

Abstract
Visible-light photoredox-catalyzed fragmentation of methyl N-phthalimidoyl oxalate allows the direct construction of a 1,4-dicarbonyl structural motif by a conjugate addition of the methoxycarbonyl radical to reactive Michael acceptors. The regioselectivity of the addition of this alkoxyacyl radical species to electron-deficient olefins is heavily influenced by the electronic nature of the acceptor, behavior similar to that exhibited by nucleophilic alkyl radicals.
Graphical Abstract

The normal reactivity of carbonyl compounds renders 1,3- or 1,5-dicarbonyl functionality much easier to incorporate into organic molecules than 1,4-dicarbonyl functionality.1 The 1,4-addition of acyl-anion equivalents to α,β-unsaturated carbonyl compounds is a general approach for constructing 1,4-dicarbonyl products. However, several steps are needed to introduce an alkoxycarbonyl group in this way.1 Transitionmetal catalyzed alkoxycarbonylation is a widely practiced and immensely important method to incorporate carbonyl functionality into alkenes;2 however the use of this chemistry to alkoxycarbonylate electron-deficient alkenes has not been widely developed.3 A potentially attractive approach for preparing γ-ketoesters would be the direct 1,4-addition of an alkoxycarbonyl radical to α,β-unsaturated carbonyl compounds.4 Although intramolecular additions of alkoxycarbonyl radicals to alkenes are well known and used productively to construct 5- and 6-membered lactones,5 there are only a few examples of synthetically useful bimolecular coupling reactions of alkoxycarbonyl radicals with alkenes.6 In these cases, the alkoxycarbonyl radical is generated by Fe- or Pd-catalyzed oxidation of carbazate precursors.
Computational studies suggest that alkoxycarbonyl radicals are less-nucleophilic than acyl radicals, leading them to be termed either as ambiphilic or in some contexts electrophilic radicals.7 These studies raise some concern about whether an alkoxycarbonyl radical would be sufficiently nucleophilic to add efficiently to an electron-deficient C–C π-bond. However, in our recent investigations on the generation of tertiary radicals from tertiary alkyl N-phthalimidoyl oxalate precursors, we observed that the intermediate alkoxycarbonyl radical formed from adamantanol precursor 1 reacted efficiently with methyl vinyl ketone to give γ-ketoester 2 (eq 1).8,9 As a result, we became interested in the possibility of using an oxalate precursor and visible-light photoredox catalysis to conveniently generate alkoxycarbonyl radicals in the context of their conjugate addition to α,β-unsaturated carbonyl compounds and related electron-deficient alkenes. Two potential precursors for producing alkoxycarbonyl radicals by visible-light photoredox catalysis would be alkyl N-phthalimidoyl oxalates8 or a salt of an alkyl hemioxalate (Scheme 1).10,11 For this method to be successful, β-scission of the alkoxycarbonyl radical B to give an alkyl radical must be slower than its reaction with the radical acceptor.12 The rate of decarboxylation of alkoxycarbonyl radicals is known to reflect the stability of the forming alkyl radical with the rate of decarboxylation of the tert-butoxycarbonyl radical estimated to be ~500 times faster than that of a primary alkoxycarbonyl radical.13 As the methoxycarbonyl radical would be expected to decarboxylate even more slowly, our studies focused on developing a convenient method to generate this carbon radical and surveying its reactivity with alkenes.
 |
(1) |
Scheme 1.

Two Potential Precursors of Alkoxycarbonyl Radicals
Methyl N-phthalimidoyl oxalate (4) was obtained by acylation of methanol with N-phthalimidoyl chlorooxalate (3) using a modification of a procedure developed earlier in our laboratory for the preparation of tertiary alkyl N-phthalimidoyl oxalates (Scheme 2).8 The reaction was carried out in the absence of DMAP at 0 °C to prevent formation of dimethyl oxalate. Additionally, the use of pyridine in place of Et3N led to more reproducible results. Phthalimidoyl oxalate 4 was not stable to silica gel chromatography; however, upon careful trituration it was isolated on multigram scale as a colorless solid in high yield and acceptable purity. Reagent 4 is stable to light and can be stored in the −20 °C freezer for prolonged periods of time without decomposition. Cesium methyl oxalate 6 was generated from commercially available methyl hemioxalate 5 upon reaction with 0.5 equiv of Cs2CO3 in water, followed by concentration to give 6 as a colorless solid.
Scheme 2.

