

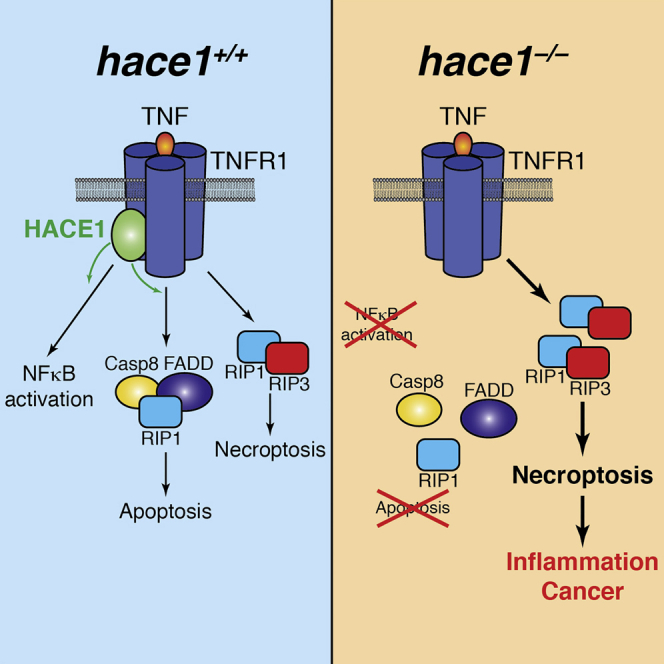
Summary
The HECT domain E3 ligase HACE1 has been identified as a tumor suppressor in multiple cancers. Here, we report that HACE1 is a central gatekeeper of TNFR1-induced cell fate. Genetic inactivation of HACE1 inhibits TNF-stimulated NF-κB activation and TNFR1-NF-κB-dependent pathogen clearance in vivo. Moreover, TNF-induced apoptosis was impaired in hace1 mutant cells and knockout mice in vivo. Mechanistically, HACE1 is essential for the ubiquitylation of the adaptor protein TRAF2 and formation of the apoptotic caspase-8 effector complex. Intriguingly, loss of HACE1 does not impair TNFR1-mediated necroptotic cell fate via RIP1 and RIP3 kinases. Loss of HACE1 predisposes animals to colonic inflammation and carcinogenesis in vivo, which is markedly alleviated by genetic inactivation of RIP3 kinase and TNFR1. Thus, HACE1 controls TNF-elicited cell fate decisions and exerts tumor suppressor and anti-inflammatory activities via a TNFR1-RIP3 kinase-necroptosis pathway.
Graphical Abstract
Highlights
-
•
Hace1 deficiency impairs TNF-driven NF-κB activation and apoptosis
-
•
Necroptosis via RIP1/RIP3/MLKL is still functional in the absence of Hace1
-
•
Hace1–/– animals show enhanced severity of colitis and colon cancer
-
•
Genetic inactivation of RIP3 and TNFR1 reverts the phenotype of hace1–/– mice
Tortola et al. report that the E3 ubiquitin ligase HACE1 is a gatekeeper of TNFR1-mediated cell fate. Hace1 deficiency impairs TNF-driven NF-κB activation and apoptosis and predisposes cells to necroptosis. Consequently, hace1–/– mice show enhanced colitis and colon cancer, which can be reverted by inactivation of pro-necroptotic kinase RIP3 and TNFR1.
Introduction
Posttranslational modifications play a key role in the regulation of protein activity, localization, and stability, as well as in the control of protein-protein interactions. Among them, ubiquitylation counts as one of the most prominent and best-characterized modifications. Ubiquitin is a small protein that can be covalently attached to lysine residues of other proteins through the interplay of different ubiquitin-activating (E1) and -conjugating (E2) enzymes and ubiquitin ligases (E3) (Hershko and Ciechanover, 1998). The analysis of mice lacking various components of the ubiquitylation machinery highlighted the importance of ubiquitylation in several cellular and physiological processes, spanning from autophagy, transcription factor regulation, and DNA repair to the control of cell cycle and cell death (Bergink and Jentsch, 2009, Bernassola et al., 2010, Hershko and Ciechanover, 1998, Mukhopadhyay and Riezman, 2007), which in turn can affect complex processes such as the immune response (Bhoj and Chen, 2009, Corn and Vucic, 2014), the maintenance of tissue homeostasis (Kumari et al., 2014), and tumorigenesis (Hoeller and Dikic, 2009, Paolino et al., 2014).
