

Abstract
The evolutionary trace (ET) is the single most validated approach to identify protein functional determinants and to target mutational analysis, protein engineering and drug design to the most relevant sites of a protein. It applies to the entire proteome; its predictions come with a reliability score; and its results typically reach significance in most protein families with 20 or more sequence homologs. In order to identify functional hot spots, ET scans a multiple sequence alignment for residue variations that correlate with major evolutionary divergences. In case studies this enables the selective separation, recoding, or mimicry of functional sites and, on a large scale, this enables specific function predictions based on motifs built from select ET-identified residues. ET is therefore an accurate, scalable and efficient method to identify the molecular determinants of protein function and to direct their rational perturbation for therapeutic purposes. Public ET servers are located at: http://mammoth.bcm.tmc.edu/.
Keywords: Evolutionary trace, Protein design, Protein engineering, Function annotation, Phylogenomics, Protein–protein interaction
1. Introduction
1.1. Basics of Evolutionary Trace: Phylogenetic Residue Variation
The evolutionary trace (ET) is a phylogenomic method to identify important amino acids in protein sequences. The approach conceptually mimics experimental mutational scanning: Whereas in the laboratory a sequence residue is deemed important when its mutation changes the response of an assay, ET infers that a residue is important when its variations during evolution correlate with major divergences (1, 2). Thus, ET aims to measure the impact of a residue not by its conservation or through its co-variations, but rather by its associated evolutionary changes and the functional perturbations and adaptation that they presumably represent.
The ET approach to measure the correlation between residue and phylogenetic variations is still under refinement. But the basic hypothesis is that residues that vary among widely divergent branches of evolution are more likely to have a larger functional impact than other residues that vary even among closely related species (see Fig. 1). Taking initially an absolute view of variation patterns (1), the ET rank ri of sequence residue i in a query protein was:
Fig. 1.
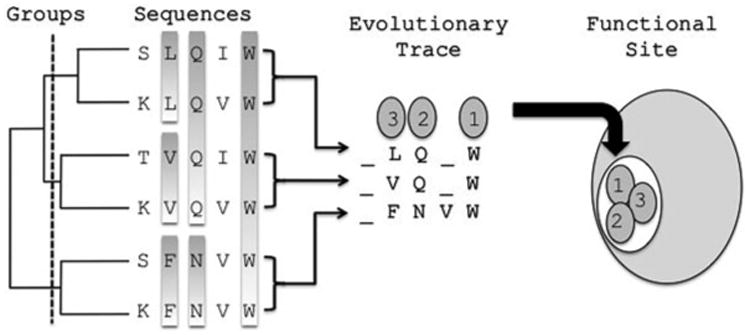
The Evolutionary Trace method. The proteins making up the multiple sequence alignment are divided into groups based on the phylogenic tree. Each group has a representative sequence with the invariant residues. The ET method extracts the relative evolutionary importance of the residues in example where the top ranked residues are marked 1, 2 and 3. These residues are then mapped onto the protein structure in order to visualize functional site.
| (1) |
where the summation is over the phylogenetic tree nodes (total of N – 1 branches); N is the number of homologs in the multiple sequence alignment. The value of δn is equal to 0 if residue position i is invariant within the sequences making up node n, while δn is equal 1 otherwise. The exact magnitude of ri is less important than its relative percentile rank compared to all residues in the protein: those with smaller percentile ranks being considered more important. In practice, (1) ranks best the sequence positions that vary among the most evolutionary divergent branches and that are also invariant within small branches of closely related species.
Following this scheme, top-ranked ET residues (or ET residues for short, usually defined as those residues ranked in the top 30th percentile) can be singled out in a sequence or structure. As expected, completely invariant residues are the most important and highly variable one tend to be least so. However, top-ranked residues can be surprisingly variable as long as these variations are between rather than within large branches. Conversely, some relatively invariant amino acids can be ranked poorly if the variations they do exhibit are within small evolutionary branches. The phylogenetic tree therefore allows ET to infer which patterns of variations are more or less important. Moreover, the use of the tree also naturally takes into account the bias due to overrepresentation of some branches, a difficult aspect for conservation or co-variation approaches.
