

Abstract
Purpose
The effectiveness of Tenofovir based HIV pre-exposure prophylaxis (PrEP) is proven, but hinges on correct and consistent use. User compliance and therapeutic effectiveness can be improved by long acting drug delivery systems. Here we describe a thin-film polymer device (TFPD) as a biodegradable subcutaneous implant for PrEP.
Methods
A thin-film polycaprolactone (PCL) membrane controls drug release from a reservoir. To achieve membrane controlled release, TAF requires a formulation excipient such as PEG300 to increase the dissolution rate and reservoir solubility. Short-term In vitro release studies are used to develop an empirical design model, which is applied to the production of in vitro prototype devices demonstrating up to 90-days of linear release and TAF chemical stability.
Results
The size and shape of the TFPD are tunable, achieving release rates ranging from 0.5–4.4 mg/day in devices no larger than a contraceptive implant. Based on published data for oral TAF, subcutaneous constant-rate release for HIV PrEP is estimated at < 2.8mg/day. Prototype devices demonstrated linear release at 1.2mg/day for up to 90 days and at 2.2mg/day for up to 60 days.
Conclusions
We present a biodegradable TFPD for subcutaneous delivery of TAF for HIV PrEP. The size, shape and release rate of the device are tunable over a > 8-fold range.
Defined Key Terms: Polycaprolactone, biodegradable implant, HIV pre-exposure prophylaxis, antiretroviral, membrane controlled release, tenofovir alafenamide fumarate
Introduction
When taken daily as an oral dose, tenofovir (TFV)-based antiretrovirals show high efficacy for HIV pre-exposure prophylaxis (HIV PrEP) in men and women, but poor adherence to the regimen drastically decreases effectiveness. [1] Clinical studies with tenofovir disoproxil fumarate (TDF)/emtricitabine (FDC) and TDF alone have highlighted a strong correlation between the level of HIV PrEP effectiveness and product adherence. [2] [3] The iPrEx [4] and Partners PrEP [5] studies investigated the prophylactic effectiveness of orally administered Truvada in gay/bisexual men and heterosexual HIV-serodiscordant couples, respectively. In these studies, Truvada reduced the risk of infection by at least 90% when taken regularly, but this level of protection declined to only 44% and 75%, respectively, when lower product adherence resulting in sub-prophylactic dosing regimens were taken into consideration. Outcomes from the VOICE and FemPrEP trials in Sub-Saharan Africa [6] [7] [8] investigating the effectiveness of TDF or TDF/FTC oral dosing regimens -further demonstrated that poor adherence to the product regimen stands as a barrier to an otherwise efficacious HIV PrEP strategy. In light of these outcomes, there is a strong interest in the development of alternative delivery strategies for antiretrovirals as HIV PrEP, that are less burdensome to the user and do not depend on user compliance.
When poor adherence is the major hurdle to an otherwise efficacious prevention approach, sustained release delivery systems can offer an alternative solution. The first long-acting pre-exposure prophylaxis (LA-PrEP) antiretroviral treatments for HIV to enter into clinical trials are both nanocrystal depot injections targeting a minimum dosing frequency of once every 2–3 months. [9] While this approach significantly decreases dosing frequency and is user independent, neither of the two products currently in Phase II trials for an HIV PrEP indication, cabotegravir (an integrase inhibitor) and rilpivirine (a non-nucleoside reverse transcriptase inhibitor (NNRTI)), have yet been proven to be efficacious for HIV PrEP in humans. These depot formulations also have a first order dissolution profile, resulting in a pharmacokinetic profile with the potential for a significant sub-therapeutic tail. [10] [11] Additionally, as a nanocrystal solution, there is no potential for retrieval after administration in the event of adverse reactions or seroconversion. This is a particularly relevant consideration in the case of HIV where low doses of antiretroviral monotherapies given to infected individuals may result in the development of viral resistance. [12]
Here we introduce a tunable biodegradable subcutaneous implant for long-acting constant rate delivery of tenofovir alafenamide fumarate (TAF) for HIV PrEP. TAF, like TDF, is a tenofovir pro-drug (Gilead Sciences, Foster City, CA), but unlike TDF which is converted to TFV upon absorption into the blood stream, TAF is primarily converted to TFV intracellularly, resulting in significantly more potency and lower systemic concentrations of TFV which, when taken systemically and chronically, can have side effects such as reduction in bone demineralization or renal toxicity [13] [14]. Since efficacy of tenofovir-based ARV for HIV PrEP has been confirmed and approved through the oral route [15] [4] it is highly likely that another tenofovir pro-drug, TAF will also be efficacious for HIV PrEP and is currently investigated in clinical studies. [16] Approved as an alternative tenofovir antiretroviral for HIV treatment, results show that only 8mg of TAF taken orally is equivalent in the AUC (area under the curve) of intracellular tenofovir as well as in antiviral activity, to 300mg of TDF, the dose commercially available as a standalone and as a TDF/FDC combination product [17]. However, as constant-rate subcutaneous delivery for HIV PrEP has not yet been explored in humans for tenofovir derived APIs, the efficacious dose of TAF from this novel route of administration is as yet unknown. Simply taking into consideration the bioavailability of TAF (35%) [18] and comparing oral TAF efficacy to TDF efficacy, subcutaneous delivery of 2.8 mg/day TAF is estimated as an upper bound for effective PrEP. However, this estimate does not take into consideration the pharmacokinetic benefits of constant rate release nor potential differences in distribution and tissue sequestration between TDF and TAF. Recently published results for subdermal delivery of TAF delivered from a non-biodegradable constant-rate implant in beagle dogs suggest that human doses as low as 0.15 mg/day may be sufficient to maintain effective concentrations of TFV-diphosphate in peripheral blood mononuclear cells. [19] In order to support device development and testing in pre-clinical models as well as later clinical dose escalation studies, a highly tunable subcutaneous TAF implant is desirable.
