

Abstract
Chromatin remodeling proteins are frequently dysregulated in human cancer, yet little is known about how they control tumorigenesis. Here, we uncover an epigenetic program mediated by the NAD+-dependent histone deacetylase Sirtuin 6 (SIRT6) that is critical for suppression of pancreatic ductal adenocarcinoma (PDAC), one of the most lethal malignancies. SIRT6 inactivation accelerates PDAC progression and metastasis via upregulation of Lin28b, a negative regulator of the let-7 microRNA. SIRT6 loss results in histone hyperacetylation at the Lin28b promoter, Myc recruitment, and pronounced induction of Lin28b and downstream let-7 target genes, HMGA2, IGF2BP1 and IGF2BP3. This epigenetic program defines a distinct subset with a poor prognosis, representing 30–40% of human PDAC, characterized by reduced SIRT6 expression and an exquisite dependence on Lin28b for tumor growth. Thus, we identify SIRT6 as an important PDAC tumor suppressor, and uncover the Lin28b pathway as a potential therapeutic target in a molecularly-defined PDAC subset.
INTRODUCTION
PDAC remains one of the most lethal of all human malignancies and is responsible for hundreds of thousands of deaths each year. Thus, there is an urgent need to improve our understanding of the molecular underpinnings that drive PDAC initiation, progression and metastasis and to leverage that understanding toward better therapeutic options. The current model proposes that a series of genetic alterations results in a stepwise progression through increasingly dysplastic precursor lesions, or pancreatic intraepithelial neoplasias (PanINs), toward invasive and finally metastatic PDAC. Initiating events identified in early PanIN lesions (PanIN I) include mutations and/or amplification of the KRAS oncogene and the loss of the CDKN2A (p16INK4A) tumor suppressor gene, present in >90% and >50% of PDAC/PanINs respectively (Ryan et al., 2014). Higher grade lesions (PanIN III) and invasive PDAC may accumulate additional genetic lesions, including inactivation of TP53 and TGFβ pathway components (SMAD4, TGFβR1, and TGFβR2) (Ryan et al., 2014). However, this fundamental model of PDAC pathogenesis, which is recapitulated in genetically engineered mice, has failed to identify either critical pathways that may be effectively targeted in the clinic or relevant molecular subsets for improved prognosis and stratification of patients toward a more effective therapy.
In addition to the above well-characterized genetic aberrations, it is becoming increasingly apparent that the dysregulation of epigenetic modifiers is central to the initiation and progression of human PDAC as well as many other tumors. Genomic deletions, mutations, and rearrangements recurrently targeting genes encoding many components of the SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeling complex have recently been identified in at least 10–15% of PDAC cases. Additionally, mutations in the histone methyltransferase mixed-lineage leukemia protein 2 & 3 (MLL2 & MLL3) and the histone demethylase KDM6A have been identified in 5–10% of PDAC (Ryan et al., 2014). However, since these chromatin-modifying enzymes may simultaneously regulate the transcription of thousands of genes by altering chromatin structure throughout the genome or may be involved in other cellular functions such as DNA repair and replication, the mechanisms by which these proteins control tumorigenesis have been difficult to elucidate. Specifically, whether these enzymes regulate an individual target gene or set of genes to drive survival, proliferation, cellular transformation, metabolic adaptations or invasive functions in PDAC is unknown; yet this understanding is critical to our ability to leverage data from the molecular profiling of human tumors to identify new therapeutic opportunities in molecularly-defined subsets of disease.
Sirtuin 6 (SIRT6) is a nicotinamide adenine dinucleotide (NAD)+-dependent histone deacetylase which removes acetyl groups from histone 3 lysine 9 (H3K9) and histone 3 lysine 56 (H3K56) motifs and has pleiotropic functions including glucose homeostasis, maintenance of genome stability, and suppression of cellular transformation (Mostoslavsky et al., 2006; Sebastian et al., 2012; Zhong et al., 2010). These functions are exemplified in both Sirt6-deficient mice, which exhibit complete loss of subcutaneous fat and lethal hypoglycemia, as well as SIRT6-deficient cells, which show increased glucose uptake, enhanced glycolysis, anchorage independent growth and tumor formation in an in vivo model of colon cancer (Mostoslavsky et al., 2006; Sebastian et al., 2012). Intriguingly, we observed copy number loss (CNL) of the SIRT6 locus in ~60% of pancreatic cancer cell lines, while its expression was downregulated in a dataset of 36 individual cases of human PDAC compared to their matched normal tissue (Sebastian et al., 2012; Ying et al., 2012). Therefore, the SIRT6 histone deacetylase is a chromatin-modifying enzyme, capable of directly reprogramming the epigenome in response to nutrient availability, which appears dysregulated in a large fraction of human PDAC.
These observations prompted us to explore the unique functions of SIRT6 in PDAC biology. Indeed, we found that SIRT6 acts a potent tumor suppressor in genetically-engineered mouse models (GEMMs) of oncogenic Kras-driven PDAC, regardless of p53 status. To our surprise, loss of SIRT6 did not accelerate PDAC tumorigenesis by enhancing aerobic glycolysis, as we had seen in colon cancer. Instead, using an unbiased and genome-wide analysis of chromatin changes in PDAC, we determined that the loss of SIRT6 results in the reactivation of the oncofetal protein Lin28b in both human and murine PDAC. Importantly, this de-repression results in the upregulation of numerous let-7 target genes and is critical for the survival of SIRT6-deficient PDAC. Thus, our findings highlight a paradigm where the loss of a pleiotropic chromatin-modifying enzyme drives tumorigenic growth through the dysregulation of a single target gene. Finally, our results define the SIRT6/Lin28b axis as a major pathway in PDAC carcinogenesis and identify a molecularly defined subset that may benefit from therapeutic intervention.
RESULTS
Loss of SIRT6 Cooperates with Oncogenic Kras to Accelerate PDAC
To determine the tissue expression pattern of SIRT6 in human PDAC tumors, we generated tissue microarrays containing 120 pathologist-verified and clinically annotated PDAC samples. Staining of these samples using a validated antibody for SIRT6 revealed that ~30–40% of PDAC tumors demonstrated reduced SIRT6 expression compared to normal pancreas (Figure 1A). Although the prognosis for this disease is already quite poor, patients who underwent surgical resection of a SIRT6low PDAC tumor had an even worse prognosis in this retrospective analysis, with a median overall survival of 17.5 months compared to 33 months in the SIRT6high tumors (Figure 1B). We next evaluated the functional role of SIRT6 by knocking down SIRT6 in human pancreatic ductal epithelial (HPDE) cells. These studies revealed that SIRT6 actively represses both global levels of acetylated H3K56 and cellular proliferation in pancreatic ductal cells (Figures S1A and S1B), prompting us to further explore the role of SIRT6 in the pathogenesis of PDAC in a physiologic context.
Figure 1. Loss of SIRT6 Cooperates with Oncogenic Kras to Accelerate PDAC.