In practice, ET residues have remarkable structural and functional properties:
They cluster together spatially in the protein structure (3)
These clusters map out on the protein surface possible functional sites for catalysis or ligand binding (4)
Internal clusters of ET residues presumably form the folding core of the protein, and, in some cases, play a critical role in allosteric regulation and specificity (5)
Mutations directed to ET residues will alter function in a variety of ways (6–8)
Mimicry of ET residues leads to peptides with functional properties (9)
And in silico mimicry of top-ranked ET residues identifies functional similarity (10, 11)
For example, this early version of ET detected functional residues and directed mutational studies into the molecular basis of G protein signaling (12–14). One hundred mutations of the Galpha-protein confirmed prior ET predictions of binding sites to the G beta gamma subunits and to the G protein-coupled receptor (15). Likewise, ET clusters of evolutionary important residues in the regulators of G protein signaling (RGS) were subsequently confirmed—one at an RGS-Galpha binding interface and another that mediates cGMP phosphodiesterase (PDE) interactions (13, 14). Moreover, these early studies ET also guided the successful transfer of function between RGS7 and RGS9 by mutationally swapping a few, select ET residues. These results suggested therefore that ET could identify a protein's binding sites and its key residues.
1.2. ET Refinements: Phylogenetic-Entropy Hybrid and Clustering z-Score
A number of refinements were added to the basic ET algorithm to increase its robustness. One issue addressed was the fact that (1) leads to ET ranks that are over-sensitive to errors, gaps, insertions, deletions and polymorphisms or natural variations among sequence. Each of these may break the perfect patterns that ET searches for, namely, variations between branches but invariance within them.
First, the Shannon Entropy (16) was introduced to measure invariance within the individual branches. This led to a hybrid entropy-phylogenetic method (17) called the real-value ET (rvET) because it produces absolute ranks that are not whole integers. By contrast, the original ET method and (1) yields integer ranks and is now referred to as integer-value ET (ivET).
To be clear, the Shannon Entropy, si, for a given residue position i is:
| (2) |
where fia is the frequency that an amino acid type, a, appears in the column containing residue position i. This Shannon Entropy is first calculated for the entire alignment, and then for every subsequent node defined by the phylogenetic tree. Finally, the rank ρi of residue i is:
| (3) |
where fia is the frequency of the amino acid of type a within the sub-alignment of group g. The number of possible nodes in the evolutionary tree is (N – 1) where N is the number of sequences in the alignment. The nodes in the phylogenetic tree are numbered in the order of increasing distance from the root. A key achievement of rvET (thereafter simply ET) is that it requires little manual curation, and thus lends itself to large-scale automation and allows for web server application.
A second important improvement quantified the notion of ET residue clusters (1, 2). Studies on numerous proteins showed that ET clusters were common and statistically significant (3), then that they significantly overlapped functional sites (4), and finally, that the extent of clustering was predictively correlated with the extent of overlap (18). In other words, the clustering z-score is a measure of ET quality such that it can be maximized in order to optimize functional site predictions (19–21).
To derive the clustering z-score, the structure provides an adjacency matrix between residues: A matrix element Aij is equal to 1 if two amino acids (labeled i and j) are within 4 Å of each other and equal to zero otherwise. If a residue meets a given ET threshold of importance, the parameter Si = 1. If that residue i does not meet this importance cut-off, then Si = 0. With these definitions, the cluster weight at a particular importance threshold is
| (4) |
where (j − i) is a weighting function that favors residues that are near in structure but far in sequence. Finally, the clustering z-score is determined, as usual:
| (5) |
The average, 〈w〉, and standard deviation, σ, in the ensemble of random residue choices are found through repeated sampling or analytically (18).
These improvements were experimentally tested in different proteins through a number of protein engineering studies that included: rewiring functional specificity (22), separating functions (6), designing of peptide inhibitors and redesigning allosteric specificity (5) (see Notes 1–4).