The implant presented here is a thin-film polymer device (TFPD), and is comprised of a biodegradable and biocompatible polycaprolactone membrane less than 50μm thick encapsulating a solid drug core. As a basic membrane-controlled reservoir system, release rates can be tuned by altering membrane thickness or surface area, and while solid drug remains within the device reservoir providing a constant activity source, the release rate remains constant. [20] [21] [22] Short term, small scale studies are used to develop an appropriate formulation for overcoming dissolution-limited release and to increase the rate of release. Additionally, these studies investigate the relationships between TAF release and device design parameters, providing an understanding of the achievable range of release and the necessary information to design implants with target specifications. The ability to accurately and reproducibly produce prototype devices targeting specific and relevant release rates based on the results of these small scale studies is demonstrated with the design, fabrication, and in vitro evaluation of relevant prototype devices. Linear release and stability of TAF in these prototype devices is demonstrated for up to 90 days. With a simple and flexible fabrication protocol and multiple approaches to tuning release, this implant can be easily tuned to achieve release rates up to 4.4 mg/day in a device comparable in size to commercially available contraceptive implants.
Materials & Methods
Film Fabrication
Films were draw-cast onto a glass surface using a multiple clearance square applicator (Paul N. Gardner Company, Inc., Pompano Beach, FL) from a 100 to 200 mg/ml solution of polycaprolactone (average Mn 80kDa, Cat#440744 Sigma, St. Louis, MO) in dichloromethane. After drying, PCL was annealed by heating to just past melting with a heat gun and cooling at room temperature. The solution concentration and height of the draw-casting rectangle were used to obtain films of varying thickness.
Device Fabrication
For each device, the PCL film thickness at multiple points (> 9) was measured using a micrometer. A NiChrom wire sandwiched between two thin slabs of polydimethylsiloxane (PDMS) and attached to a DC power supply constituted a heat-sealing apparatus through which controlled current was applied (1.2A) to heat the wire. PCL film was placed over the wire, and when the wire heats, the PCL melts. To fabricate a rod-shaped device, the PCL thin film was reinforced on each end with thicker (>30um) pieces of PCL film to provide structural support to the thin-film cylinder during device loading and later sampling. This reinforcement was done by sealing the thicker piece of PCL along the edge of the membrane film, folding over the added piece of PCL and sealing again along the same line. The cylindrical device was fabricated by wrapping the PCL around a rod-shaped mold of the desired diameter and sealing the film together along the lengthwise axis first and then along one reinforced end to create a PCL tube with one end open. Empty devices were weighed prior to loading. Device dimensions and rod geometry were based on commercially available contraceptive implants. The acceptable maximum sized device was defined as a 2mm diameter × 40mm long rod with 250mm2, based on the commercially available implants Nexplanon/Implanon (Merck, Whitehouse Station, NJ).
TAF was graciously provided by Gilead Sciences (Foster City, CA). When formulated with PEG300, TAF was mixed with PEG300 at desired ratios (1:2, 1:1, or 2:1 w/w) immediately prior to device loading. Devices were loaded using either a 1mL insulin syringe with the needle removed or with a P100 pipette tip. After loading, devices were laid flat on the PDMS of the heat sealing apparatus and sealed. Once sealed devices were weighed to determine the total payload and device dimensions were recorded.