A, Immunohistochemistry of SIRT6 in human PDAC samples (left & center) compared to normal pancreas (right) and quantification of IHC scoring (bottom right). B, Kaplan–Meier analysis of the indicated PDAC patient samples based on SIRT6 IHC score (n=120). C, Kaplan–Meier analysis of the indicated genetically engineered mouse models (GEMMs) showing time until signs of illness necessitated euthanasia. All animals euthanized had pancreatic tumors. D, Necropsy of Sirt6f/f;KrasG12D;p53f/+;p48-Cre (SIRT6 KO) GEMM euthanized at 13 weeks. Top left, Image of abdominal contents showing pancreatic mass and splenomegaly. Top middle, extracted SIRT6 KO tumor. Upper right, haematoxylin and eosin (H&E) staining showing PDAC histology. Bottom left, Gross image of liver with metastases. Bottom middle, H&E stain of liver metastasis. Bottom right, H&E stain of lung metastasis. E, Quantification of the metastatic potential of SIRT6 KO and Sirt6+/+;KrasG12D;p53f/+;p48-Cre (SIRT6 WT) GEMMs from the Kaplan-Meier analysis in Fig. 1C to the livers or the lungs. F, Kaplan–Meier analysis of the indicated genetically engineered mouse models (GEMMs) showing time until signs of illness necessitated euthanasia. All animals euthanized had pancreatic tumors. G, Necropsy of Sirt6f/f;KrasG12D;p53+/+;p48-Cre GEMM euthanized at 55 weeks. Top left, Image of abdominal contents showing pancreatic mass and splenomegaly. Top middle, extracted Sirt6f/f;KrasG12D;p53+/+;p48-Cre pancreatic tumor with spleen attached. Upper right, haematoxylin and eosin (H&E) staining showing PDAC histology. Bottom left, H&E of liver metastasis. Bottom right, H&E stain of lung metastasis. H, Quantification of the metastatic potential of Sirt6f/f;KrasG12D;p53+/+;p48-Cre and Sirt6+/+;KrasG12D;p53+/+;p48-Cre GEMMs from the Kaplan-Meier analysis in Fig. 1F to the livers or the lungs. Scale bars, black 50 μm, blue 20 μm. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. See also Fig S1.
To determine whether SIRT6 delays the development of PDAC in a genetically engineered mouse model (GEMM), we crossed Sirt6 conditional knockout mice (Sirt6fl/fl) with mice harboring a pancreas-specific Cre recombinase (p48-Cre), a floxed p53 allele (p53f/+), and a lox-STOP-lox (LSL) KrasG12D allele to generate LSL-KrasG12D; p48-Cre mice with specific loss of one or both Sirt6 and p53 alleles in the pancreas. Remarkably, in the context of activated Kras in the pancreas, loss of Sirt6 greatly accelerated the development of lethal pancreatic tumors regardless of p53 status (Figures 1C–H and S1C). In addition to developing PDAC and high-grade pancreatic intraepithelial neoplasia (PanIN) at an earlier age (Figures S1D and S1E), Sirt6-deficient tumors had a much greater propensity to metastasize to the lung, compared to their Sirt6 wild-type (WT) counterparts (Figures 1D–E and 1G–H). Importantly, these results demonstrate that SIRT6 suppresses both the formation and metastasic spread of KRASG12D-driven PDAC and establish SIRT6 as a critical tumor suppressor in this disease.
SIRT6 Suppresses Proliferation of Established PDAC Though Histone Deacetylation
To understand how the loss of this bioenergetic sensor influences the biology of established tumor cells, we derived cell lines from Sirt6f/f;KrasG12D;p53f/+;p48-Cre (SIRT6 KO) and Sirt6+/+;KrasG12D;p53f/+;p48-Cre (SIRT6 WT) murine tumors. Interestingly, we noted that SIRT6 KO PDAC cell lines were highly enriched for tumor sphere forming cells compared to SIRT6 WT cells grown under restrictive culture conditions, which suggested an enhanced tumorigenic potential (Figure 2A). In accordance with the role of SIRT6 as a histone deacetylase and repressor of Myc-mediated transcription (Sebastian et al., 2012), PDAC cells lacking SIRT6 had increased global levels of H3K56Ac as well as increased chromatin-bound Myc compared to SIRT6 WT cell lines, although total levels of Myc in the whole cell lysate were similar between the two genotypes (Figures 2B and S1F–G). The direct role of SIRT6 histone deacetylase activity in regulating these phenotypes was confirmed by the fact that WT but not catalytically inactive SIRT6 (S6HY) reduced global levels of H3K56Ac, chromatin-bound Myc, cell proliferation and tumor sphere formation (Figures 2B–D). Finally, restoration of SIRT6 expression in SIRT6 KO PDAC cells also slowed tumor growth in vivo (Figure 2E). We next sought to validate our findings in human PDAC. We first analyzed SIRT6 levels in a panel of patient-derived PDAC lines. Consistent with our analysis of human PDAC tissues, we found that SIRT6 protein levels were reduced in 6 out of 13 (46%) of the human PDAC cell lines we surveyed when compared to HPNE cells (Figure 2F). Restoration of SIRT6 expression in human PDAC cell lines that exhibit low levels of SIRT6 (SIRT6low) reduced H3K9Ac and H3K56Ac levels, cell cycle progression and cellular proliferation, while inducing apoptosis and robustly inhibiting tumor sphere formation in a manner that required SIRT6 deacetylase activity (Figures 2G–J and S1H–I). Thus, the loss of this NAD+-dependent histone deacetylase leads to hyperacetylation of chromatin and increased cellular proliferation in both normal pancreatic ductal cells and PDAC.
Figure 2. SIRT6 Suppresses Proliferation of Established PDAC Though Histone Deacetylation.

A, Murine PDAC cells were grown under restrictive, nonadherent conditions to induce tumor sphere formation, photomicrographs (left) and quantified (right). Two independent cell lines are represented. B–E, Murine PDAC cells were engineered to express empty vector (vector), SIRT6 WT (S6 WT) or the SIRT6 HY (S6HY) catalytically inactive mutant. B, Immunoblot (IB) of chromatin extract and whole cell lysate (WCL). C, Growth curve of a representative SIRT6 KO PDAC line. D, Quantification of tumor spheres formed by two independent SIRT6 KO PDAC lines and grown as in A. E, Tumor weights (left) and gross image of SIRT6 KO PDAC cell line grown for 3 weeks as a subcutaneous xenograft. F, IB of WCL in human PDAC cell lines with quantification of SIRT6/actin ratios below. G–J, Panc3.27 and Panc-1 cells were engineered to express Vector, S6WT or S6HY under a doxycycline (Dox)-inducible system. G, IB of chromatin extract. H, IB of chromatin extract from Panc3.27 cells treated with Dox for the indicated times. The partial effect of S6HY on H3K56Ac levels after 4 days of overexpression likely relates to its partial catalytic activity. I, Proliferation was quantified by trypan blue exclusion assay. J. Photomicrographs (left) and quantification of Panc3.27 tumor spheres. K–M, HPDE cells were engineered to express empty vector (shCtl) and shSIRT6 under a Dox-inducible system. K, IB of WCL, please see Figure S1A for SIRT6 levels. L, Quantitative polymerase chain reaction with reverse transcription (qRTPCR) analysis of SIRT6 and glycolytic genes. M, FDG-Glucose uptake in HPDE cells. N–P, Panc3.27 and Panc-1 cells were engineered to express empty Vector, S6WT or S6HY under a Dox-inducible promoter. N, IB of WCL. O, qRTPCR analysis of glycolytic genes in Panc3.27 cells. P, FDG-Glucose uptake in Panc3.27 cells after treatment with dox for the indicated times. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. See also Fig S1 and S2.