Preparation of Methyl N-Phthalimidoyl Oxalate (4) and Cesium Methyl Oxalate 6
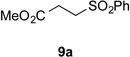
Using conditions optimized earlier for the coupling of tertiary radicals generated from related precursors with electrondeficient alkenes,8,10 the reaction of radical precursors 4 and 6 with phenyl vinyl sulfone (8a) was examined (Table 1). Coupling of N-phthalimidoyl oxalate 4 (1.5 equiv) with phenyl vinyl sulfone in the presence of 1.5 mol % of [Ru(bpy)3](PF6)2, 1.5 equiv of diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate (7), and 1 equiv of i-Pr2NEt·HBF4 in 1:1 THF/CH2Cl2 with irradiation at room temperature with low-intensity blue LEDs gave product 9a in 50% yield (Table 1, entry 1). In contrast, the reaction of methyl cesium oxalate 6 (1.5 equiv) with phenyl vinyl sulfone in the presence of 2 mol % of Ir[dF(CF3)ppy]2(dtbbpy)PF6, 10 equiv of water in 3:1 DME:DMF irradiated with 35 W blue LEDs provided adduct 9a in only 2% yield (Table 1, entry 2). As low yields were also obtained in further screening of the reaction of oxalate salt 6 with benzyl acrylate,14 we chose to focus on optimizing the coupling of phthalimidoyl oxalate 4 with acceptor 8a. The major by-product of the reaction of entry 1 was identified as the product of addition of the 2-tetrahydrofuryl radical to phenyl vinyl sulfone (~30%). To suppress this unwanted reactivity, a solvent screen was performed, which identified CH2Cl2 as the optimal solvent (entry 3). The choice of a polar aprotic solvent was important, as reactions run in nonpolar solvents such as benzene led to lower yields, likely because of the low solubility of Hantzsch ester 7 (entry 4). Employing alternative reductive quenchers such as 1,3-dimethyl-2-arylbenzimidazolines,15 or 2-phenylbenzothiazoline,16 led to greatly diminished yields of addition product 9a. The yield of 9a was improved substantially by increasing the amounts of phthalimidoyl oxalate 4 and Hantzsch ester 7 to 3 equiv (entry 5). Raising the temperature of the reaction proved to be detrimental (entry 6), whereas omission of the ammonium additive resulted in a slight increase in yield (entry 7). Finally, by varying the concentration of the reaction mixture, we were able to reduce the excess of the radical precursor 4 and the Hantzsch ester 7 from 3 to 2 equiv without compromising the isolated yield of 9a (entry 8). In the absence of visible light, no product was formed (entry 9), whereas reactions carried out in absence of the photocatalyst or for 6 h instead of 18 h led to greatly reduced formation of coupled product 9a (entries 10,11).17
Table 1.
Initial Studies and Reaction Optimization
 | |||||
|---|---|---|---|---|---|
| entrya | radical precursor (equiv) |
solvent (M) | t (°C) | 7 (equiv) |
yield (%)b |
| 1c | 4 (1.5) | 1:1 CH2Cl2:THF (0.1) | 23 | 1.5 | 50 |
| 2 | 6 (1.5) | 3:1 DME:DMF (0.1) | 40 | - | 2 |
| 3c | 4 (1.5) | CH2Cl2 (0.1) | 23 | 1.5 | 55 |
| 4c | 4 (1.5) | benzene (0.1) | 23 | 1.5 | 38 |
| 5c | 4 (3.0) | CH2Cl2 (0.1) | 23 | 3.0 | 90 |
| 6c | 4 (3.0) | CH2Cl2 (0.1) | 80 | 3.0 | 50 |
| 7 | 4 (3.0) | CH2Cl2 (0.1) | 23 | 3.0 | 94d |
| 8 | 4 (2.0) | CH2Cl2 (0.6) | 23 | 2.0 | 94d |
| 9e | 4 (2.0) | CH2Cl2 (0.6) | 23 | 2.0 | 0 |
| 10f | 4 (2.0) | CH2Cl2 (0.6) | 23 | 2.0 | 16 |
| 11g | 4 (2.0) | CH2Cl2 (0.6) | 23 | 2.0 | 56 |
Reaction conditions for radical precursor 4: 1 equiv 8a, 1.5 mol % [Ru(bpy)3](PF6)2, low-intensity blue LEDs, 18 h; reaction conditions for radical precursor 6: 1 equiv 8a, 2 mol % Ir[dF(CF3)ppy]2(dtbbpy)PF6, 35 W blue LEDs, 18 h.
