which are not extensions of a primary bone osteosarcoma.2 It represents <1% of all de novo sarcomas, and is historically believed to be an aggressive tumor connoting a poor prognosis.3
Researchers have long known that ESOS behaves differently than its primary counterpart of osseous origin. ESOS seems to affect more elderly patients, and it does not respond well to chemotherapy. Local and distant failures are common.4 However, due to the rarity of ESOS, there have been limited advances in trimodality therapies or new biological agent testing. It has been estimated that no more than 350 cases have been recorded,5 with most being limited to single case reports or small case series. The purpose of this project is to present a recent and relatively large series of ESOS and their associated treatment and survival outcome.
MATERIALS AND METHODS
The Rare Cancer Network (RCN) is a consortium of international oncology investigators dedicated to advancing the knowledge of care for patients with rare tumors.6 A project was initiated to study the collective multi-institutional experience of ESOS. Inclusion criteria were confirmed pathologic diagnosis and a minimum of 6-month follow-up after treatment completion. Pathology was reviewed in each case to reconfirm the diagnosis of ESOS. The local medical records were reviewed in full, and anonymized data were sent to 1 author (T.T.S.) for further analysis. Every effort has been made to protect the integrity and confidentiality of the collected data. Patient and tumor characteristics and multimodality treatment information were included. If ESOS originated from an organ site, the exclusion of carcinoma as a diagnosis had to be confirmed. Clinically, the sequence and choices of surgery, chemotherapy, and radiotherapy were determined according to each hospital’s local policy.
Thirty-seven eligible patients were analyzed. The patients were diagnosed between 1998 and 2010, with an additional long-term survivor who was initially diagnosed in 1972. All patients received definitive combined modality therapies, including surgery, radiotherapy, or chemoradiotherapy, except for 4 patients with initially metastatic disease. The patients with metastases at diagnosis were all included in the overall survival (OS) but not in the disease-free survival (DFS) analyses. DFS, calculated for the 33 patients who initially presented with nonmetastatic ESOS, was calculated from the date of diagnosis to the date of last follow-up or local/metastatic relapse, whichever was earliest. OS was calculated from the date of diagnosis to the date of last follow-up or death from any cause. The work-up of studied patients included a complete history and physical, laboratory examination, computed tomography, and/or magnetic resonance imaging, and tissue biopsy. For patients who experienced relapse, salvage therapy was given and information collected. This study was approved by the Institutional Review Board at all participating institutions.
Data Analysis
Descriptive statistics were used to summarize patient, tumor, and treatment characteristics. DFS and OS plots were generated using the Kaplan-Meier method.7 Log-rank and Cox score P values were used. Cox proportional hazards models were used to obtain likelihood ratio P values for univariate and multivariate significance. Multivariate models were selected using stepwise models and were verified using backward elimination. Univariate and multivariate differences were considered significant if the P value was <0.05 using 2-tailed t tests. All statistical analysis was performed using JMP 9.0.1 (SAS Institute Inc., Cary, NC).
RESULTS
Characteristics of Patients and Tumors
Through RCN, clinical data of 37 eligible patients with ESOS from 4 institutions were reviewed. Table 1 summarizes patient characteristics and demographics along with tumor information at initial diagnosis. The median age of the 37 patients was 55 years (range, 13 to 81 y). Twenty-one (57%) patients were male. The histology included chondroblastic and fibroblastic types. Thirty-three (89%) patients had osteosarcoma in a soft-tissue site, and 4 (11%) had osteosarcoma in non–soft-tissue sites (hard palate, ethmoid sinus, and breast). Previous radiotherapy is a known risk factor for sarcoma as a secondary malignancy, and 5 (14%) patients had prior radiation to the pelvis, thigh, thorax, or retroperitoneum.
TABLE 1.
Characteristics of 37 Patients With Extraskeletal Osteosarcoma

The most common site for soft-tissue ESOS was the lower extremity (14 patients [38%]), followed by intrapelvic area and retroperitoneum (13 patients [35%]). Three (8%) patients had ESOS in the thorax, and 2 (5%) had it in the upper extremities. A 4-grade system in pathologic grading was used; of the 37 patients, most (31 [84%]) had high-grade (grade 3/4) ESOS (Table 1). Tumors from 5 (14%) of the 37 patients were evaluated as intermediate grade (grade 2). There were no low-grade tumors, although in 1 (3%) case the grade was unknown. Using the American Joint Committee on Cancer staging system (7th ed), we classified 5 (14%) of the 37 cases as stage II; 28 (76%) as stage III; and 4 (11%) as stage IV.