Release Studies
TAF concentrations in all samples were measured by UV absorbance at 260nm using a SpectraMax M5 (Molecular Devices, Sunnyvale, CA) plate reader. All release studies were done in phosphate buffered saline (PBS), pH 7.4 at 37°C in an incubator on an agitator at 120 rpm. TAF solubility in release buffer under these conditions was determined to be 8 mg/ml by measuring the TAF concentration in the supernatant of a solution with excess drug powder. Release buffer volume and time intervals between samples were chosen to fully submerge device and to maintain sink conditions in release buffer at less than 1.6 mg/ml. Volumes ranged from 1 – 10mL with sample time points ranging from 1–5 days. At each time point, the device was transferred to an aliquot of fresh release buffer. For each sample TAF concentration was measured and mass of TAF released during the time interval was calculated. For each device a cumulative mass versus time profile was used to determine the linear release rate. Linear release profiles were normalized to membrane area by dividing the release rate (mg/day) by device surface area (mm2).
Stability Analysis
TAF stability in the device reservoir was evaluated by opening up a device with remaining TAF, dissolving the entire reservoir contents into release buffer, and measuring TAF purity using RP-HPLC. This HPLC method separates TAF from process impurities associated with TAF production as well as TAF degradation products. TAF purity was calculated as % peak area associated with TAF relative to total peak area of TAF related degradation products and product impurities. This method was provided by Gilead Sciences. TAF was analyzed from device reservoirs after incubation in PBS, pH 7.4 at 37°C for 0, 27, 49, and 89 days.
Results
Short-term screening studies
In these small-scale studies, release data was collected over 5–10 days. Linear release rates were obtained while solid drug remained in the device reservoir and were compared to device parameters to verify membrane controlled release, to demonstrate the relationship between device parameters and TAF release, to explore the achievable range of release rates, and to develop an empirical model to design devices targeting specific release rates.
TAF Release
TAF release from 8 different devices was evaluated. In each of these devices TAF was loaded in solid form with no additional excipients into the device reservoir. At the end of the study, all devices still visibly contained solid TAF in the device reservoir and cumulative release profiles appeared linear. Figure 1 illustrates the relationships between release rate and surface area.
Figure 1.
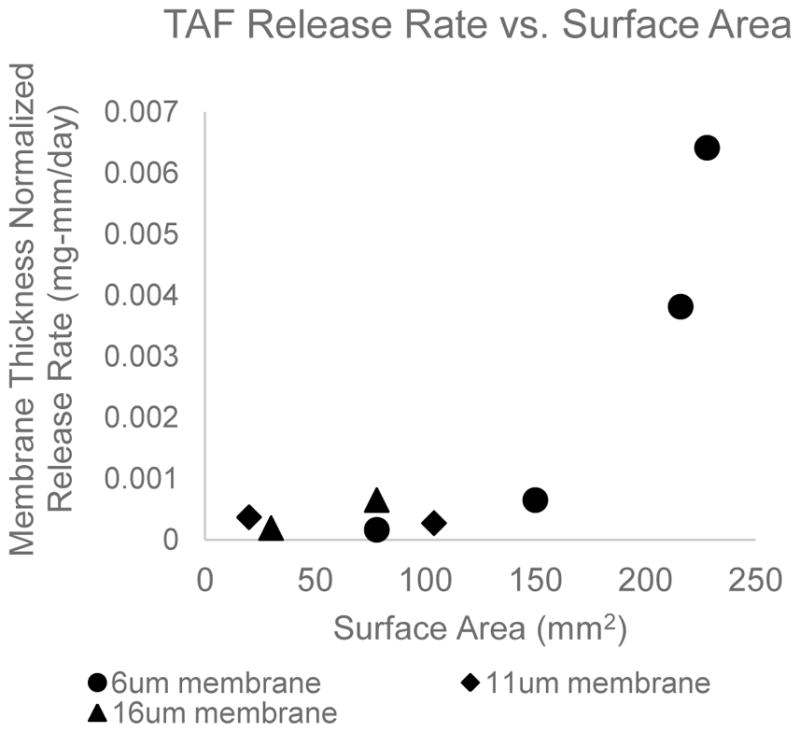
Linear release rate of TAF from PCL reservoir device has a non-linear dependence on surface area. This is not characteristic of membrane controlled release. Release rates are normalized for membrane thickness and each data point represents an individual device.