Yield determined by 1H NMR analysis of the crude reaction mixture using 1,4-dimethoxybenzene as an internal standard.
Reaction was performed in the presence of i-Pr2NEt•HBF4 (1 equiv) as an additive.
Isolated yield after silica gel chromatography.
Reaction performed in the absence of visible light.
Reaction performed in the absence of photocatalyst.
Reaction was stopped after 6 h.
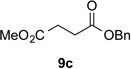
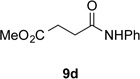
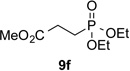
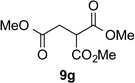
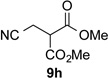
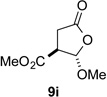
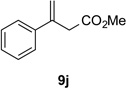
With optimal reaction conditions identified, the scope of the conjugate addition of the methoxycarbonyl radical to a range of alkene coupling partners was investigated (Table 2). Acceptors containing a terminal double bond and activated by sulfone, ketone, ester, amide, nitrile, or phosphonate functional groups performed best in the reaction, furnishing the corresponding products in moderate to high yields (entries 1−6). However, introduction of either an α methyl or α phenyl substituent to methyl vinyl ketone resulted in no detectable formation of the coupled product, as did incorporation of such substituents into benzyl acrylate.18 Cyclopent-2-en-1-one was a poor coupling partner, yielding the desired product in 27% yield. Whereas 5-oxocyclopent-1-ene-1-carbonitrile underwent conjugate hydride reduction by the Hantzsch ester 7.18 However, acyclic enones containing a second electron-withdrawing substituent at the β-carbon, such as dimethyl fumarate or trans-3-cyanoacrylate, did react in high yield (entries 7,8). The outcome of the latter reaction is noteworthy, as acceptor 8h possessing two electron-withdrawing groups of different steric and electronic properties underwent exclusive addition α to methyl ester substituent. This sense of regioselectivity, which was attributed by Giese to larger LUMO coefficient at C-2,4a has been observed previously; however, in these previous cases the magnitude of regioselection was much lower 5–6:1.20 4-Methoxybutenolide (8i), which was shown previously to react in good yield with a nucleophilic tertiary carbon radical,8,10 coupled in low yield with the methoxycarbonyl radical (entry 9). Coupling of the methoxycarbonyl radical with 2-phenylallyl bromide (8j) gave allylic substitution product 9j in 47% yield. To our surprise, methyl 2-(bromomethyl)acrylate was unreactive.18
Table 2.
Acceptor Scope with N-phthalimidoyl Oxalate 4
 | |||||
|---|---|---|---|---|---|
| entrya | acceptor | product | yield (%) | ||
| 1 | 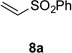 |
 |
94 | ||
| 2 |  |
 |
74 | ||
| 3 | 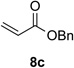 |
 |
89 | ||
| 4 | 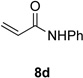 |
 |
60 | ||
| 5 | 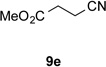 |
74 | |||
| 6 | 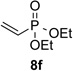 |
 |
66 | ||
| 7 | 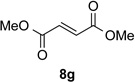 |
 |
80 | ||
| 8 | 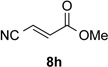 |
 |
91 | ||
| 9 | 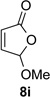 |
 |
28 | ||
| 10 | 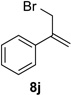 |
 |
47 | ||
Reaction performed using the optimized conditions (see Supporting Information).
All yields are yields of pure products isolated after silica gel chromatography.
As alkoxycarbonyl radicals had been suggested to be ambiphilic or electrophilic, we examined the reactivity of the methoxycarbonyl radical generated from N-phthalimidoyl oxalate 4 with electron-rich alkenes and styrenes (Scheme 3).18 For example, performing the reaction in the presence of a prototypical electron-rich alkene, butyl vinyl ether (8k), led to no detectable coupled product. Reactions carried out in the presence of styrene derivatives (8l–8n) led to broad peaks in 1H NMR spectra of crude reaction mixtures, indicating likely polymerization of the intermediate stabilized benzylic radicals formed upon addition of the methoxycarbonyl radical.
Scheme 3.