1.3. ET Optimization and Future Directions
A third generation of improvements originates from the fact that the clustering among top-ranked residues can be treated as a measure of ET quality. The greater the clustering z-scores the better the “fitness” among the selection of sequences making up the alignment, the phylogenetic tree and the 3D structure of the protein. This held true when extended for selecting structures among a set of decoy models of protein folds where the structures closer to native (18) were more likely to be chosen. This idea was also extended in order to select the most relevant sequences for ET analysis. Specifically, a Metropolis Monte Carlo algorithm was tested in 50 diverse proteins to choose sequences that maximized the clustering z-scores. The greater these z-scores, the better the clusters predicted functional sites (19). Another and structure-free quality measure, Rank Information, can likewise identify problematic “misfit” sequences during analysis (23). More recently, multiple ET quality measures were formally defined, such that maximizing their value optimizes the prediction of functional sites and annotations (21). Together these studies further confirm a quantitative relationship among evolutionary pressure (the ET rank), the protein fold and functional site locations; and they point to a common feature of ET quality: the rank distribution that best reflects evolutionary history and functional pressures appear to maximize “rank continuity,” namely the similarity of ET ranks among structurally neighboring residues within the structure (21).
1.4. Large Scale Validation: Protein Function Annotation
ET was also validated on a large scale in the context of protein function prediction. This application is motivated by Structural Genomics (SG) which solves many protein structures that cannot be annotated by homology-based annotation transfer (24). Since typically a few residues are essential for binding or catalytic activities it may be possible instead to rely on local structural similarities (25): different structures may perform similar bio-chemical function if they share a common spatial organization of experimentally verified functional motifs (26) or, lacking those, key functional residues as defined by ET.
A series of technical studies developed these ideas into an Evolutionary Trace Annotation (ETA) pipeline to predict the function of novel protein structures. ET rankings proved useful to define small structure-function motifs called 3D-templates (27), to identify meaningful geometric and evolutionary matches of these templates to other protein structures based on reciprocity (10), and voting plurality (28) in order to infer function in enzymes and non-enzymes alike (10, 11). ETA was extensively benchmarked; for example, its positive predictive value was 93% (10) in 1218 SG enzymes (whose functions were described the first three digits of the Enzyme Commission classification, EC numbers). ETA matches further create a network of local structural and evolutionary similarities among the entire structural proteome, in which edges between protein nodes indicate reciprocal ETA matches (11), and such that a diffusion algorithm can then transfer annotations globally over the entire network. Every combination of protein and function receives a confidence score, and the highest one defines the functional prediction. This competitive annotation diffusion strategy yields predictions at the most detailed (fourth) EC level. For example, false positives fell fourfold, at 97% sensitivity, against a recent method (29). On a large-scale SG set, accuracy rose 6% and false positives fell twofold at 65% coverage, compared to ETA.
In practice, ETA predictions are being validated experimentally (30). For example, ETA suggested carboxylesterase activity (EC3.1.1.1) for a bacterial protein of unknown function (Uniprot accession Q99WQ5, gene name SAV0321, PDB 3h04 chain A) found in a vancomycin resistant strain of the bacteria Staphylococcus aureus (31). The ETA annotation was based on template matches to three other carboxylesterases with only 10% to 13% sequence identity to the query. In vitro biochemical assays then showed that SAV0321 has carboxylesterase activity at a level similar to the positive control.
This work is notable for two reasons. First, it improves function discovery in proteins of known structure by formulating reliable hypothesis for efficient experimental validation. This supports the general aim of SG, which is to inform on function through structural knowledge. Second, since ET ranks, the 3D templates and matches they define are at the heart of ETA, it provides a direct and proteomic scale test of ET identification of key functional residues.
2. Methods
2.1. Functional Site and Functional Residue Predictions by Evolutionary Trace
To ensure that only the most relevant proteins are analyzed, a custom database of sequences removes from NCBI's non-redundant protein sequence database any sequence with “synthetic construct,” “artificial,” “fragment” and “partial” in the sequence header.
To identify homologs to the protein being traced, a BLAST (BLAST Local Alignment Search Tool) (32) search is done on the custom database. Typically, the default number of homologs is limited to 500 sequences and the maximum E-value threshold is set to 0.05 (see Note 5).