Life or death cell fate decisions are critical for development, against infectious diseases, or cancer formation. Apoptosis was considered the sole form of programmed cell death during development or disease (Elmore, 2007). However, besides apoptosis, alternative programmed necrosis or necroptosis can be induced by death receptors (Vandenabeele et al., 2010). Survival, apoptosis, and necroptosis can all be triggered by the same cell-surface receptors. There has been intense interest in how these fate decisions are made by identifying the gatekeepers of apoptotic versus necroptotic cell fate and the signaling cascades involved (Christofferson and Yuan, 2010). One of the most paradigmatic signaling pathways that regulates cellular fate is triggered by the tumor necrosis factor receptor 1 (TNFR1). TNFR1 engagement elicits cell survival pathways via NF-κB induction (Beg and Baltimore, 1996, Van Antwerp et al., 1996, Wang et al., 1998). When NF-κB activation is inhibited, TNF stimulation triggers caspase-8-mediated apoptosis via a defined death effector complex (Micheau and Tschopp, 2003, Wang et al., 1996). In specific conditions, blockade of both NF-κB and apoptosis may result in programmed necrotic cell death (Cho et al., 2009, He et al., 2009, Vanden Berghe et al., 2014, Zhang et al., 2009). Necroptosis is mediated via the RIP1/RIP3/MLKL kinase pathway and recently has been implicated in normal development and in pathogenesis of diseases, including ischemic injury, neurodegeneration, and intestinal inflammation (Duprez et al., 2011, Kaiser et al., 2011, Kang et al., 2013, Linkermann et al., 2012, Welz et al., 2011).
HECT domain and ankyrin repeat-containing E3 ligase 1 (HACE1) is an E3 ligase initially identified in the context of sporadic Wilms’ tumor (Anglesio et al., 2004). A number of reports showed that HACE1 is downregulated in various types of tumors (Anglesio et al., 2004, Hibi et al., 2008, Küçük et al., 2013, Zhang et al., 2007), and a previous study from our group highlighted the important function of HACE1 as a tumor suppressor: mice lacking HACE1 showed an increased propensity to develop spontaneous tumors as well as a reduced survival in different cancer models (Zhang et al., 2007). In addition to this function, HACE1 has been implicated in Golgi membrane dynamics (Tang et al., 2011), control of oxidative stress (Daugaard et al., 2013, Rotblat et al., 2014), and heart failure following cardiac injury (Zhang et al., 2014). However, the complexity of the molecular and cellular processes potentially regulated by HACE1 is still largely unexplored.
Here we report that HACE1 is a key regulator of TNFR1-mediated cell fate. Genetic inactivation of hace1 impairs NF-κB activation and apoptosis downstream of TNFR1, whereas RIP3-regulated necroptotic cell fate is not affected. Importantly, we found that hace1 mutant mice exhibit enhanced intestinal inflammation and colon cancer following epithelial injury, which can be rescued by simultaneous genetic inactivation of either RIP3 or TNFR1.
Results
HACE1 Controls TNFR1-Induced NF-κB Activation
To identify pathways relevant to HACE1 function, we scanned the signaling pathways downstream of multiple ligand/receptor pairs, using hace1 mutant (hace1–/–) mouse embryonic fibroblasts (MEFs). Intriguingly, hace1–/– MEFs stimulated with TNF showed compromised phosphorylation and incomplete degradation of the NF-κB inhibitor IκBα. As a consequence, TNF-induced phosphorylation and activation of the NF-κB subunit p65 was strongly reduced in hace1–/– MEFs (Figure 1A). Impaired NF-κB activation was confirmed using an electrophoretic mobility shift assay (EMSA) filter plate assay (Figure 1B). Re-expression of wild-type (WT) HACE1 in hace1–/– MEFs, but not that of the E3 ligase inactive HACE1 C876S mutant (Anglesio et al., 2004), restored TNF-induced p65 NF-κB activation (Figure 1B; Figure S1A). Moreover, TNF-induced NF-κB-dependent expression of interleukin-6 (IL-6) (Bollrath and Greten, 2009) also was markedly inhibited in hace1-deficient MEFs (Figure 1C).
Figure 1.

HACE1 Is Essential for the TNFR1-NF-κB Response
(A) Hace1+/+ and hace1–/– MEFs were stimulated with murine TNF (10 ng/ml) to induce the activation of TNFR1 downstream pathways. β-actin protein levels are shown as controls. Results are representative of three independent experiments.
(B) NF-κB activation of TNF (10 ng/ml) stimulated (+) and untreated (−) hace1+/+ MEFs, hace1–/– MEFs, hace1–/– MEFs re-expressing WT HACE1, and hace1–/– MEFs re-expressing the E3 ligase-dead C876S mutant. Triplicate filter plate assays of nuclear extracts are shown as mean values ± SD. HACE1 expression and re-expression were confirmed by western blot.