We have previously established SIRT6 as a central regulator of glycoytic metabolism (Sebastian et al., 2012; Zhong et al., 2010). Consistent with this finding, knockdown of SIRT6 in HPDE cells resulted in increased expression of HIF1α target genes involved in glycolytic metabolism, such as pyruvate dehydrogenase 1 (PDK1), lactate dehydrogenase a (LDHA), and the glucose transporter (GLUT1) (Figures 2K and 2L). These gene expression changes corresponded with an increase in uptake of the fluorescently labeled glucose analog 2-(N-7-nitrobenz-2-oxa-1,3,diazol-4-yl)amino)-2-deoxyglucose (2-NBDG) (Figure 2M). Conversely, when SIRT6 levels were restored in SIRT6low PDAC cell lines, glycolytic gene expression and glucose uptake were all repressed (Figures 2N–P). Likewise, SIRT6 KO PDAC cells demonstrated relatively high expression of Pdk1, Ldha and Pfkm as well as 2-NBDG uptake compared to SIRT6 WT cells (Figures S2A and S2B), and expression of SIRT6 reduced glycolytic gene expression (Figure S2C). However, despite these increased levels of glucose uptake and glycolytic gene expression, knocking down Pdk1 or Ldha, both central regulators of glycolytic metabolism, had equivalent effects on SIRT6 WT and KO PDAC cells (Figures S2D–K). In addition, pharmacologic inhibition of PDK1 with the small-molecule PDK1 inhibitor, dichloroacetate (DCA), inhibited growth of both SIRT6 WT and KO PDAC cell lines with similar potency (Figure S2L). These results suggested that lack of SIRT6 does not render PDACs more sensitive to glycolysis inhibition. To fully evaluate the role of glycolysis in the accelerated formation of SIRT6 KO PDAC, SIRT6 KO and WT mice were treated with DCA in their drinking water from 4 weeks of age and monitored for the development of lethal PDAC tumors. Consistent with our in vitro results, DCA treatment minimally delayed the onset of SIRT6 KO PDAC (Figure S2M). Overall, our results indicate that enhanced glycolysis plays a modest role in the increased aggressiveness of these SIRT6-deficient tumors, in contrast to what we previously observed in colon cancer (Sebastian et al., 2012).
SIRT6 Suppresses Expression of the Oncofetal Protein Lin28b in Human and Murine PDAC
The lack of differential sensitivity of SIRT6 WT and KO PDAC cells to Pdk1 and Ldha knockdown and the failure to reverse the SIRT6 KO phenotype with DCA treatment prompted us to investigate alternative pathways regulated by SIRT6 that could limit the growth of PDAC cells. Since expression of WT but not catalytically inactive SIRT6 slowed the growth of both human and murine PDAC cells, we hypothesized that these pathways would be regulated by the histone deacetylase activity of SIRT6. We therefore sought to identify novel genes regulated by SIRT6 histone deacetylase activity by performing chromatin immunoprecipitation (ChIP) of H3K56Ac marks (the main chromatin substrate of SIRT6) followed by next generation sequencing (ChIP-seq) on SIRT6 WT and SIRT6 KO murine PDAC cells, as well as SIRT6 KO cells engineered to express WT SIRT6 (SIRT6 KO + SIRT6 WT). In SIRT6 KO cells, we identified a total of 12,049 genes that were decorated with H3K56Ac within 1 kilobase (kb) of their transcription start site. To identify genes that were dynamically regulated by SIRT6, we isolated genes which were only marked in SIRT6 KO cells but not SIRT6 WT cells, and which lost this mark upon reexpression of SIRT6 (Figure 3A). We then ranked the remaining 184 gene promoters based on fold change of H3K56Ac in SIRT6 KO compared to SIRT6 WT cells. Intriguingly, the RNA-binding protein Lin28b was the top candidate in this list (Figure 3B; Table S1).
Figure 3. SIRT6 Suppresses Expression of the Oncofetal Protein Lin28b in Human and Murine PDAC.

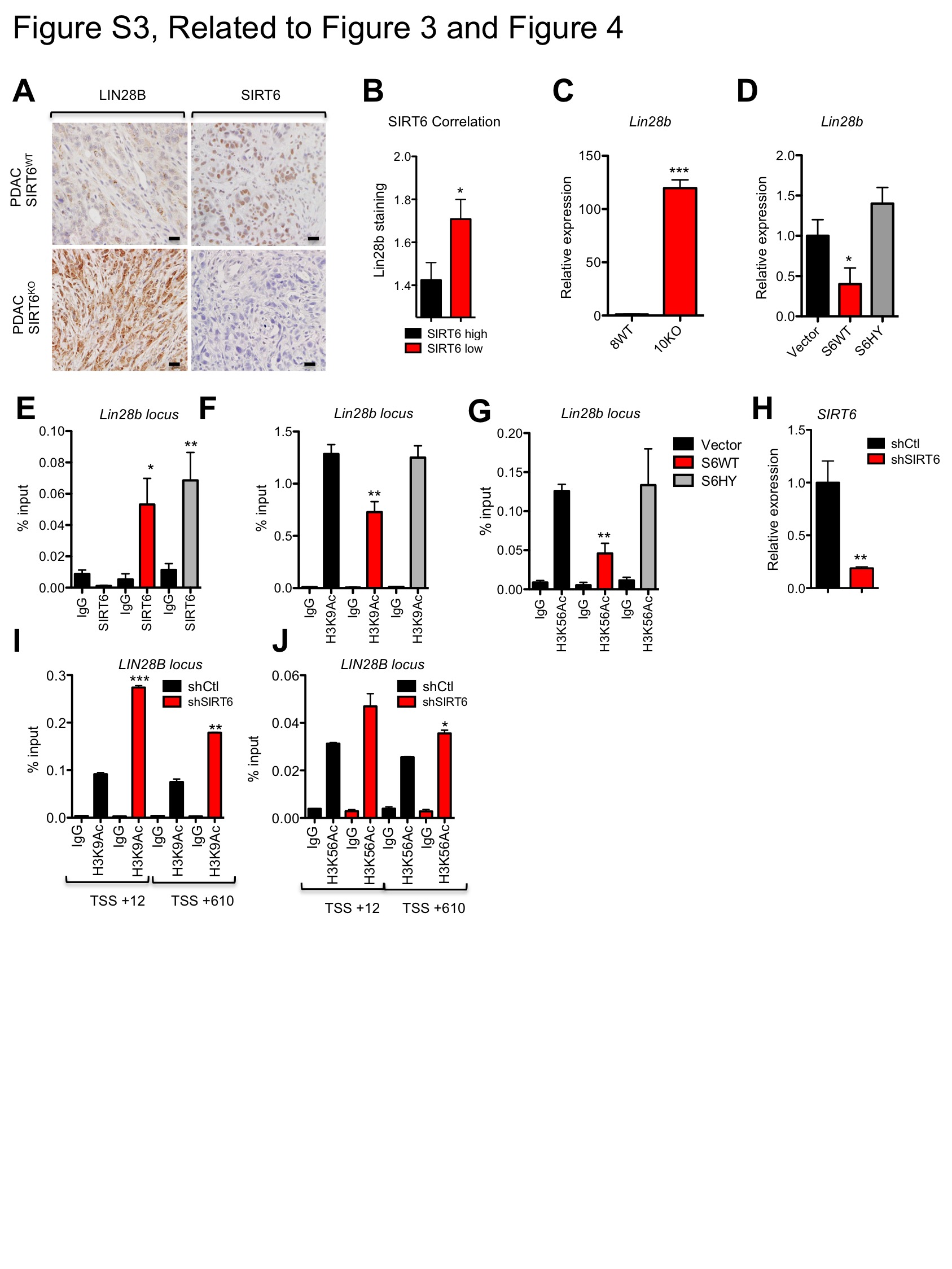
A, Venn diagram of gene promoters decorated by H3K56Ac in SIRT6 WT, SIRT6 KO and SIRT6 KO PDAC cells engineered to express SIRT6 WT as determined by Chromatin immunoprecipitation (ChIP) sequencing (Seq). B, Integrative genomics viewer track of H3K56Ac levels along the Lin28b promoter of the indicated murine PDAC cell lines. C, Expression of Lin28b in four independent SIRT6 WT and SIRT6 KO murine PDAC cell lines as measured by qRTPCR; data are presented as mean ± s.e.m. between three independent experiments. D, IB of chromatin and WCL from individual SIRT6 WT and SIRT6 KO PDAC cell lines. E,F qRTPCR analysis for expression of LIN28B and SIRT6 in human PDAC cell lines displayed as a bar graph (E) and scatter plot (F) demonstrating an inverse correlation; data represent mean ± s.e.m. between three independent experiments. G, Immunohistochemistry of LIN28B and SIRT6 in human PDAC samples (left) compared to normal pancreas (right). H, I, LIN28B levels in Panc3.27 expressing empty vector, S6WT or the S6HY catalytically inactive mutant as measured by qRTPCR (H) and IB (I). J, Lin28b levels in two independent SIRT6 KO murine PDAC cells lines expressing empty vector, S6WT or S6HY. Scale bars, black 50 μm. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. See also Fig S3.