Coupling of N-phthalimidoyl Oxalate with Electron-Rich Alkenes and Styrenes
In summary, methyl N-phthalimidoyl oxalate (4) was shown to be a convenient precursor of the methoxycarbonyl radical under visible-light photoredox conditions. It reacts in good yield with terminal alkenes harboring a variety of electron-withdrawing substituents, thus providing a convenient method for the direct construction of γ-ketoesters and related products. It also reacts in high yield with 1,2-disubstituted alkenes activated by two electron-withdrawing substituents, and in one relevant case with regioselectivity higher than that of alkyl radicals. We attribute the somewhat limited scope of reactivity of the methoxycarbonyl radical to it being less nucleophilic than alkyl carbon radicals. No indication that the methoxycarbonyl radical shows ambiphilic reactivity was observed.
Supplementary Material
Acknowledgments
This research was supported by the National Science Foundation (CHE1265964), F.S. and Joan Rowland Fellowship to YS and by a graduate fellowship award to YS (1F31GM113494). NMR and mass spectra were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH Shared Instrumentation grants.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: XXXX.
Experimental procedures, characterization data of new compounds, and copies of 1H and 13C NMR spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.(a) Lever OW., Jr Tetrahedron. 1976;32:1943. [Google Scholar]; (b) Seebach D. Angew. Chem., Int. Ed. 1979;18:239. [Google Scholar]; (c) Yus M, Nájera C, Foubelo F. Tetrahedron. 2003;59:6147. [Google Scholar]; (d) Wyatt PJ, Warren SG. Organic Synthesis: Strategy and Control. Chap. 1–14. West Sussex: Wiley; 2006. [Google Scholar]; (e) Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]
- 2.For selected reviews, see: Kiss G. Chem Rev. 2001;101:3435. doi: 10.1021/cr010328q. El Ali B, Alper H, Beller M, Bolm C. Transition Metals for Organic Synthesis. Weinheim: Wiley–VCH; 2008. pp. 46–67. Brennnführer A, Neumann H, Beller M. ChemCatChem. 2009;1:28. Wu X-F, Fang X, Wu L, Jackstell R, Neumann H, Beller M. Acc. Chem. Res. 2014;47:1041. doi: 10.1021/ar400222k.
- 3.(a) Hong P, Mise T, Yamazaki H. Chem. Lett. 1982;11:361. [Google Scholar]; (b) Consiglio G, Nefkens SCA, Pisano C, Wenzinger F. Helv. Chim. Acta. 1991;74:323. [Google Scholar]; (c) Jiménez-Rodriguez C, Eastman GR, Cole-Hamilton DJ. Inorg. Chem. Commun. 2005;8:878. [Google Scholar]; (d) Suleiman R, El Ali B. Tetrahedron Lett. 2010;51:3211. [Google Scholar]; (e) Fang X, Jackstell R, Beller M. Angew. Chem. Int. Ed. 2013;52:14089. doi: 10.1002/anie.201308455. [DOI] [PubMed] [Google Scholar]
- 4.For selected reviews of radical conjugate additions reactions, see: Giese B. Angew. Chem., Int. Ed. Engl. 1983;22:753. Renaud P, Gerster M. Angew. Chem., Int. Ed. 1998;37:2562. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2562::AID-ANIE2562>3.0.CO;2-D. Sibi MP, Porter NA. Acc. Chem. Res. 1999;32:163. Sibi MP, Manyem S, Zimmerman J. Chem. Rev. 2003;103:3263. doi: 10.1021/cr020044l. Srikanth GSC, Castle SL. Tetrahedron. 2005;61:10377.
- 5.From (2-thioxopyridin-1(2H)-yl) oxalates: Barton DHR, Crich D. J. Chem. Soc., Perkin 1. 1986:1603. Togo H, Yokoyama M. Heterocycles. 1990;31:437. Togo H, Fujii M, Yokoyama M. Bull. Chem. Soc. Jpn. 1991;64:57. From chlorocarbonates or selenocarbonates: Bachi MD, Bosch E. Tetrahedron Lett. 1986;27:641. Singh AK, Bakshi RK, Corey EJ. J. Am. Chem. Soc. 1987;109:6187. Bachi MD, Bosch E. J. Org. Chem. 1992;57:4696. Trost BM, Waser J, Meyer A. J. Am. Chem. Soc. 2008;130:16424. doi: 10.1021/ja806724x. Lu P, Mailyan A, Gu Z, Guptill DM, Wang H, Davies HML, Zakarian A. J. Am. Chem. Soc. 2014;136:17738. doi: 10.1021/ja510573v. From S-alkoxycarbonyl xanthates: Forbes JE, Saicic RN, Zard SZ. Tetrahedron. 1999;55:3791. From xanthates: Davy JA, Mason JW, Moreau B, Wulff JE. J. Org. Chem. 2012;77:6332. doi: 10.1021/jo300939f. From xanthenoic esters: Plessis C, Derrer S. Tetrahedron Lett. 2001;42:6519. From N,S-dimethyldithiocarbamoyl oxalates: Kyne SH, Schiesser CH. Chem. Commun. 2014;50:12040. doi: 10.1039/c4cc06132b.