Sequences with less than half the length of the query protein are eliminated, as are those with greater then 98% or less than 28% sequence identity (see Note 6).
A ClustalW alignment is generated (www.clustal.org) with default parameters set at gap open penalty (10) and gap extension penalty (0.05). For the ET web servers (see Note 7). The current ET code accepts MSF format.
The alignment is rescanned for sequences that are too short. After these are removed, the remaining sequences are then aligned again.
To generate an evolutionary tree, a pairwise sequence similarity matrix is constructed and the UPGMA method is applied. Any phylogenetic tree that represents the family of proteins can be used as input into the ET code.
Integer or rvET ranks are computed as described above: sub-alignments that correspond to nodes in the evolutionary tree are formed and (1), or (2) and (3) are applied (see Note 8).
If a structure is provided: structural clusters of highly ranked residues in the query structure are identified and their statistical significance is measured as described in Subheading 3.2. These clusters indicate likely functional hot spots and provide a suitable hypothesis to direct mutational studies in order to identify functional regions and determinants and drug target sites.
Direct visualization of ET results can be obtained via two programs: the ET Viewer and the PyETV application (33). ET servers and viewers are available at http://mammoth.bcm.tmc.edu/ETserver.html.
2.2. Protein Function Prediction by Evolutionary Trace Annotation
rvET is applied to a query protein structure of unknown function to rank the evolutionary importance of its residues.
The first cluster with ten evolutionarily important surface residues is identified. A residue is defined to be on the surface if its solvent accessibility is at least 2 Å (2) as calculated by DSSP (34).
The six most evolutionarily important residues in that cluster define the query template. Their alpha carbon coordinates define the template geometry. If ties arise between candidate residues, those closest to a point halfway between the center of mass of the growing template are chosen.
The template is allowed to vary in keeping with the side chain variations found in multiple sequence alignment used by ET, provided an amino acid appears at least twice.
The templates are matched to target proteins of known structure and function (the current target set is 2008PDB90 (24)). Functions are described by the Enzyme Commission (EC) numbers (35) or Gene Ontology (GO) molecular terms (36). Geometric matches are obtained hierarchically, employing a distance cutoff of 2.5Å (28). Finally, a root-mean-square-distance (RMSD) is calculated.
It is important to filter nonspecific geometric matches. First, only those with RMSD below 2Å are considered for further analysis. Second, a support vector machine (SVM) chooses matches that are both geometrically and evolutionarily significant (it combines RMSD and evolutionary similarity between the template and the matched sites in the target structures). Third, these steps are repeated by reversing the role of the query and of the target structure in order to assess reciprocity: reciprocal ETA matches between two protein structures are much less likely to be due to chance. Fourth, all-against-all matches enable to tally how often a query matches to different proteins with the same function. A plurality rule is then applied to transfer to the query the one function annotation that is matched the most often. In the case of a tie, no prediction is suggested.
For GO annotations, ETA takes into account all known GO terms and their parent terms for each match. ETA votes at each GO depth in such a way that the most voted or tied terms are considered to be predictions. Voting continues until a GO term has no more child terms. Once a term or terms are considered to be predictions, their child terms are also suggested as predictions. In the voting procedure, self-matches are excluded.
An ETA server is available at http://mammoth.bcm.tmc.edu/ETA
3. Tools
3.1. ET Servers
A summary of ET tools is reported in Table 1. There are a number of servers that provide ET results:
The first server (http://mammoth.bcm.tmc.edu/ETserver.html) requires the users to enter a PDB ID (e.g., 2phy). The web output includes links that launch ETV and PyMOL with which to view a structural mapping of every trace. This output also packages zipped versions of all the files used or generated by ET.
The Evolutionary Trace Report Maker is a second server (37), which produces a fully automated ET report in a pdf document (http://mammoth.bcm.tmc.edu/report_maker). It pools data on protein sequence, structure and elementary annotation from several sources, and adds to that background inference on functional sites and residues obtained from rvET. It requires either a Protein Data Bank (PDB) identifier or a UniProt accession number for a sequence. Report Maker utilizes HSSP alignments when available.