Although highly expressed in embryonic tissues, Lin28b is fully silenced during differentiation and in healthy adult cells (Moss and Tang, 2003; Rybak et al., 2008; Yang and Moss, 2003), but may be aberrantly reactivated in a variety of human cancers (Iliopoulos et al., 2009; Thornton and Gregory, 2012; Viswanathan et al., 2009). While Lin28b has been correlated with advanced disease and poor prognosis (King et al., 2011; Lu et al., 2009; Viswanathan et al., 2009), its functional role and mechanism of reactivation in human cancer remain poorly understood. Furthermore, neither Lin28b expression, its regulation nor its functional role in PDAC have previously been explored. Although the Myc transcription factor can bind to consensus sequences within the Lin28b promoter (Chang et al., 2009), overexpression of Myc does not seem sufficient to drive its expression, suggesting that additional cofactors or epigenetic modifications are required (Viswanathan and Daley, 2010). The high levels of H3K56Ac over the Lin28b gene promoter in SIRT6 KO PDAC cells prompted us to explore whether loss of the epigenetic modifier SIRT6 may be one such mechanism of reactivation and whether the expression of Lin28b may drive the growth of a specific subset of PDAC.
Strikingly, all SIRT6 KO PDAC mouse lines analyzed exhibited far higher Lin28b expression than SIRT6 WT PDAC lines, both at the RNA and protein level (Figures 3C and 3D). Similar differences were seen in vivo, as PDAC tumors from SIRT6 KO mice were also universally positive for LIN28B, while SIRT6 WT PDAC tumors demonstrated only background levels of staining for LIN28B by immunohistochemistry (Figure S3A). Remarkably, expression of SIRT6 and LIN28B were also inversely correlated in human PDAC cell lines by quantitative real-time PCR (qRT-PCR) (Figures 3E and 3F). To define the physiological significance of these observations, we analyzed expression of LIN28B directly in our panel of 120 human PDAC patient samples. Consistently, tumors with low or undetectable levels of SIRT6 exhibited robust staining for LIN28B (Figures 3G and S3B). Lastly, ectopic expression of WT, but not catalytically inactive SIRT6, suppressed expression of LIN28B in Panc3.27 cells (Figures 3H and 3I) and in 2 independent murine SIRT6 KO PDAC lines (Figure 3J) confirming that loss of SIRT6 leads to the reactivation of Lin28b in both human and murine PDAC. Interestingly, SIRT6 may also regulate Lin28b expression in non-epithelial tissues as restoration of SIRT6 reversed the elevated levels of Lin28b expression observed in an immortalized murine embryonic fibroblast (MEF) cell line (Mostoslavsky et al., 2006) (Figures S3C and S3D).
SIRT6 Co-represses Myc-driven Transcription of Lin28b Through Histone Deacetylation
We have previously shown that SIRT6 represses Myc-dependent transcription by deacetylating histone marks, resulting in an inaccessible chromatin structure (Sebastian et al., 2012). Therefore, we decided to use chromatin immunoprecipitation (ChIP) assays to interrogate whether SIRT6 may co-repress Myc at the Lin28b locus. Interestingly, SIRT6 KO cells had significantly increased levels of H3K56Ac and H3K9Ac compared to SIRT6 WT cells at two known Myc binding sites within the Lin28b promoter, suggesting an open and permissive chromatin conformation (Figures 4A–C). Direct binding of SIRT6 to these Myc binding sites within the Lin28b promoter was confirmed in SIRT6 KO MEFs transfected with either WT SIRT6 or S6HY, whereas only WT SIRT6 could remove H3K56Ac and H3K9Ac marks in this region (Figures S3E–G). Furthermore, we found that this was a dynamic and constitutive process in human PDAC cells with high levels of SIRT6 (SIRT6high), such as COLO357 cells, where acute loss of SIRT6 by shRNA-mediated knockdown resulted in increased H3K9 and H3K56 acetylation in a homologous region of the human LIN28B promoter (Figures S3H–J). We then confirmed the critical role of Myc in driving Lin28b expression in PDAC by knocking down Myc in three independent SIRT6 KO murine and SIRT6low human PDAC cell lines, which resulted in a proportional reduction in Lin28b expression (Figures 4D and 4E). Consistent with their antagonistic relationship, Myc knockdown phenocopied restoration of SIRT6 expression in both SIRT6 KO murine and SIRT6low human PDAC cells, where we observed reduced cellular proliferation rates and tumor sphere formation (Figures 4F–J). Taken together, these data strongly support a mechanism by which SIRT6 actively co-represses Myc-dependent transcription in both human and murine PDAC specifically at the Lin28b locus, through deacetylation of H3K56 and H3K9 chromatin marks.
Figure 4. SIRT6 Co-represses Myc-driven Transcription of Lin28b Through Histone Deacetylation.

A, Schematic representation of the human genomic region near the transcription start site of LIN2B. Putative Myc binding sites are indicated (CACGTG or CATGTG); both sites are conserved between human and mouse. B,C, ChIP of H3K56Ac (B) and H3K9Ac (C) marks followed by amplification of the regions surrounding the Myc binding sites in the LIN28B promoter. D,F&I Analysis of three independent SIRT6 KO murine PDAC cell lines expressing the either shMyc or control hairpins for expression of Myc (left) and Lin28b (right) by qRT-PCR (D), cell proliferation (F) and tumor sphere forming ability (I). E,G,H&J Analysis of three independent SIRT6low PDAC cell lines expressing the either shMyc or control hairpins for expression of MYC (left) and LIN28B (right) by qRT-PCR (E), IB of MYC knockdown (G), cell proliferation (H) and tumor sphere forming ability (J). For E, H&J, data are representated as mean ±std between triplicates. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. See also Fig S3.
SIRT6low PDAC Cells are Addicted to Lin28b
We next examined the functional role of Lin28b in SIRT6 KO murine PDAC cells and SIRT6low human PDAC cells. Knocking down Lin28b with both shRNA and siRNA resulted in potent suppression of cell proliferation and tumor sphere formation in two independent murine SIRT6 KO cell lines, while two independent SIRT6 WT PDAC lines were completely insensitive to the same treatment (Figures S4A–F). More importantly, both shRNA and siRNA against LIN28B also markedly reduced the proliferation, tumor sphere forming ability and in vivo xenograft growth of several human SIRT6low PDAC lines without any discernable effect on the growth of human PDAC lines with normal levels of SIRT6 (SIRT6high) (Figures 5A–H and S4G–I). As with restoration of SIRT6 expression, knockdown of Lin28b led to both G1 cell cycle arrest and induction of apoptosis in two independent SIRT6low lines (Figures S4J–M). Thus, LIN28B is both upregulated and critically required for the growth and survival of this subset of PDAC cancers, as defined by loss of SIRT6 expression.
Figure 5. SIRT6low Human PDAC cells are Addicted to Lin28b.

A–H, Human PDAC cell lines with either high or low levels of SIRT6 expression were treated with shLIN28B versus a control hairpin. A, IB of WCL for SIRT6 and LIN28B. B, Number (left) and size (right) of tumor spheres. C, Photo-micrographs of tumor spheres. D,F, Growth curve of SIRT6low (D) and SIRT6high (F) human PDAC cells, quantified by MTT assay. E&G, show visualization of day 6 results in SIRT6low (E) and SIRT6high (G) human PDAC cells. H, Tumor weights of SIRT6low and SIRT6high PDAC lines grown as subcutaneous xenografts (n=5). * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. See also Fig S4 and S5.