TAF Formulation with PEG300
To investigate the effect of adding PEG300 to the device reservoir and to determine the relationship between TAF release and membrane surface area, TAF release from triplicate rod-shaped devices in three different sizes (57 mm2 +/− 6, 109 mm2 +/− 18, 260mm2 +/−62) loaded with a 2:1 TAF to PEG mass ratio was evaluated for up to 5 days. All devices had a membrane thickness of 8.5μm. Figure 2A shows the average cumulative release profile for each device size. The average cumulative release profile is only calculated while solid drug remains in all three device reservoirs and the release profile is linear. As expected for membrane controlled release, the rate of TAF release scales proportionally with increasing membrane surface area (Figure 2B).
Figure 2.

(A) Cumulative release profile of TAF from PCL TFPDs loaded with TAF-PEG300 (2:1 w/w ratio). All devices have 8.5μm thick membrane. Each series represents an average of triplicate devices with error bars representing 1 standard deviation. Average values are only calculated for time points while all devices retain solid TAF in device reservoir and release profile is linear. (B) TAF linear release rates taken from the data shown in (A) scale linearly with membrane surface area. Error bars represent 1 standard deviation.
To further investigate the relationship between device design and TAF release, TAF release from triplicate rod-shaped devices, 294 mm2 +/− 36 surface area, with three different membrane thicknesses ( 0.009, 0.015, 0.026 mm) loaded with a 2:1 TAF to PEG ratio was evaluated for up to 7 days. Figure 3A shows the average cumulative release profile for each membrane thickness. The average cumulative release profile is only calculated while solid drug remains in the reservoir of all three devices and the release profile is linear. Figure 3B illustrates the relationship between linear release rate normalized for surface area and membrane thickness, including data represented in Figure 3A as well as three additional data points taken from the prototype device studies described later in this manuscript (Figure 6). Additionally, release rates from devices with 8.5 μm thick membranes loaded with mass ratios of TAF to PEG (2:1 (n=3), 1:1 (n=3) and 1:2 (n=6)) were evaluated to determine if TAF release depends on excipient content within this range (Figure 4).
Figure 3.

Reference source not found.: (A) Cumulative release profile of TAF from PCL TFPDs loaded with TAF-PEG300 (2:1 w/w ratio). Each series represents average of triplicate devices with error bars representing 1 standard deviation. Average values are only calculated for time points while all devices retain solid TAF in device reservoir and release profile is linear. All devices are 2–2.5mm diameter × 40mm long rods (294 mm2 +/− 26 surface area). (B) TAF linear release rates taken from the data shown in (A) and from prototype devices in [Figure 6]. Error bars represent 1 standard deviation.
Figure 6. TAF TFPD Prototypes.

(A) Device parameters and predicted release rates for TAF TFPD prototype devices. Each group consists of n=3 devices and measurements represent average values +/− 1 standard deviation. (B) Cumulative release profiles for prototype device groups. Prototype devices are loaded with a 1:1 TAF:PEG300 (w/w) formulation. Release profile is linear (solid data points) until device nears depletion (outlined data points). Linear regression determines average release rate for each prototype group (mg/day). (C) Purity of TAF remaining in device reservoir after incubation under physiological conditions (PBS pH 7.4, 37°C).
Figure 4.
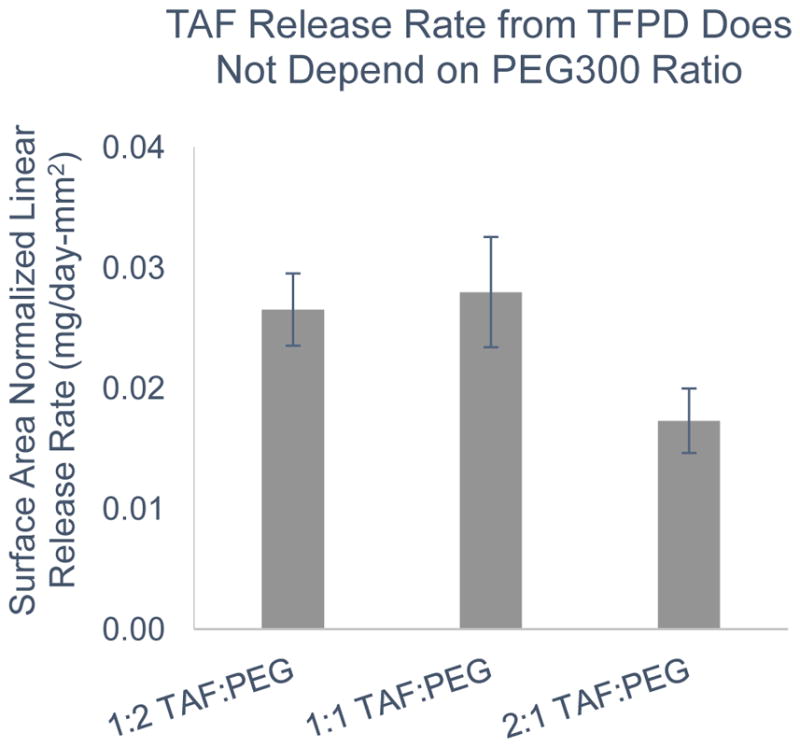
Reference source not found.: Surface area normalized release rates for TAF from PCL TFPD (8.5 μm membrane) with a range of w/w ratios of TAF to PEG300. No statistically significant difference in release rates (p=0.16).