The median follow-up was 45 months, with 6 (16%) local and 11 (30%) distant recurrences as first event, respectively. For patients who were still alive at follow-up, the median follow-up was 80 months (quartiles, 62 to 96 mo; with or without disease). Including the 4 stage IV cases (patients who initially presented with metastases), 16 (43%) patients had died at follow-up, including 13 (35%) who died with disease.
Treatment Characteristics
Table 2 summarizes the treatment of the 37 patients. Most patients (29 [78%]) had tumors >5 cm clinically; 14 (38%) of these patients had tumors >10 cm. Fourteen (42%) of 33 patients received neoadjuvant chemotherapy, with 3 partial responses and 2 complete responses at the time of surgery. Of 33 patients, 28 (85%) underwent curative surgery. None of the metastatic patients received surgery.
TABLE 2.
Treatment of 37 Patients With Extraskeletal Osteosarcoma

Only 3 (19%) of 16 evaluable surgically evaluable specimens had tumor necrosis of >90%. Sixteen (48%) patients received external beam radiotherapy, often concurrent with chemotherapy neoadjuvantly; adjuvant chemotherapy was given to 13 (39%) patients. Six patients received neoadjuvant radiation therapy (median 50.0 Gy; range, 41.4 to 50.4 Gy). Ten patients received adjuvant radiation therapy (median, 53.7 Gy; range, 30.0 to 70.0 Gy). No patients received RT alone. The neoadjuvant chemotherapy regimens were: ifosfamide and doxorubicin (n=3); ifosfamide and doxorubicin alternating with cisplatin and doxorubicin (n=1); ifosfamide, mitomycin/methotrexate, doxorubicin, and cisplatin (n=6); and mitomycin C, doxorubicin, and cisplatin (n=1). The adjuvant chemotherapy regimens were: doxorubicin alone (n=2); ifosfamide and etoposide (n=2); ifosfamide, mitomycin/methotrexate, doxorubicin, and cisplatin-based (n=8); and doxorubicin and cisplatin (n=3). Gemcitabine-based chemotherapy was the most common salvage therapy and appeared effective as a second-line regimen. The median prescribed radiation dose was 50.4 Gy (range, 40 to 70 Gy). The significant heterogeneity in chemotherapy regimens limits the ability to assess the efficacy of chemotherapy in these patients.
Survival Analysis
The median follow-up was 45 months. For our cohort of patients with ESOS, Figures 1A and B show the Kaplan-Meier plots for OS and DFS, respectively. The DFS analyses excluded 4 patients who had metastatic disease at presentation. At the end of the follow-up period, 20 (55%) patients were alive, and 13 (43%) remained disease free. Median OS has not been reached yet; 75% and 60% of the patients were alive at 26 and 54 months, respectively. At 80 and 120 months, 58% and 52% of the patients were alive, respectively. When the 4 patients with metastases at presentation were excluded, the median DFS for 33 patients was estimated to be 90 months. Of these 33 patients, 65% and 57% remained disease free and alive at 20 and 40 months, respectively. In patients with nonmetastatic disease, 27 (82%) of them had known local control (LC) status in follow-up. The 1-year LC rate was 88% (95% confidence interval, 68% to 95%). Both of the actuarial 3- and 5-year LC rates were 80%. On univariate analyses, LC was not associated with any RT (P=0.17), neoadjuvant RT or chemoradiation (P=0.95), adjuvant RT (P=0.15), or surgery (P=0.63).
FIGURE 1.
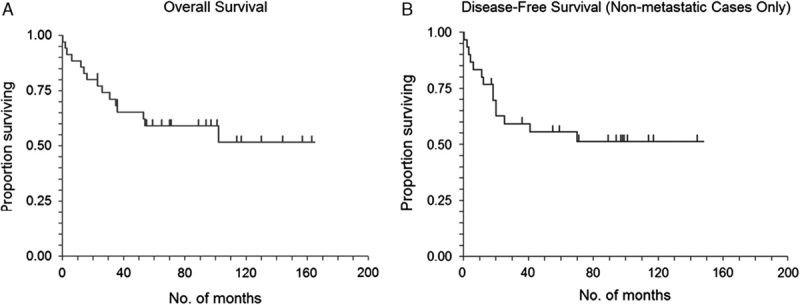
Kaplan-Meier survival graphs. Overall survival of all patients with extraskeletal osteosarcoma (A). Disease-free survival of patients with extraskeletal osteosarcoma who initially presented with localized (nonmetastatic) disease (B).