Let-7 suppresses Igf2bps and Hmga2 expression and PDAC cell growth
The most well-characterized function of Lin28b is to inhibit the biogenesis of a family of 12 tumor suppressor microRNAs (miRNAs), collectively referred to as let-7 (Heo et al., 2008; Newman et al., 2008; Pasquinelli et al., 2000; Rybak et al., 2008; Viswanathan et al., 2008). Mature let-7 miRNAs are found in a reciprocal pattern with Lin28b, suppressed in embryonic tissues and highly expressed in normal adult cells (Bussing et al., 2008; Moss and Tang, 2003; Rybak et al., 2008; Yang and Moss, 2003), where it can promote the degradation of a number of targets involved in carcinogenesis (Johnson et al., 2005; Mayr et al., 2007; Sampson et al., 2007), including Insulin Growth Factor 2 Binding Proteins (IGF2BPs) and High Mobility Group AT-Hook 2 (HMGA2) (Boyerinas et al., 2008; Mayr et al., 2007; Nguyen et al., 2014; Park et al., 2007; Polesskaya et al., 2007). To determine whether Lin28b may drive the growth and survival of PDAC cells through the inhibition of let-7, we measured the levels of let-7 miRNA family members following Lin28b knockdown in our human SIRT6low PDAC cells. Indeed, the expression of almost all let-7 family members increased following Lin28b knockdown (Figures S5A; Table S2). To assess the functional significance of this let-7 reactivation, we transfected exogenous mimetics of the let-7c and let-7d family members into human PDAC cells and found that they specifically inhibited the growth of the SIRT6low cell lines BxPc3 and Panc-1 without any significant effect on the growth of the SIRT6high cell line COLO357 (Figures S5B and S5C). We also obtained a miRNA which mimics let-7g, but has been altered so that it is unable to be bound and degraded by Lin28b (Piskounova et al., 2008). Ectopic expression of this let-7g mimetic (7S21L) potently inhibited both proliferation and tumor sphere forming ability of SIRT6low PDAC cell lines (Figures S5D–F). Importantly, growth inhibition following ectopic expression of let-7 mimetics was also accompanied by a reduction in the expression of let-7 target genes, IGF2BP1, IGF2BP3, and HMGA2 as well as LIN28B, which is also directly inhibited by let-7 as a part of a feedback loop (Rybak et al., 2008) (Figures S5G–I). Thus, multiple let-7 family members potently and selectively inhibit the growth of SIRT6low PDAC cells, potentially through the suppression of let-7 target genes.
SIRT6low PDAC are dependent on let-7 target genes for growth
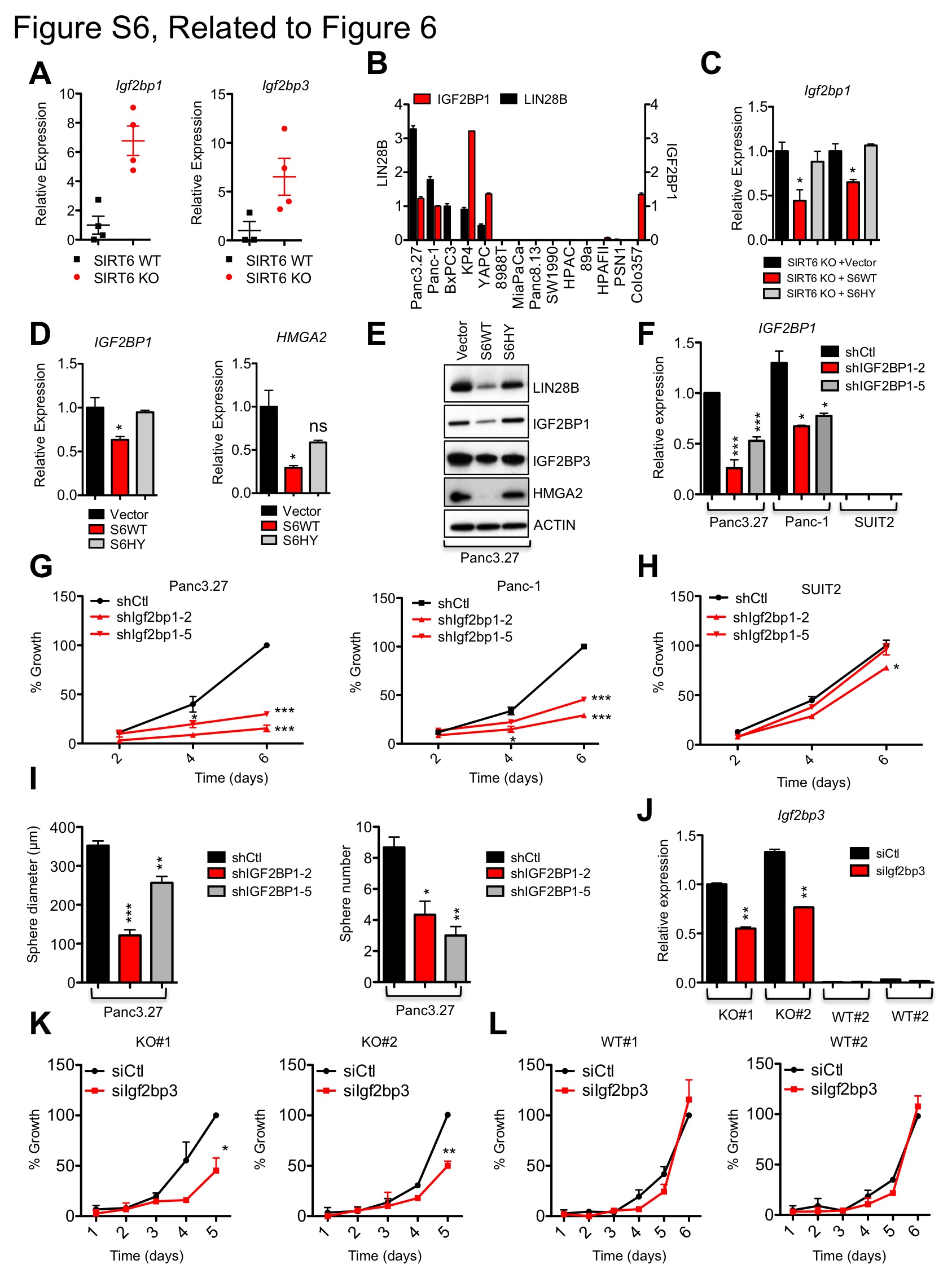
Interestingly, high expression of Igf2bp1 and Igf2bp3, which are both directly inhibited by let-7, is correlated with increased agressiveness and metastasis in pancreatic tumors (Thakur et al., 2008; Yantiss et al., 2005), and in support of a causal role in transformation, transgenic overexpression of murine Igf2bp3 (IMP3/KOC) leads to increased cell proliferation and metaplasia of pancreatic acinar cells (Wagner et al., 2003). Both Igf2bp1 and Igf2bp3 were highly upregulated in our SIRT6 KO cells relative to SIRT6 WT cells (Figure S6A) and in our human SIRT6low PDAC cells (Figures 6A and S6B). In addition, HMGA2 is another let-7 target gene that is associated with advanced tumor grade and lymph node metastasis in PDAC (Piscuoglio et al., 2012) and strikingly, was universally expressed in SIRT6low lines, but almost completely absent in all SIRT6high lines analyzed (Figure 6A). Restoration of WT but not catalytically inactive SIRT6 in SIRT6 KO murine and SIRT6low human PDAC cells reduced expression of both Lin28b and let-7 target genes (Figures S6C–E), confirming the direct role of SIRT6 in regulating this pathway. While these findings are consistent with a model whereby SIRT6 acts upstream of the Lin28b/let-7 axis to suppress Lin28b expression, enhance let-7 levels and inhibit expression of let-7 target genes, the functional role of each of these let-7 target genes in driving the growth of PDAC cells has not yet been clearly established. Therefore, we knocked down either HMGA2 or IGF2BP3 in a panel of human PDAC cell lines. Remarkably, although the Lin28b/let-7 pathway has many known targets, knock-down of either HMGA2, IGF2BP1 or IGF2BP3 was sufficient to inhibit both proliferation and tumor sphere formation in SIRT6low PDAC cells without any discernable effect on SIRT6high PDAC cells (Figures 6B–H and S6F–I). Further, knock-down of Igf2bp3 with siRNA (Figure S6J) specifically slowed growth of SIRT6 KO cells but had no effect on SIRT6 WT murine PDAC cells (Figures S6K and S6L). Thus, multiple let-7 target genes may cooperate to drive the growth of SIRT6low PDAC.