The “ET Wizard” server is accessible directly through the evolutionary trace viewer (ETV), launched separately in the “Utils” menu, and useful for generating user-controlled traces (see below).
Table 1. Available ET tools.
| Name/URL | Type | Purpose | Input | Output |
|---|---|---|---|---|
| Evolutionary Trace Results http://mammoth.bcm.tmc.edu/ETserver.html | Web server | Functional site prediction | PDB ID | ET analyses files |
| Evolutionary Trace Report maker http://mammoth.bcm.tmc.edu/report_maker | Web server | Functional site prediction | PDB ID or Uniprot accession number | PDF report, ET analyses files |
| Evolutionary Trace Viewer (ETV) http://mammoth.bcm.tmc.edu/traceview | Molecular viewer, Web application, Web server | Functional site prediction, visualization | ET analyses (.etvx file), PDB ID | 3D molecular graphics, ET analyses files, multiple sequence alignment, evolutionary tree |
| PyMOL ETV http://mammoth.bcm.tmc.edu/traceview/HelpDocs/PyETVHelp/pyInstructions.html | Molecular viewer | Functional site prediction, visualization | ET rank data, PDB, PyMOL scripts | 3D molecular graphics |
| Evolutionary Trace Annotation (ETA) server http://mammoth.bcm.tmc.edu/eta | Web server | Functional annotation | PDB ID | EC and GO annotations, 3D templates, PDB matches |
3.2. Evolutionary Trace Viewer: A Tool to Run ET and View Results
The ETV (38) (http://mammoth.bcm.tmc.edu/traceview) is a one-stop environment to run, visualize and interpret ET predictions of functional sites in protein structures. It is implemented in Java and runs across different operating systems utilizing Java Web Start Technology for self-installation.
A key ETV feature is an interactive molecular graphics display that reads in the results of an ET analysis in the form of an .etvx file. This file is selected in the “File” menu command: “Open ETV Results.” It produces a colored structural map of the ET rank of every protein residue. Evolutionary and functional hot spots become readily apparent in the form of structural clusters of top-ranked residues, and the statistical z-score of these clusters is shown. The threshold of percentile rank to color top-ranked residues can be adjusted by moving a slider (horizontal scrollbar) prominently shown on top of the graphics window, or a rainbow coloring over all residues is also available to display at once a heatmap of evolutionary importance.
A second feature of ETV is that the evolutionary tree used to compute the ET rank of every residue can be viewed: select “ET Tree” under the “View” menu.
Critically, an ET Wizard is integrated into ETV (under the “Utils” menu”) to let users launch customized ET analyses. The ET Wizard accepts either a PDB ID, or a PDB formatted file provided directly by the user as input. Users may then also choose to provide their own custom alignments or set of input sequences. Alternately, they can allow the ET Wizard to build its own alignments (see Note 9).
A database of pre-generated ET analysis results for all unique chains in the PDB is maintained and regularly updated.
3.3. PyMOL ETV: A High-Resolution ET Viewer for Protein Chains and Complexes
The ET Viewer (ETV) displays just one single chain at a time. Since protein–protein interactions are an emerging target for design and therapeutics, an alternative system was developed to trace multi-protein interfaces. This PyETV (for PyMOL Evolutionary Trace Viewer) (33) provides a high graphics quality interface to map evolutionary forces and identify functional sites in complexes.
The PyETV is a plug-in that builds on the popular and extensible PyMOL molecular graphics package (39). Information for its installation, and instructional videos, are available at http://mammoth.bcm.tmc.edu/traceview/HelpDocs/PyETVHelp/pyInstructions.html. PyETV is also integrated into the web server http://mammoth.bcm.tmc.edu/ETserver.html through web links to PyMOL scripts.
PyMOL (39) (www.pymol.org) is a versatile molecular graphics package developed by Bill DeLano to view, select, label, and perturb any number of structures or substructures (such as groups of atoms or residues) in many ways (e.g., cartoon, surface, stereo etc.). Moreover, it is easily extended with plug-ins—scripts that can add to PyMOL's user interface and can overlay complementary information to a protein structure, such as electrostatics maps.