Table 3 summarizes the univariate and multivariate analyses for OS in all patients. Univariate analysis showed that poor OS was related to primary size >10 cm (P=0.002), no surgery (P<0.001), and older age at diagnosis (P=0.002). In the final multivariate analysis, pathologic size and age at diagnosis were dropped as variables in the best-fitting Cox model. Primary size >10 cm (P=0.005) remained prognostic for OS in the multivariate Cox model.
TABLE 3.
Summary of Univariate and Multivariate Analyses for Overall Survival
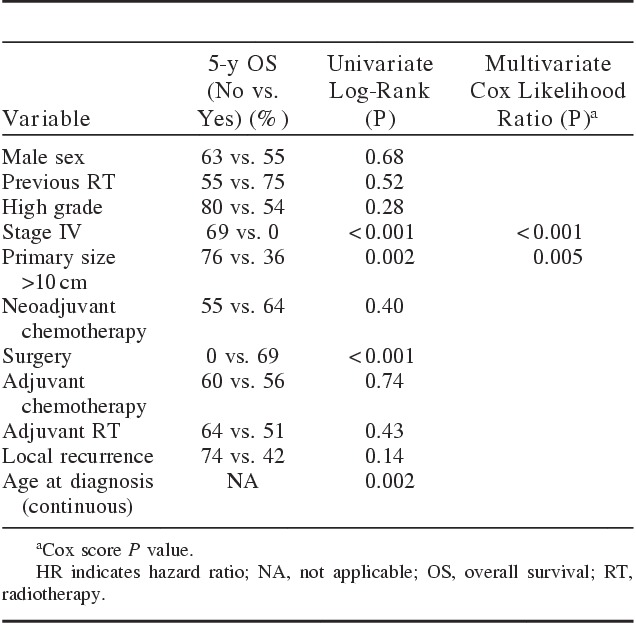
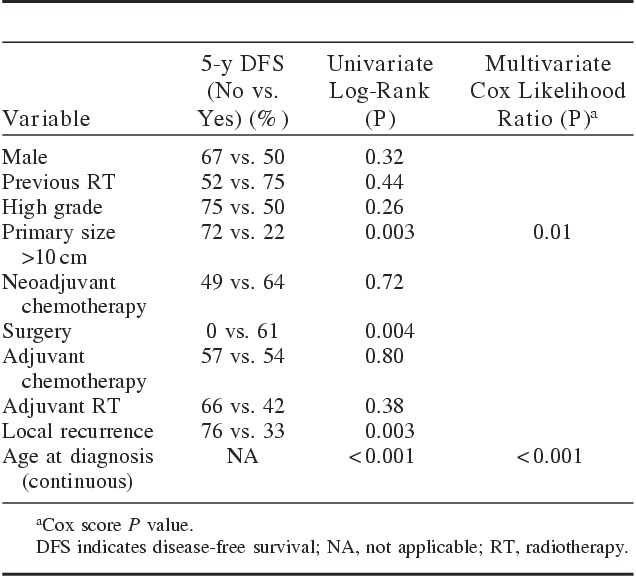
Table 4 shows the univariate and multivariate analyses for the 33 patients with initially nonmetastatic disease. For patients without metastases, univariate analysis showed that DFS was related to primary size >10 cm (P=0.003), surgery (P=0.004), local recurrence (P=0.003), and age at diagnosis (P<0.001). In multivariate analysis for DFS, primary size >10 cm (P=0.01) and age (P<0.001) remained significant. Similar to the findings of the OS analysis, the DFS analysis showed older age to be associated with poor DFS.
TABLE 4.