- 6.(a) Taniguchi T, Sugiura Y, Zaimoku H, Ishibashi H. Angew. Chem. Int. Ed. 2010;49:10154. doi: 10.1002/anie.201005574. [DOI] [PubMed] [Google Scholar]; (b) Su Y-H, Wu Z, Tian S-K. Chem. Commun. 2013;49:6528. doi: 10.1039/c3cc42994f. [DOI] [PubMed] [Google Scholar]; (c) Xu X, Tang Y, Li X, Hong G, Fang M, Du X. J. Org. Chem. 2014;79:446. doi: 10.1021/jo402529r. [DOI] [PubMed] [Google Scholar]; (d) Wang G, Wang S, Wang J, Chen S-Y, Yu X-Q. Tetrahedron. 2014;70:3466. [Google Scholar]; (e) Zong Z, Lu S, Wang W, Li Z. Tetrahedron Lett. 2015;56:6719. [Google Scholar]
- 7.(a) Kyne SH, Schiesser CH, Matsubara H. Org. Biomol. Chem. 2007;5:3938. doi: 10.1039/b714324a. [DOI] [PubMed] [Google Scholar]; (b) Kyne SH, Schiesser CH. Aust. J. Chem. 2009;62:728. [Google Scholar]
- 8.(a) Lackner GL, Quasdorf KW, Overman LE. J. Am. Chem. Soc. 2013;135:15342. doi: 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lackner GL, Quasdorf KW, Pratsch G, Overman LE. J. Org. Chem. 2015;80:6012. doi: 10.1021/acs.joc.5b00794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carbazate-derived alkoxycarbonyl radicals have been shown to couple with α,β-unsaturated amides see ref 6c.
- 10.Nawrat CC, Jamison CR, Slutskyy Y, MacMillan DWC, Overman LE. J. Am. Chem. Soc. 2015;137:11270. doi: 10.1021/jacs.5b07678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.These methods build on pioneering early contributions of Barton and co-workers, see ref 5a.
- 12.The decarboxylation of carboxyl radical A should be very fast the rate constant for the loss of CO2 from a carboxyl radical is estimated to be on the order of 109 s–1 see: Togo H. Advanced Free Radical Reactions for Organic Synthesis. Elsevier Science; 2004.
- 13.(a) Pfenninger J, Hueberger C, Graf W. Helv. Chim. Acta. 1980;63:2328. [Google Scholar]; (b) Simakov PA, Martinez FN, Horner JH, Newcomb M. J. Org. Chem. 1998;63:1226. [Google Scholar]; (c) Morihovitis T, Schiesser CH, Skidmore MA. J. Chem. Soc. Perkin Trans. 2. 1999:2041. [Google Scholar]; (d) Bucher G, Halupka M, Kolano C, Schade O, Sander W. Eur. J. Org. Chem. 2001:545. [Google Scholar]
- 14.See the Supporting Information for details.
- 15.Zhu X-Q, Zhang M-T, Yu A, Wang C-H, Cheng J-P. J. Am. Chem. Soc. 2008;130:2501. doi: 10.1021/ja075523m. [DOI] [PubMed] [Google Scholar]
- 16.Chikashita H, Miyazaki M, Itoh K. Synthesis. 1984:308. [Google Scholar]
- 17.Formation of the coupled product in the absence of the photocatalyst is attributed to electron transfer from the photoexcited Hantzsch ester to methyl N-phthalimidoyl oxalate. For similar observation see: Okada K, Okamoto K, Morita N, Okubo K, Oda M. J. Am. Chem. Soc. 1991;113 9401 and ref 8b.
- 18.A summary of acceptors that did not react under the conditions of Table 1 is provided in the Supporting Information.
- 19.Ouellet SG, Walji AM, MacMillan DWC. Acc. Chem. Res. 2007;40:1327. doi: 10.1021/ar7001864. [DOI] [PubMed] [Google Scholar]
- 20.(a) Giese B, Lachhein S. Chem. Ber. 1985;118:1616. [Google Scholar]; (b) Drescher M, Hammerschmidt F. Monatsh. Chem. 1997;128:713. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.