Through the PyETV plug-in, any number of user-generated and pre-generated ET analysis results can be mapped to any number of structures and displayed in PyMOL. In particular, predicted biological assemblies from PISA (40) and ET analysis for each component in the assembly can be loaded directly through PyETV using the “Assembly” tab. As with ETV, PyETV provides a colored structural map of the importance of each residue in a protein.
3.4. Evolutionary Trace Annotation Server: Automated Function Prediction in Protein Structures Using 3D Templates
ETA analysis starts with the PDB code of the protein structure of unknown function, including a 1-digit chain identifier. Click “Submit.” An ET analysis then provides information on the evolutionary importance of each residue. If this ET analysis is cached, the server goes to step 2. If not, it launches automatically a new trace with default parameters. One may gain control over this process by uploading a custom ET analysis that was run before through the ET Wizard. Clicking “Browse” to locate such an ET file and “Upload” to submit it to the ETA server (http://mammoth.bcm.tmc.edu/ETA).
Next, the server predicts a functional site template by identifying a cluster of evolutionarily important residues on the surface of the protein, picking the six most important ones. It renders an image of the template. This template can be explored in depth by clicking on the image to download a PyMOL session file. The template may be customized if alternate choices of residues are of interest. Click “Submit Template” to continue with the analysis.
The server next identifies possible amino acid types for each template residue based on the multiple sequence alignment used by ET. Each unique combination is listed, along with the number of times it occurs in the alignment. Combinations may be turned on or off using their check boxes. Custom amino acid labels can also be added. Click “Find Matches” to begin the template search.
The results page contains GO and EC predictions based on reciprocal matches (highly reliable) and non-reciprocal matches (less reliable). The GO terms and EC numbers are hyperlinked to web pages containing more information about that GO term or EC number.
4. Notes
Rewiring functional specificity: Top-ranked residues were exchanged to rewire transcriptional specificity in evolutionary divergent helix-loop-helix proneural transcription factors from the frog and the fly, and vice versa (22).
Separating functions: Alanine mutations of ET-predicted functional residues confirmed predictions of new functional sites and led to selective loss of function in the Ku70/80 heterodimer. One site was found to be responsible for telomere maintenance and another site, that was structurally diametrically opposite and facing the centromere, was responsible for end-joining of double-strand DNA break repair (6).
Design of peptide inhibitors: Helical peptides were engineered to mimic ET-predicted sites composed mostly of solvent exposed helices. The top-ranked residues were left intact while the lesser-ranked amino acids were chosen to favor helix formation. These peptides disrupted in vitro binding among nuclear receptors (41) and, in another case, G protein-coupled receptor phosphorylation by G protein receptor kinase (9).
Redesigning allosteric specificity: ET residues in the transmembrane domain of Class A GPCRs (42) were targeted for mutations. Some selectively uncoupled beta-arrestin-mediated signaling from G protein-mediated signaling (43). Others rewired a dopamine receptor to become serotonin responsive not by altering ligand binding specificity, but rather by altering the response of the allosteric pathway to either ligands (5).
ET analysis can be done for any reasonable set of sequences. Typically 15–20 sequences are needed but this depends on the validity and diversity of the set. When structural information is known, HSSP alignments can also be an option.
The parameters for filtering sequences were optimized for better functional site prediction. They are often adjusted on a case-by-case basis, for example, when studying an entire family, it is important to ignore cut-offs like sequence identity.
For cases where homologues are close, the quicktree option in ClustalW dramatically decreases computational time.
In sequence analysis, gaps are treated as a 21st amino acid. This is simply a computational tool and has no relevance.
In the ET Wizard tool, the user can control the number of sequences to be included in the alignment, after a BLAST search, and the thresholds for acceptable sequence identity and sequence length.
Acknowledgments
The authors gratefully acknowledge grant support from the National Institute of Health through NIH-GM079656, NIH-GM066099, T90 DA022885, R90 DA023418, NLM 5T15LM07093, and of the National Science Foundation through NSF CCF-0905536.