Summary of Univariate and Multivariate Analyses for Disease-free Survival
DISCUSSION
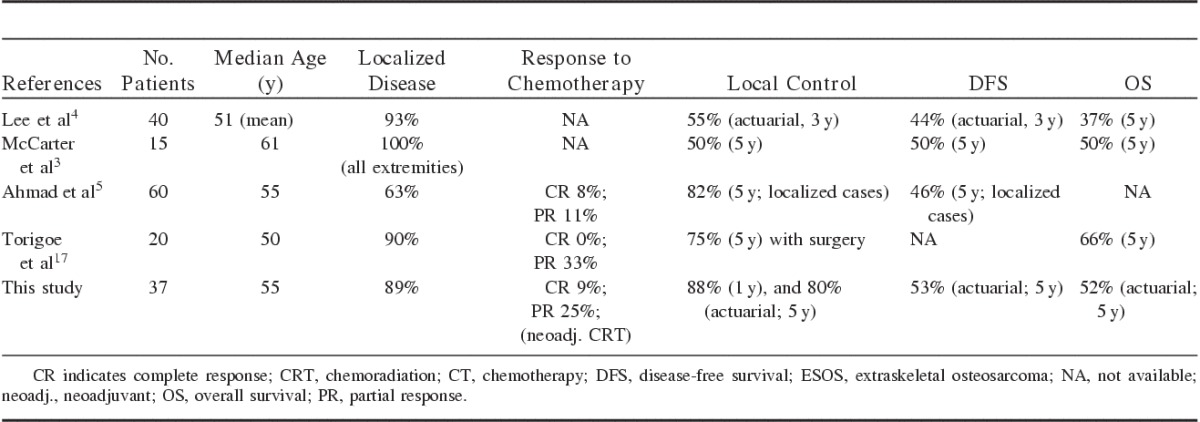
Researchers2,8 have long recognized the clinical identity of the osteosarcoma variant that exists in extraosseous locations. However, due to the rarity of this tumor, most publications have been limited to case reports only.8–14 Pediatric patients have also been reported, for example, in the soft thigh tissue of a 6-year-old boy15 and a high-grade ESOS in the breast of a 16-year-old female.16 Several larger studies have been reported,3–5,17,18 to which the results of our study in this series are comparable (Table 5).
TABLE 5.
Outcome Summary of Previous ESOS Studies
The use of surgery is a strongly statistically significant univariate for improved OS and DFS in our analysis. The RCN group believes that multimodality therapy remains important in the treatment of ESOS, which is also a common choice of practice in our current patient series. Although the use of RT and surgery were not statistically significant in our variate analyses for LC, the interpretation of these results are limited due to the limited number of ESOS cases we could find regarding this very unusual variant of osteosarcoma.
In contrast, indeed, the use of radiotherapy and chemotherapy for ESOS had been questioned before, especially in the older studies. In 1995, Lee et al4 from Mayo Clinic reported one of the earliest series of 40 patients with ESOS. The reported 5-year survival was 37%, and the size of the initial lesion did not correlate with longer survival. McCarter et al3 also reported their experience with 15 ESOS patients, most of whom underwent surgical resection. These authors concluded that the use of adjuvant chemotherapy or radiotherapy did not affect survival in ESOS patients, however, this finding could be related to the small series reported in the literature on ESOS. Ahmad et al5 reported the outcomes of 60 consecutive patients with ESOS. Their 5-year LC rate was excellent at 82% for localized cases. They also reported a respectable rate of response to chemotherapy, with 19% of their patients achieving a complete or partial response to neoadjuvant chemotherapy. Their chemotherapy was doxorubicin-based, which was similar to our regimens. In 2007, the Japanese Musculoskeletal Oncology Group17 reported on a 20-patient ESOS series. Their 5-year LC was 75% with surgery, which resulted in a 66% five-year survival. Most (75%) of their patients received doxorubicin and/or cisplatin-based chemotherapy together, and 33% achieved partial response. Although toxicities from these aggressive chemotherapy regimens were not reported, no grade 5 toxicity (death) was found. The use of chemotherapy, especially for high-grade sarcoma and ESOS, remains a subject for further exploration.
The limitations of this study included its small number of patients, retrospective design, and heterogeneity of treatments across hospitals and continents. The strength of our study was the pooling of cases from both US and international medical institutions, which added additional insights to the nature and treatment of this rare type of osteosarcoma. Every effort was made for the clarification of these selected patients’ treatment and their data if questions arose. Because of the low incidence of ESOS, no prospective study has been conducted; as a result, retrospective studies remain an important tool for clinical investigation. Our series has added to and updated the current literature in the treatment and outcome of ESOS in the relatively modern period of era investigated.