Prototype Studies
Using the empirical correlations obtained in the short-term screening studies, three groups of triplicate prototype devices were designed to target three different release rates relevant to the estimated range for effective TAF dosing in humans (2.5 mg/day, 1.4 mg/day, 0.4 mg/day). These devices were fabricated by hand at a lab-scale as previously described. All devices were evaluated for TAF release until device depletion, demonstrating linear release profiles, reproducibility between replicate devices, and accuracy in device designs targeting specific release rates. Additionally, the chemical stability of TAF remaining in the device reservoir was evaluated at 0, 27, 49, and 89 days.
Prototype devices targeting specific release rates
Three groups of triplicate rod-shaped devices were fabricated according to different device designs targeting three release rates. Images of Group II devices and a representative device from Group III are shown in Figure 5. Figure 6A contains the device parameters for each group and the target release rates based on these parameters (2.5 mg/day, 1.4 mg/day, 0.4 mg/day). Devices were loaded with a 1:1 w/w TAF and PEG300 paste and total TAF the total TAF load for each device is presented in Figure 6A. Based on the target release rate and TAF loading, the expected device duration was calculated and provided in Figure 6A. Figure 6B shows the linear release profiles for all three groups of prototype devices until the device approaches depletion. Prototype release rates for Groups I and II are within 15% of target and within 25% of target for Group III. Variability in release rates within each group is less than 10%.
Figure 5. TAF Thin Film Polycaprolactone Device Prototypes.

(A) 2.5mm diameter, 40mm long prototypes loaded with 230mg 1:1 TAF:PEG300 (w/w). (B) 0.6mm diameter, 20mm long prototype loaded with 26mg 1:1 TAF:PEG300 (w/w).
TAF Stability in Device Reservoir
A single device from Group I or Group II was sacrificed for analysis of TAF remaining in the device reservoir at 27, 49, and 89 days. Figure 6C shows the purity of TAF remaining in the device reservoir over the course of the in vitro release study out to 89 days. TAF purity remains unchanged within the device reservoir with up to 49 days. Between days 49 and 89, TAF purity decreases by 19% with TFV and monophenyl TFV being the major degradation products.
Discussion
Short-term screening studies
TAF Release
For membrane-controlled release, release rates are expected to increase proportionally with membrane surface area. The non-linear relationship between surface area and release rate shown in Figure 1 suggests that membrane controlled release is not achieved and TAF release from the device may be dependent on the rate of TAF dissolution in the device reservoir. Furthermore, based on these preliminary observations, a maximum device area of 250mm2 and a minimum membrane thickness of 6um will result in TAF release rates around 1 mg/day. In attempts to achieve membrane controlled release and a broader range of release rates, TAF was formulated in the device reservoir with excipients known to increase solubility and dissolution rate.
TAF Formulation with PEG300
PEG300, a commonly used solubilizing excipient, was evaluated to increase dissolution and solubility of TAF in the device reservoir with the goals of achieving membrane controlled release and of increasing TAF release up to 3 mg/day for a maximum sized (260 mm2) device. When formulated with PEG300 in a 2:1 mass ratio, we achieved TAF release rates up to 4.4 mg/day in a rod shaped device 2mm in diameter and 4.1cm long with a 260 mm2 surface area (Figure 2). Furthermore, when formulated with PEG300, we observed membrane controlled release of TAF from the device as exhibited by a proportional relationship between release rate and membrane surface area (Figure 2B). Compared to the non-linear relationship between release rate and membrane surface area observed in the TFPD with TAF and no excipient, these results suggest that addition of PEG300 to the device reservoir increases TAF dissolution such that diffusion through the membrane and not dissolution is the rate-limiting step in drug release. Additionally, the release rate of TAF from a maximum sized device with an 8.6μm thick membrane increases from 0.8 mg/day with no excipient to 4.4 mg/day with PEG300. As Figure 3B shows, there was an inverse linear correlation between release rate and membrane thickness until the membrane reaches a critical thickness (0.015 mm) at which point increasing membrane thickness no longer impacted release rate. These results demonstrate that membrane thickness can be used to control the rate of TAF release in a range of 1.6–4.4 mg/day for a maximum sized device.