References
- 1.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 2.Lichtarge O, Yamamoto KR, Cohen FE. Identification of functional surfaces of the zinc binding domains of intracellular receptors. J Mol Biol. 1997;274:325–337. doi: 10.1006/jmbi.1997.1395. [DOI] [PubMed] [Google Scholar]
- 3.Madabushi S, et al. Structural clusters of evolutionary trace residues are statistically significant and common in proteins. J Mol Biol. 2002;316:139–154. doi: 10.1006/jmbi.2001.5327. [DOI] [PubMed] [Google Scholar]
- 4.Yao H, et al. A Sensitive, Accurate, and Scalable Method to Identify Functional Sites in Protein Structures. J Mol Biol. 2003;326:255–261. doi: 10.1016/s0022-2836(02)01336-0. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez GJ, Yao R, Lichtarge O, Wensel TG. Evolution-guided discovery and recoding of allosteric pathway specificity determinants in psychoactive bioamine receptors. Proc Natl Acad Sci U S A. 107:7787–7792. doi: 10.1073/pnas.0914877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribes-Zamora A, Mihalek I, Lichtarge O, Bertuch AA. Distinct faces of the Ku heterodimer mediate DNA repair and telomeric functions. Nat Struct Mol Biol. 2007;14:301–307. doi: 10.1038/nsmb1214. [DOI] [PubMed] [Google Scholar]
- 7.Rajagopalan L, Pereira FA, Lichtarge O, Brownell WE. Identification of functionally important residues/domains in membrane proteins using an evolutionary approach coupled with systematic mutational analysis. Methods Mol Biol. 2009;493:287–297. doi: 10.1007/978-1-59745-523-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobayashi H, Ogawa K, Yao R, Lichtarge O, Bouvier M. Functional rescue of beta-adrenoceptor dimerization and trafficking by pharmacological chaperones. Traffic. 2009;10:1019–1033. doi: 10.1111/j.1600-0854.2009.00932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baameur F, et al. Role for the regulator of G-protein signaling homology domain of G protein-coupled receptor kinases 5 and 6 in beta 2-adrenergic receptor and rhodopsin phosphorylation. Mol Pharmacol. 77:405–415. doi: 10.1124/mol.109.058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward RM, et al. De-orphaning the structural proteome through reciprocal comparison of evolutionarily important structural features. PLoS ONE. 2008;3:e2136. doi: 10.1371/journal.pone.0002136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erdin S, Ward RM, Venner E, Lichtarge O. Evolutionary trace annotation of protein function in the structural proteome. J Mol Biol. 396:1451–1473. doi: 10.1016/j.jmb.2009.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onrust R, et al. Receptor and betagamma binding sites in the alpha subunit of the retinal G protein transducin. Science. 1997;275:381–384. doi: 10.1126/science.275.5298.381. [DOI] [PubMed] [Google Scholar]
- 13.Sowa ME, He W, Wensel TG, Lichtarge O. A regulator of G protein signaling interaction surface linked to effector specificity. Proc Natl Acad Sci U S A. 2000;97:1483–1488. doi: 10.1073/pnas.030409597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sowa ME, et al. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat Struct Biol. 2001;8:234–237. doi: 10.1038/84974. [DOI] [PubMed] [Google Scholar]
- 15.Lichtarge O, Bourne HR, Cohen FE. Evolutionarily conserved Galphabetagamma binding surfaces support a model of the G protein-receptor complex. Proc Natl Acad Sci U S A. 1996;93:7507–7511. doi: 10.1073/pnas.93.15.7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shenkin PS, Erman B, Mastrandrea LD. Information-theoretical entropy as a measure of sequence variability. Proteins. 1991;11:297–313. doi: 10.1002/prot.340110408. [DOI] [PubMed] [Google Scholar]
- 17.Mihalek I, Res I, Lichtarge O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J Mol Biol. 2004;336:1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 18.Mihalek I, Res I, Yao H, Lichtarge O. Combining inference from evolution and geometric probability in protein structure evaluation. J Mol Biol. 2003;331:263–279. doi: 10.1016/s0022-2836(03)00663-6. [DOI] [PubMed] [Google Scholar]
- 19.Mihalek I, Res I, Lichtarge O. Evolutionary and structural feedback on selection of sequences for comparative analysis of proteins. Proteins. 2006;63:87–99. doi: 10.1002/prot.20866. [DOI] [PubMed] [Google Scholar]