Figure 6. Lin28b Decreases let-7 Levels and Increases Igf2bp1/3 and Hmga2 Levels in PDAC.

A, IB of WCL and chromatin of human PDAC cell lines. SIRT6 and ACTIN have been reproduced from Figure 2F for comparison as these are from the same blot. B–E, SIRT6low and SIRT6high human PDAC cells treated with either shHMGA2 or control hairpin. Confirmation of HMGA2 knockdown by qRTPCR (B) and IB (C). D, Growth curves of SIRT6low (Panc3.27, BxPc3 and Su86.86) and SIRT6high (COLO357) human PDAC cell lines. E, Quantification of sphere diameter of SIRT6low (Panc3.27 and BxPc3) and SIRT6high (COLO357) human PDAC cell lines (left) and representative photo pictomicrographs (right). F–H, SIRT6low and SIRT6high human PDAC cells treated with either shIGF2BP3 or control hairpins. F, Confirmation of IGF2BP3 knockdown by qRTPCR. G, Growth curves of SIRT6low (Panc3.27, BxPc3 and Su86.86) and SIRT6high (SUIT2) human PDAC cell lines. H, Quantification of sphere diameter and number of SIRT6low (Panc3.27 and BxPc3) and SIRT6high (COLO357) human PDAC cell lines (left) and representative photo pictomicrographs (right). For B & D–H, data are representated as mean ±std between triplicates. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. See also Fig S6.
Increased expression of LIN28B and let-7 target genes correlates with poor survival in PDAC
These observations prompted us to investigate the relevance of this pathway to the human disease. As shown previously, loss of SIRT6 expression in human PDAC tumors defined a subset of patients with a worse prognosis (Figure 1B). Strikingly, elevated expression of LIN28B also correlated with poor prognosis in the same cohort of 120 patient samples (Figure 7A). Moreover, gene set enrichment analysis (GSEA) comparing PDAC tumors (Badea et al., 2008; Biankin et al., 2012; Pei et al., 2009; Perez-Mancera et al., 2012; Zhang et al., 2012) and cell lines (Barretina et al., 2012) (Table S3) with high versus low expression of LIN28B revealed that LIN28Bhigh tumors were strongly enriched for the expression of Myc targets (Figure S7A), as well as for let-7 targets, curated in three independent gene sets (Figure 7B). This finding was further validated in the CCLE dataset (Figure 7C). More specifically, the oncofetal targets of let-7, which includes the IGF2BPs and HMGA2, were upregulated in LIN28Bhigh tumors in three independent datasets (Figure 7D). Accordingly, loss of let-7 expression, as measured by in-situ hybridization (ISH) for let-7a, also corresponded to a shorter overall survival (Figure S7B). Finally, expression of these oncofetal targets IGF2BP3 and HMGA2 correlated both with each other and a worse prognosis in the cancer genome atlas (TCGA) dataset (Figures 7E and 7F). Taken together, our findings are consistent with a model whereby loss of SIRT6 in PDAC allows for aberrant hyperacetylation of the Lin28b promoter, enhancing Myc-driven transcription of Lin28b, which then inhibits the let-7 family of miRNA. This allows for the reactivation of let-7 target genes such as HMGA2 and IGF2BPs, which serve to drive the growth and survival of a highly aggressive form of pancreatic cancer (Figure 7G).
Figure 7. Increased expression of LIN28B and let-7 target genes correlates with poor survival in PDAC.

A, Kaplan–Meier analysis of the indicated PDAC patient samples based on LIN28B IHC score (n=120). B–C, Gene Set Enrichment Analysis (GSEA) plots showing that human PDAC tumors (B) and PDAC cell lines from the Cancer Cell line Encyclopedia (CCLE) (C) with high levels of LIN28B (LIN28Bhigh) overexpress many of the genes that are regulated by let-7. D, GSEA plots showing that human LIN28Bhigh PDAC tumors overexpress targets of let-7 which are oncofetal genes. E, Correlation of HMGA2 and IGF2BP3 RNA expression in human PDAC samples from the TCGA pancreatic cancer dataset. F, Kaplan-Meier survival curves based on expression of HMGA2 (left) and IGF2BP3 (right) in human pancreatic cancer datasets from the TCGA. G, Model for SIRT6 loss in PDAC pathogenesis. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. See also Fig S7.
DISCUSSION
Alterations in epigenetic control are an important hallmark of cancer. Such alterations are thought to endow cells with the plasticity to override normal differentiation and growth control programs. Due to their poor vascularity and dense stroma, PDAC cells must acquire multiple metabolic adaptations to grow in a hypoperfused microenvironment. SIRT6 is a nutrient sensor and histone deacetylase that reprograms the epigenome in response to nutrient stress. We show that SIRT6 is downregulated in PDAC relative to normal tissue and that loss of SIRT6 leads to dysregulation of the PDAC epigenome to drive its growth. By developing novel GEMMs, we demonstrate that ablation of SIRT6 potently cooperates with activated Kras (which is mutated in >90% of human PDAC) to accelerate PDAC onset and promote metastasis. Mechanistically, loss of SIRT6 results in hyperacetylation of H3K9 and H3K56 at the promoter of the LIN28B gene, creating a more permissive chromatin state and allowing for the Myc transcription factor to drive its expression. We further show that this aberrant Lin28b expression is required for the growth of SIRT6-deficient tumor cells, thus identifying Lin28b as an oncogenic driver in this distinct subset, representing ~ 30–40% of human PDAC.
The Lin28/let-7 axis is now recognized as central to maintaining proper cell fate and coordinating proliferation, growth, and energy utilization at the cellular level, as well as growth, developmental timing, tissue homeostasis and metabolism in whole organisms (Thornton and Gregory, 2012). While Lin28b is silenced during embryonic development (Moss and Tang, 2003; Rybak et al., 2008; Yang and Moss, 2003), it may be aberrantly reactivated in a variety of human cancers (Iliopoulos et al., 2009; Thornton and Gregory, 2012; Viswanathan et al., 2009) by mechanisms that remain poorly understood. We recently identified eight loss-of-function tumor-associated SIRT6 point mutations, several of which specifically abrogated SIRT6 deacetylase activity, and we had previously found that many human cancer cell lines demonstrate copy number loss of the SIRT6 locus (Kugel et al., 2015). Thus, our findings that loss of SIRT6 allows for the reactivation of Lin28b in PDAC may have important implications for other cancers as well. In addition, future studies will be needed to determine what role SIRT6 plays in the silencing of Lin28b during embryonic development and whether Lin28b expression may contribute to the early post-natal lethality observed in SIRT6 KO mice (Mostoslavsky et al., 2006).