Release rates for different mass ratios of TAF to PEG300 were explored in order to determine the impact of these different formulations on release rate. While a 2:1 mass ratio is ideal in order to maximize TAF loading capacity, the thick paste is challenging to load by hand into a maximum sized device when loading to full capacity. We therefore explored the use of formulations with more PEG300 which results in a more fluid formulation that is easier to load by hand. Results demonstrate that release rates for TAF-PEG300 formulations with 2:1, 1:1, and 1:2 mass ratios were not significantly different (p=0.16) (Figure 4).
Prototype Studies
Prototype devices targeting specific release rates
Release profiles from prototype devices demonstrate the potential of this system to deliver relevant constant-rate doses of TAF. TAF release is linear until devices near depletion. When TAF depletes from the device to levels below the solubility of TAF in the device reservoir, there is no longer a constant TAF concentration in the device reservoir and TAF release is proportional to TAF reservoir concentration. This results in the non-linear region of the release profiles represented by open data points in Figure 6B. The mass of TAF released in this non-linear phase will depend on the device size and volume of the device reservoir; larger reservoir volumes will result in larger quantities of soluble TAF. Group I and II prototype devices are maximum sized devices and released on average a total of 10 mg of TAF in the non-linear phase.
All devices presented in this manuscript were fabricated and loaded by hand at a lab-scale. Hand making devices can lead to error in both the accuracy and precision of device fabrication relative to device design. To account for error in the accuracy of device fabrication, target release rates were calculated from the actual device parameters of fabricated devices. This is why target release rates were 2.5 mg/day, 1.4 mg/day, and 0.4 mg/day rather than say 2.5 mg/day, 1.5 mg/day, and 0.5 mg/day. Error in the precision of device fabrication is reported in Figure 6A as +/− 1 standard deviation. Additionally, all prototype devices were loaded with a 1:1 mass ratio of TAF to PEG300 rather than the 2:1 mass ratio used in the screening studies. This was due to the difficulty of fully loading a 40mm long device with the 2:1 formulation due to the high viscosity of the formulation. As the TFPD system is translated from lab scale fabrication to a manufacturing process, accuracy, precision and loading are expected to be improved. In addition to improving accuracy and precision in device fabrication, a manufacturing process with the capacity to fully load devices with a 2:1 mass ratio of TAF to PEG300 will result in significantly higher loading capacities for TAF in the device reservoir. In the prototype devices presented here, the duration of release for each group of devices is dependent on the loading capacity of TAF into the device reservoir. With a 1:1 mass ratio of TAF to PEG300, a fully loaded maximum sized device release 1.2 mg/day has a linear release profile out to 75 days with full device depletion at around 100 days. With a higher loading capacity, the duration of linear release from these devices will be longer.
TAF Stability in Device Reservoir
Results from the analysis of TAF chemical stability in the device reservoir indicate that under physiological conditions in a hydrated device, solid TAF contained in the device reservoir remained stable (Figure 6C). The loss of chemical purity between days 49 and 89 was likely due to the depletion of TAF solid from the device reservoir. The decrease in TAF purity was due to degradation of TAF via hydrolysis to mono-phenyl TFV, a form of TFV that may still be metabolized into active TFV-DP in vivo. Ongoing and future studies will further investigate the in vivo activity of TAF released from the TFPD in animal models.
Conclusion
Development and in-vitro testing of a TFPD is presented here as a subcutaneous implant for delivery of the ARV TAF, for HIV PrEP. As a reservoir device with a thin-film membrane made from PCL, a biocompatible and biodegradable polymer [23] [24], this system should be retrievable throughout the duration of administration if needed, but will not require removal upon drug depletion. This is a highly desirable feature for HIV PrEP in particular due to the possibility of seroconversion after device implantation and the potential for developing resistance with low doses of a single ARV post-seroconversion.
Exhibiting membrane controlled release up to 3 months, release rates from the TAF TFPD are highly tunable with membrane surface area and membrane thickness. The device development work here presents a thorough analysis of the relationships between device design and TAF release rates which provides the basis for designing devices to accurately target specific release rates. These results show that without additional solubilizing excipients, TAF release from the TFPD is dissolution limited. However, with PEG300 as a solubilizing excipient, TAF exhibits membrane controlled release and increased release rates.