CONCLUSIONS
The survival outcome in this international cohort of patients with ESOS was similar to those reported in patients with osteogenic sarcomas. Stage IV at diagnosis and large primary tumor size were strong prognostic factors for poor OS, whereas older age and larger primary tumor size predicted poorer DFS. A multimodality treatment approach remains standard in patients with localized disease; however, the indications for neoadjuvant therapy are less clear for ESOS. Future research for novel chemotherapeutic agents and safe dose-escalation radiation techniques for both systemic controls and LCs, respectively, may result in breakthrough treatments for ESOS and osseous sarcomas in general.
Footnotes
The authors declare no conflicts of interest.
REFERENCES
- 1.Meyers PA, Gorlick R. Osteosarcoma. Pediatr Clin North Am. 1997;44:973–989. [DOI] [PubMed] [Google Scholar]
- 2.Sordillo PP, Hajdu SI, Magill GB, et al. Extraosseous osteogenic sarcoma. A review of 48 patients. Cancer. 1983;51:727–734. [DOI] [PubMed] [Google Scholar]
- 3.McCarter MD, Lewis JJ, Antonescu CR, et al. Extraskeletal osteosarcoma: analysis of outcome of a rare neoplasm. Sarcoma. 2000;4:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JS, Fetsch JF, Wasdhal DA, et al. A review of 40 patients with extraskeletal osteosarcoma. Cancer. 1995;76:2253–2259. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad SA, Patel SR, Ballo MT, et al. Extraosseous osteosarcoma: response to treatment and long-term outcome. J Clin Oncol. 2002;20:521–527. [DOI] [PubMed] [Google Scholar]
- 6.Patel A, Ozsahin M, Mirimanoff R-O, et al. The Rare Cancer Network: achievements from 1993 to 2012. Rare Tumors. 2012;4:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 8.Mirra JM, Fain JS, Ward WG, et al. Extraskeletal telangiectatic osteosarcoma. Cancer. 1993;71:3014–3019. [DOI] [PubMed] [Google Scholar]
- 9.Mavrogenis AF, Papadogeorgou E, Papagelopoulos PJ. Extraskeletal osteosarcoma: a case report. Acta Orthop Traumatol Turc. 2012;46:215–219. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura T, Matsumine A, Nishimura K, et al. Extraskeletal subcutaneous osteosarcoma of the upper arm: a case report. Oncol Lett. 2011;2:75–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uccello M, Malaguarnera M, Giordano M, et al. A large calcified retroperitoneal extraskeletal osteosarcoma with consequent bilateral hydronephrosis. Eur Rev Med Pharmacol Sci. 2012;16:977–982. [PubMed] [Google Scholar]
- 12.Maheshwari V, Farhan AS, Adreena KA. Extraskeletal osteogenic sarcoma: a rare entity. Indian J Pathol Microbiol. 2008;51:56–57. [DOI] [PubMed] [Google Scholar]
- 13.Matono R, Maruyama R, Ide S, et al. Extraskeletal osteosarcoma of the pleura: a case report. Gen Thorac Cardiovasc Surg. 2008;56:180–182. [DOI] [PubMed] [Google Scholar]
- 14.Salamanca J, Dhimes P, Pinedo F, et al. Extraskeletal cutaneous chondroblastic osteosarcoma: a case report. J Cutan Pathol. 2008;35:231–235. [DOI] [PubMed] [Google Scholar]
- 15.Liu ZJ, Zhao Q, Zhang LJ. Extraskeletal osteochondroma near the hip: a pediatric case. J Pediatr Orthop B. 2010;19:524–528. [DOI] [PubMed] [Google Scholar]
- 16.Zils K, Ebner F, Ott M, et al. Extraskeletal osteosarcoma of the breast in an adolescent girl. J Pediatr Hematol Oncol. 2012;34:e261–e263. [DOI] [PubMed] [Google Scholar]
- 17.Torigoe T, Yazawa Y, Takagi T, et al. Extraskeletal osteosarcoma in Japan: multiinstitutional study of 20 patients from the Japanese Musculoskeletal Oncology Group. J Orthop Sci. 2007;12:424–429. [DOI] [PubMed] [Google Scholar]
- 18.Alonge TO, Obamuyide HA, Ogun GO. Extraosseous osteosarcoma in Ibadan: case series over a 20-year period. Rare Tumors. 2009;1:6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]