Given the critical roles for Lin28b in stem cell pluripotency, one can speculate that overexpression of oncofetal proteins reactivate programs of embryonic growth to promote a more “undifferentiated” and thereby aggressive form of pancreatic cancer. Consistently, upregulated genes downstream of Lin28b, includes the oncofetal RNA-binding proteins Igf2bp1 & 3 that have been associated with poorly differentiated PDAC. Expression of Igf2bps increase progressively with PDAC tumor stage (Yantiss et al., 2005) and high levels of Igf2bps in PDAC correlate with increased metastasis and extremely poor survival outcome (Schaeffer et al., 2010; Taniuchi et al., 2014). In this context, we observed signs of both accelerated initiation (increased number of PanIN) as well as increased metastatic potential in mice expressing high levels of Lin28b and Igf2bps. Igf2bps also have functions in binding and stabilizing IGF2 and Myc transcripts, thus increasing their translation (Bell et al., 2013; Nielsen et al., 2004; Noubissi et al., 2006). Reinforcing Myc signaling and increasing IGF2 signaling could both serve to encourage proliferation and survival of our PDAC cells. Strikingly, knockdown of Igf2bp3 in multiple independent SIRT6low and SIRT6 KO cell lines was sufficient to significantly inhibit their growth, while having no effect on the growth of SIRT6high and SIRT6 WT lines. Similarly, elevated protein expression of HMGA2 in PDAC has been associated with a more advanced tumor grade, epithelial to mesenchymal transition, and lymph node metastases, and this protein also promoted the growth of SIRT6low but not SIRT6high PDAC cells. Thus, we propose a model whereby Lin28b drives the growth of SIRT6-deficient PDAC through the inhibition of multiple let-7 isoforms, resulting in a coordinated upregulation of a large number of Lin28b/let-7 target genes, including oncofetal proteins like IGF2BPs and HMGA2 (Figure 7G).
There is some evidence that reactivation of Lin28b may be the result of a more general mechanism that follows loss of epigenetic barriers. When human embryonic stem cells were used to model pediatric gliomas with H3.3K27M histone mutations, the gene that was reactivated to the highest extent in response to global H3K27 hypomethylation was LIN28B (Funato et al., 2014). Additionally, prolonged inhibition of the methyltransferase EZH2 in glioblastoma lead to upregulation of Lin28b expression (de Vries et al., 2015). EZH2 acts mainly through trimethylation of histone H3 lysine27, which is associated with transcriptional repression, thus loss of H3K27 trimethylation in two different contexts lead to upregulation of Lin28b expression. The activity of SIRT6 may provide a previously unrecognized epigenetic barrier, suppressing the expression of Lin28b specifically in PDAC. The H3K9 and H3K56 hyperacetylation of the Lin28b gene in response to SIRT6 loss may function to inhibit the reciprocal methylation of this histone residue, preventing H3K9Me3-mediated gene silencing, thereby licensing the aberrant re-expression of Lin28b to drive this fatal disease.
Therapeutic strategies for Kras-driven cancers such as PDAC have been limited by a failure to identify pathways that are specifically required in cancer cells but dispensable in normal tissues. Oncofetal proteins represent attractive targets for such strategies, as they are highly expressed in embryonic tissues but silenced in normal adult cells. Thus, our critical findings highlight Lin28b as a novel oncogene in PDAC and identify a clinically-relevant and molecularly-defined subset of PDAC, which may benefit from therapeutic approaches aimed at targeting components of the Lin28b/let-7 pathway and provide new insights into the epigenetic mechanisms governing the reactivation of these developmental programs in cancer.
EXPERIMENTAL PROCEDURES
All experimental procedures are described in detail in the Supplemental Experimental Procedures.
Mice
Mice were housed in pathogen-free animal facilities. All experiments were conducted under protocol 2007N000200 approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital. Mice were maintained on a mixed 129SV/C57BL/6 background. Data presented include both male and female mice. All mice included in the survival analysis were euthanized when criteria for disease burden were reached.
Sirt6flox/flox conditional strain (Sebastian et al., 2012) were crossed with the p48-Cre strain (Kawaguchi et al., 2002), the conditional p53flox strain (Marino et al., 2000) and the LSL-KrasG12D strain (Jackson et al., 2001) which consists of a mutant KrasG12D allele knocked into the endogenous Kras locus, preceded by an LSL cassette.
Tumor Sphere Assay
Cells were plated as single-cell suspension in ultralow attachment 24-well plates (Corning) and grown in DMEM/F12 medium (serum free) supplemented with 20 μl ml−1 B27 (Invitrogen), 20 ng ml−1 EGF and 20 ng ml−1 bFGF. Fresh media (300 μl) was added every 3 days. Tumor spheres were counted and photographed at day 10. Tumor sphere assays were performed in triplicate, and are represented as mean ± s.e.m. between three independent experiments.
Xenografts
For murine PDAC xenografts 2×104 cells were injected subcutaneously into the flanks of Severe Combined Immunodeficiency (SCID) mice (Charles River). For human PDAC xenografts 5×106 cells in a 1:1 mixture of PBS:Matrigel (Corning) were injected subcutaneously into the flanks of SCID mice (Charles River) For both models, mice were checked for the appearance of tumors twice per week, and the tumors were harvested when they reached ~100 mm in diameter.
Chromatin Immunoprecipitation
ChIP and qRTPCR were performed as previously described (Donner et al., 2007). Details are listed in Extended Experimental Procedures. The antibodies used were anti-H3K9Ac (Millipore 07-352), anti-H3K56Ac (ab76307) and the primers’ sequences for all the qRTPCRs are included in Table S4. Quantitative RTPCR for ChIP analyses were performed in duplicate and are represented as mean ± s.e.m. between two independent experiments.
GEO Accession Numbers
The GEO number for the ChIP-Seq and small RNA-Seq data sets in this manuscript is GSE79505.
Statistics
For qRTPCR analysis, proliferation assays, glucose uptake, tumor sphere formation, and tumor size, significance was analyzed using 2-tailed Student’s t test. A p-value of less than 0.05 was considered statistically significant. A log-rank test was used to determine significance for Kaplan-Meier analyses. Fisher’s exact t test was performed for comparison of metastatic disease burdens.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Richard Gregory for the pBabe let-7g 7S21L mutant construct and members of the Mostoslavsky and Bardeesy labs for invaluable discussions and advise. This work was supported in part by NIH grants CA175727-01, R21CA198109-01 (to R.M.), P50CA1270003, P01 CA117969-07, R01 CA133557-05, The National Pancreas Foundation (to R.M.), and the Linda J. Verville Cancer Research Foundation (to NB). R.M. is a Kristine and Bob Higgins MGH Research Scholar, a Warshaw Institute Fellow, a Howard Goodman Awardee, and the recipient of the Glenn Award for Research in Biological Mechanisms of Aging. N.B. holds the Gallagher Endowed Chair in Gastrointestinal Cancer Research. N.B. is a member of the Andrew Warshaw Institute for Pancreatic Cancer Research. S.K. is the recipient of a Canadian Institutes of Health Research postdoctoral fellowship. S.K.S. is supported by a NCI Mentored Clinical Scientist Research Career Development Award (1K08CA194268-01) and a DF/HCC GI SPORE Career Development Project Award (P50CA127003). C.S. is supported by the Visionary Postdoctoral Fellowship from the Department of Defense. A.G. is the recipient of the Broad Institute SPARC (Scientific Projects to Accelerate Research and Collaboration) program.