- 20.Mihalek I, Res I, Lichtarge O. A structure and evolution-guided Monte Carlo sequence selection strategy for multiple alignment-based analysis of proteins. Bioinformatics. 2006;22:149–156. doi: 10.1093/bioinformatics/bti791. [DOI] [PubMed] [Google Scholar]
- 21.Wilkins AD, Lua R, Erdin S, Ward RM, Lichtarge O. Sequence and structure continuity of evolutionary importance improves protein functional site discovery and annotation. Protein Sci. 19:1296–1311. doi: 10.1002/pro.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quan XJ, et al. Evolution of neural precursor selection: functional divergence of proneural proteins. Development. 2004;131:1679–1689. doi: 10.1242/dev.01055. [DOI] [PubMed] [Google Scholar]
- 23.Yao H, Mihalek I, Lichtarge O. Rank information: a structure-independent measure of evolutionary trace quality that improves identification of protein functional sites. Proteins. 2006;65:111–123. doi: 10.1002/prot.21101. [DOI] [PubMed] [Google Scholar]
- 24.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polacco BJ, Babbitt PC. Automated discovery of 3D motifs for protein function annotation. Bioinformatics. 2006;22:723–730. doi: 10.1093/bioinformatics/btk038. [DOI] [PubMed] [Google Scholar]
- 26.Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32:D129–133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kristensen DM, et al. Recurrent use of evolutionary importance for functional annotation of proteins based on local structural similarity. Protein Sci. 2006;15:1530–1536. doi: 10.1110/ps.062152706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kristensen DM, et al. Prediction of enzyme function based on 3D templates of evolutionarily important amino acids. BMC Bioinformatics. 2008;9:17. doi: 10.1186/1471-2105-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Redfern OC, Dessailly BH, Dallman TJ, Sillitoe I, Orengo CA. FLORA: a novel method to predict protein function from structure in diverse superfamilies. PLoS Comput Biol. 2009;5:e1000485. doi: 10.1371/journal.pcbi.1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venner E, Lisewski AM, Erdin S, Ward RW, Amin S, Lichtarge O. Accurate protein structure annotation through competitive diffusion of enzymatic functions over a network of local evolutionary similarities. PLoS One. 2010;12:e14286. doi: 10.1371/journal.pone.0014286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gill SR, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 33.Lua RC, Lichtarge O. PyETV: a PyMOL evolutionary trace viewer to analyze functional site predictions in protein complexes. Bioinformatics. 26:2981–2982. doi: 10.1093/bioinformatics/btq566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 35.International Union of Biochemistry and Molecular Biology Nomenclature Committee & Webb, E.C. Enzyme nomenclature 1992 : recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the nomenclature and classification of enzymes. Academic Press; San Diego: 1992. [Google Scholar]
- 36.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mihalek I, Res I, Lichtarge O. Evolutionary trace report_maker: a new type of service for comparative analysis of proteins. Bioinformatics. 2006;22:1656–1657. doi: 10.1093/bioinformatics/btl157. [DOI] [PubMed] [Google Scholar]
- 38.Morgan DH, Kristensen DM, Mittelman D, Lichtarge O. ET viewer: an application for predicting and visualizing functional sites in protein structures. Bioinformatics. 2006;22:2049–2050. doi: 10.1093/bioinformatics/btl285. [DOI] [PubMed] [Google Scholar]
- 39.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 40.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 41.Gu P, et al. Evolutionary trace-based peptides identify a novel asymmetric interaction that mediates oligomerization in nuclear receptors. J Biol Chem. 2005;280:31818–31829. doi: 10.1074/jbc.M501924200. [DOI] [PubMed] [Google Scholar]
- 42.Madabushi S, et al. Evolutionary trace of G protein-coupled receptors reveals clusters of residues that determine global and class-specific functions. J Biol Chem. 2004;279:8126–8132. doi: 10.1074/jbc.M312671200. [DOI] [PubMed] [Google Scholar]
- 43.Shenoy SK, et al. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]