With this TAF-PEG300 formulation, we demonstrate the ability to target a range of release rates relevant to the estimated effective rates for TAF HIV PrEP subcutaneous delivery based on clinical pharmacokinetic data from oral TAF studies for HIV treatment. The tunability of this device is key to its success in pre-clinical and clinical development. Being able to tune release with both surface area and membrane thickness within this target range allows us to decouple release rate from reservoir volume and thus loading capacity. As a result, we have the potential to maximize our loading capacity and the implant duration or the option to limit the implant duration and loading capacity, but to create a smaller and easier to insert implant, all while maintaining the same release rate. Furthermore, we are able to leverage the empirical correlations presented here between device design and TAF release to tune devices for use in future in vivo studies to be appropriate for the selected species as well as to consider dose escalation studies.
The biodegradable and subcutaneous implant described here offers great potential for user-independent and long-acting delivery of HIV PrEP. With such flexibility in design and potential to tune the device for a broad range of release rates, the device is well suited for use throughout pre-clinical and clinical trials for an API such as TAF for which the efficacious constant-rate subcutaneous dose for HIV PrEP has not yet been confirmed in humans.
Acknowledgments
This research is made possible by the generous support of the American people through the U.S. President’s Emergency Plan for AIDS Relief. The contents are the responsibility of the authors and do not necessarily reflect the views of USAID, PEPFAR, or the United States Government.
Tenofovir Alafeinimde Fumarate drug substance and analytical methods were graciously provided by Gilead Sciences, Inc.
Abbreviations
- PrEP
pre-exposure prophylaxis
- LA-PrEP
long acting pre-exposure prophylaxis
- TFV
tenofovir
- TDF
tenofovir disproxil fumarate
- TAF
tenofovir alafenamide fumarate
- FDC
emtricitabine
- TFPD
thin film Polycaprolactone device
- PCL
Polycaprolactone
- ARV
antiretroviral
- API
active pharmaceutical ingredient
- PDMS
polydimethylsiloxane
- DC
direct current
- UV
ultraviolet
- HPLC
high performance liquid chromatography
- PEG
polyethylene glycol
References
- 1.Baeten J, Celum C. Systemic and topical drugs for the prevention of HIV infection: antiretroviral preexposure prophylaxis. Annu Rev Med. 2013;64:219–232. doi: 10.1146/annurev-med-050911-163701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGowan I. An overview of antiretroviral pre-exposure prophylaxis of HIV infection. Am J Reprod Immunol. 2014;71(6):624–630. doi: 10.1111/aji.12225. [DOI] [PubMed] [Google Scholar]
- 3.Amico K, Mansoor L, Corneli A, Torjesen K, van der Straten A. Adherence support approaches in biomedical HIV prevention trials: experienes, insights and future directions from four multisite prevention trials. AIDS Behav. 2013;17:2143–2155. doi: 10.1007/s10461-013-0429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.N. I. o. A. a. I. D. (NIAID) clinicaltrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Emtricitabine/Tenofovir Disproxil Fumarate for HIV Prevention in Men (iPrEx) cited 2015 Aug 02. [Google Scholar]
- 5.U. o. Washington. Clinicaltrials.gov [internet] Bethesda (MD): National Library of Medicine (US); 2000. Pre-Exposure Prophylaxis to Prevent HIV-1 Acquisition Within HIV-1 Discordant Couples (Partners PrEP) cited 2015 Aug 02. [Google Scholar]
- 6.Marrazzo J, Ramjee G, Richardson B, Gomez K, Mgodi N, Nair G, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. NEngl JMed. 2015;372:50–518. doi: 10.1056/NEJMoa1402269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Damme L, Corneli A, Ahemd K, Agot K, Lombaard J, Kapiga S, et al. Preexposure prophylaxis for HIV infection among African women. NEngl JMed. 2012;367:411–422. doi: 10.1056/NEJMoa1202614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.N. I. o. A. a. I. D. NIAID. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Safety and Effeciveness of Tenofovir 1% Gel, Tenofovir Disproxil Fumarate, and Emtricitabine/Tenofovir Disproxil Fumarate Tablets in Preventing HIV in Women. cited 2015 Aug 02. [Google Scholar]