Footnotes
Supplemental Information includes Extended Experimental Procedures, seven figures, and five tables and can be found with this article online at http://
AUTHOR CONTRIBUTIONS: S.K. collected the data. S.K. and R.M. analyzed the data. R.M., S.K.S., C.S. and N.B. contributed to the experimental design. C.S. performed cell cycle and annexin V apoptosis assays. J.F. and Y.K. performed immunohistochemistry on tissue sections. K.N.R. and S.R. performed computational analysis on gene expression data. E.J. and R.I.S performed small RNA-seq and computational analysis. A.G and A.G. performed the ChIP-sequencing experiments. K.S.A and M.N.R performed the RNA in situ hybridization. V.D. analyzed the histology from the murine pancreas specimens and scored immunohistochemistry slides. S.K and R.M. wrote the paper. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepato-gastroenterology. 2008;55:2016–2027. [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M, Huttelmaier S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cellular and molecular life sciences : CMLS. 2013;70:2657–2675. doi: 10.1007/s00018-012-1186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyerinas B, Park SM, Shomron N, Hedegaard MM, Vinther J, Andersen JS, Feig C, Xu J, Burge CB, Peter ME. Identification of let-7-regulated oncofetal genes. Cancer research. 2008;68:2587–2591. doi: 10.1158/0008-5472.CAN-08-0264. [DOI] [PubMed] [Google Scholar]
- Bussing I, Slack FJ, Grosshans H. let-7 microRNAs in development, stem cells and cancer. Trends in molecular medicine. 2008;14:400–409. doi: 10.1016/j.molmed.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, Jung J, Gao P, Dang CV, Beer MA, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3384–3389. doi: 10.1073/pnas.0808300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries NA, Hulsman D, Akhtar W, de Jong J, Miles DC, Blom M, van Tellingen O, Jonkers J, van Lohuizen M. Prolonged Ezh2 Depletion in Glioblastoma Causes a Robust Switch in Cell Fate Resulting in Tumor Progression. Cell reports. 2015 doi: 10.1016/j.celrep.2014.12.028. [DOI] [PubMed] [Google Scholar]
- Donner AJ, Szostek S, Hoover JM, Espinosa JM. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Molecular cell. 2007;27:121–133. doi: 10.1016/j.molcel.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346:1529–1533. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Molecular cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & development. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nature genetics. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- King CE, Cuatrecasas M, Castells A, Sepulveda AR, Lee JS, Rustgi AK. LIN28B promotes colon cancer progression and metastasis. Cancer research. 2011;71:4260–4268. doi: 10.1158/0008-5472.CAN-10-4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugel S, Feldman JL, Klein MA, Silberman DM, Sebastian C, Mermel C, Dobersch S, Clark AR, Getz G, Denu JM, et al. Identification of and Molecular Basis for SIRT6 Loss-of-Function Point Mutations in Cancer. Cell reports. 2015 doi: 10.1016/j.celrep.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Katsaros D, Shaverdashvili K, Qian B, Wu Y, de la Longrais IA, Preti M, Menato G, Yu H. Pluripotent factor lin-28 and its homologue lin-28b in epithelial ovarian cancer and their associations with disease outcomes and expression of let-7a and IGF-II. European journal of cancer. 2009;45:2212–2218. doi: 10.1016/j.ejca.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes & development. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss EG, Tang L. Conservation of the heterochronic regulator Lin-28, its developmental expression and microRNA complementary sites. Developmental biology. 2003;258:432–442. doi: 10.1016/s0012-1606(03)00126-x. [DOI] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. Rna. 2008;14:1539–1549. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LH, Robinton DA, Seligson MT, Wu L, Li L, Rakheja D, Comerford SA, Ramezani S, Sun X, Parikh MS, et al. Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer cell. 2014;26:248–261. doi: 10.1016/j.ccr.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Kristensen MA, Willemoes M, Nielsen FC, Christiansen J. Sequential dimerization of human zipcode-binding protein IMP1 on RNA: a cooperative mechanism providing RNP stability. Nucleic acids research. 2004;32:4368–4376. doi: 10.1093/nar/gkh754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noubissi FK, Elcheva I, Bhatia N, Shakoori A, Ougolkov A, Liu J, Minamoto T, Ross J, Fuchs SY, Spiegelman VS. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature. 2006;441:898–901. doi: 10.1038/nature04839. [DOI] [PubMed] [Google Scholar]
- Park SM, Shell S, Radjabi AR, Schickel R, Feig C, Boyerinas B, Dinulescu DM, Lengyel E, Peter ME. Let-7 prevents early cancer progression by suppressing expression of the embryonic gene HMGA2. Cell cycle. 2007;6:2585–2590. doi: 10.4161/cc.6.21.4845. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Muller P, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer cell. 2009;16:259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KA, Grutzmann R, Aust D, Rummele P, et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012;486:266–270. doi: 10.1038/nature11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscuoglio S, Zlobec I, Pallante P, Sepe R, Esposito F, Zimmermann A, Diamantis I, Terracciano L, Fusco A, Karamitopoulou E. HMGA1 and HMGA2 protein expression correlates with advanced tumour grade and lymph node metastasis in pancreatic adenocarcinoma. Histopathology. 2012;60:397–404. doi: 10.1111/j.1365-2559.2011.04121.x. [DOI] [PubMed] [Google Scholar]
- Piskounova E, Viswanathan SR, Janas M, LaPierre RJ, Daley GQ, Sliz P, Gregory RI. Determinants of microRNA processing inhibition by the developmentally regulated RNA-binding protein Lin28. The Journal of biological chemistry. 2008;283:21310–21314. doi: 10.1074/jbc.C800108200. [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Cuvellier S, Naguibneva I, Duquet A, Moss EG, Harel-Bellan A. Lin-28 binds IGF-2 mRNA and participates in skeletal myogenesis by increasing translation efficiency. Genes & development. 2007;21:1125–1138. doi: 10.1101/gad.415007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. The New England journal of medicine. 2014;371:1039–1049. doi: 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, Wulczyn FG. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nature cell biology. 2008;10:987–993. doi: 10.1038/ncb1759. [DOI] [PubMed] [Google Scholar]
- Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, Petrelli NJ, Dunn SP, Krueger LJ. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer research. 2007;67:9762–9770. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- Schaeffer DF, Owen DR, Lim HJ, Buczkowski AK, Chung SW, Scudamore CH, Huntsman DG, Ng SS, Owen DA. Insulin-like growth factor 2 mRNA binding protein 3 (IGF2BP3) overexpression in pancreatic ductal adenocarcinoma correlates with poor survival. BMC cancer. 2010;10:59. doi: 10.1186/1471-2407-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi K, Furihata M, Hanazaki K, Saito M, Saibara T. IGF2BP3-mediated translation in cell protrusions promotes cell invasiveness and metastasis of pancreatic cancer. Oncotarget. 2014;5:6832–6845. doi: 10.18632/oncotarget.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur A, Bollig A, Wu J, Liao DJ. Gene expression profiles in primary pancreatic tumors and metastatic lesions of Ela-c-myc transgenic mice. Molecular cancer. 2008;7:11. doi: 10.1186/1476-4598-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JE, Gregory RI. How does Lin28 let-7 control development and disease? Trends in cell biology. 2012;22:474–482. doi: 10.1016/j.tcb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ. Lin28: A microRNA regulator with a macro role. Cell. 2010;140:445–449. doi: 10.1016/j.cell.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O’Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nature genetics. 2009;41:843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Kunsch S, Duerschmied D, Beil M, Adler G, Mueller F, Gress TM. Transgenic overexpression of the oncofetal RNA binding protein KOC leads to remodeling of the exocrine pancreas. Gastroenterology. 2003;124:1901–1914. doi: 10.1016/s0016-5085(03)00402-5. [DOI] [PubMed] [Google Scholar]
- Yang DH, Moss EG. Temporally regulated expression of Lin-28 in diverse tissues of the developing mouse. Gene expression patterns : GEP. 2003;3:719–726. doi: 10.1016/s1567-133x(03)00140-6. [DOI] [PubMed] [Google Scholar]
- Yantiss RK, Woda BA, Fanger GR, Kalos M, Whalen GF, Tada H, Andersen DK, Rock KL, Dresser K. KOC (K homology domain containing protein overexpressed in cancer): a novel molecular marker that distinguishes between benign and malignant lesions of the pancreas. The American journal of surgical pathology. 2005;29:188–195. doi: 10.1097/01.pas.0000149688.98333.54. [DOI] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Schetter A, He P, Funamizu N, Gaedcke J, Ghadimi BM, Ried T, Hassan R, Yfantis HG, Lee DH, et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PloS one. 2012;7:e31507. doi: 10.1371/journal.pone.0031507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.