- 9.Spreen W, Margolis D, Pottage J. Long-acting antiretrovirals for HIV treatment and prevention. Current Opinion in HIV and AIDS. 2013;8(6):565–571. doi: 10.1097/COH.0000000000000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van’t Klooster G, Hoeben E, Borghys H, Looszova A, Bouche M, van Velsen F, Baert L. Pharmacokinetics and Disoposition of Rilpivirine (TMC278) Nanosuspension as a Long-acting Injectable Antiretroviral Formulation. Antimicrobial Agents and Chemotherapy. 2010;54(5):2042–2050. doi: 10.1128/AAC.01529-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spreen W, Ford S, Chen S, Wilfret D, Margolis D, Gould E, Piscitelli S. GSK1265744 Pharmacokinetics in Plasma and Tissue After Single-Dose Long Acting Injectable Administration in Healthy Subjects. J Acquir Immun Defic Syndr. 2014;67(5):481–486. doi: 10.1097/QAI.0000000000000301. [DOI] [PubMed] [Google Scholar]
- 12.Taylor S, Boffito M, Khoo S, Smit E, Back D. Stopping antiretroviral therapy. AIDS. 2007;21:1673–1682. doi: 10.1097/QAD.0b013e3281c61394. [DOI] [PubMed] [Google Scholar]
- 13.Lee W, He G, Eisenberg E, Cihlar T, Swaminathan S, Mulato A, CK Selective Intracellular Activation of a Novel Prodrug of the Human Immunodeficiency Virus Reverse Transcriptase Inhibitor Tenofovir Leads to Preferential Distribution and Accumulation in Lymphatic Tissue. Antimicrobial Agents and Chemotherapy. 2005;49(5):1989–1906. doi: 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falutz J. Unmasking the bare bones of HIV Preexposure Prophylaxis. Clinical Infections disease. 2015:1–3. doi: 10.1093/cid/civ329. [DOI] [PubMed] [Google Scholar]
- 15.Grant R, Lama J, Anderson P, McMahan V, Liu A, Vargas L, et al. Preexposure prophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.U. o. N. C. C. H. ( NCT02357602) ClinicalTrials.gov. 2015. Dose Proportionality of TFV-DP After a Single Dose of GS-7340 in Women. [Google Scholar]
- 17.Ruane P, et al. Antiviral Activity and Safety, and Pharmacokinetics/Pharmacodynamics of Tenofovir Alafenamide as 10-day Monotherapy in HIV-1 Positive Adults. J Acquir Immune Defic Syndr. 2013;63(4):449–455. doi: 10.1097/QAI.0b013e3182965d45. [DOI] [PubMed] [Google Scholar]
- 18.Babusis D, et al. Mechanism for Effective Lymphoid Cell and Tissue Loading Following Oral Administration fo Nucleotide Prodrug GS-7340. Mol Pharm. 2013;10(2):459–466. doi: 10.1021/mp3002045. [DOI] [PubMed] [Google Scholar]
- 19.Gunawardana M, Remedios-Chan M, Miller C, Fanter R, Yang F, Marzinke M, Hendrix C, Beliveau M, Moss J, Smith T, Baum M. Pharmacokinetics of Long-Acting Tenofovir Alafenimide (GS-7340) Subdermapl Implant for HIV Prophylaxis. Antimicrobial Agents and Chemotherapy. 2015;59(7):3913–3919. doi: 10.1128/AAC.00656-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siepmann J, Siepmann F. Modeling of diffusion controlled drug delivery. Journal of Controlled Release. 2012;161:351–362. doi: 10.1016/j.jconrel.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Schlesinger E, Ciaccio N, Desai T. Polycaprolactone Thin-Film Drug Delivery Systems: Empirical and Predictive Models for Device Design. Material Science and Engineering C: Materi Bio Appl. 2015;57:232–239. doi: 10.1016/j.msec.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lance K, Goods S, Mendes T, Ishikiriyama M, Chew P, Estes L, Yamada K, Mudumba S, Bhistikul R, Desai T. In vitor and in vivo sustained zero-order delivery of Rapamycin (Sirolimus) from a biodegradable intraocular device. Investigative Opthalmology & Visual Science. 2015;56:7331–7337. doi: 10.1167/iovs.15-17757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun H, Mei L, Song C, Cui X, Wang P. The in vivo Degradation, Absoprtion, and Excetion of PCL-based Implant. Biomaterials. 2006;27:1735–1740. doi: 10.1016/j.biomaterials.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 24.Woodruff M, Hutmacher D. The Return of the Forgotten Polymer: Polycaprolactone in the 21st Century. Progress in Polymer Science. 2010;35(10):1217–1256. [Google Scholar]
- 25.Center for the AIDS Programme of Research in South Africa. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Safety and Effectiveness Study of a Candidate Vaginal Microbicide for Prevention of HIV (CAPRISA004) cited 2015 Aug 02. [Google Scholar]
- 26.Baeten J, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012;367(5):399–410. doi: 10.1056/NEJMoa1108524. [DOI] [PMC free article] [PubMed] [Google Scholar]