(C) Time course of interleukin-6 (IL-6) induction in hace1+/+ and hace1–/– MEFs following TNF (10 ng/ml) stimulation. IL-6 concentration was determined by ELISA in triplicate (mean values ± SD).
(D) In vivo Listeria monocytogenes infections in hace1+/+ and hace1–/– mice. Four days after infection animals were sacrificed, spleen extracts were plated, and bacterial loads in each spleen were determined as colony-forming units (CFUs)/spleen. Data for individual mice are shown.
(E) Kaplan-Meier survival curves of hace1+/+ and hace1–/– mice after Listeria infections are shown (p < 0.01).
See also Figure S1.
In addition to defective NF-κB induction, TNFR1-induced JNK and consequent c-Jun phosphorylation were markedly reduced in hace1–/– MEFs, a phenomenon that was accompanied by increased and prolonged ERK1/2 and p38-MAPK phosphorylation (Figure 1A). TNFR1 and NF-κB are essential for the in vivo clearance of the intracellular pathogen Listeria monocytogenes (Pfeffer et al., 1993, Sha et al., 1995). Considering the observed in vitro defects in TNFR1/NF-κB activation, we tested whether HACE1 also controls the response to L. monocytogenes in vivo. Importantly, we observed impaired clearance of L. monocytogenes (Figure 1D), accompanied by enhanced lethality of the hace1–/– mice as compared to their WT littermates (Figure 1E), a phenotype that is compatible with and might be dependent on defective TNFR1/NF-κB activation. Thus, HACE1 E3 ligase activity is essential for TNF-induced activation of both NF-κB and JNK downstream signaling in vitro, and HACE1 controls in vivo immunity to L. monocytogenes.
Impaired TNFR1-Induced Apoptosis in the Absence of HACE1
When NF-κB activation is blocked, TNF stimulation results in apoptotic cell death (Beg and Baltimore, 1996, Van Antwerp et al., 1996, Wang et al., 1996). Given that TNF-mediated NF-κB activation is impaired in hace1–/– cells, we reasoned that loss of HACE1 might sensitize cells to TNFR1-induced apoptosis. Surprisingly, TNF stimulation alone did not induce significant cell death in hace1–/– cells (Figures 2A and 2B), even when we monitored these cells for 72 hr (Figure S1B). Moreover, whereas, as expected, control WT MEFs underwent apoptosis in response to treatment with TNF and actinomycin-D for 8 hr (ActD; used to block NF-κB anti-apoptotic transcriptional activity; Lasek et al., 1996), hace1–/– MEFs remained resistant to apoptotic death (Figures 2A and 2B) and cells remained viable, albeit only partially, even at later time points (Figure S1C). Treatment with ActD alone did not induce cell death in control nor hace1–/– MEFs (Figure S1D).
Figure 2.

Loss of Hace1 Confers Resistance to TNF-Induced Apoptosis In Vitro
(A and B) Hace1–/– MEFs do not undergo apoptosis. Cell viability (A) and representative images (B) are shown for hace1+/+ and hace1–/– MEFs treated for 8 hr with either TNF alone (10 ng/ml) or TNF + ActD (1 μg/ml). Cell viability was determined in quadruplicate cultures (mean values ± SD). For images, cells were stained with crystal violet. Magnifications are ×10.
(C) Levels of cleaved caspase-8 (cC8), cleaved caspase-3 (cC3), total RIP1 kinase protein, and cleaved RIP1 kinase (cRIP1) in lysates from hace1+/+ and hace1–/– MEFs treated with TNF + ActD for the indicated time points. GAPDH is shown as a loading control.
(D) Cell viability of hace1+/+ and hace1–/– MEFs untreated (control) or exposed for 8 hr to TNF alone or TNF + cycloheximide (CHX; 10 μg/ml). Mean survival (±SD) was determined in quadruplicate cultures.
(E and F) Cell viability of hace1–/– MEFs re-expressing WT HACE1 and hace1–/– MEFs re-expressing the E3 ligase-dead C876S mutant left untreated (control) or treated for 8 hr with TNF alone and either TNF + ActD (E) or TNF + CHX (F). Mean survival (±SD) was determined in quadruplicate cultures. Results are representative of more than three independent experiments.
See also Figure S1.
Consistent with a critical role for HACE1 in mediating TNF/ActD-induced apoptosis, activation of caspase-8 and its downstream death effector caspase-3 as well as cleavage of RIP1 kinase (RIP1) were markedly impaired in hace1–/– MEFs (Figure 2C). We also failed to detect caspase-3 activation in hace1–/– MEFs using a DEVDase activity assay (Figure S1E). Similarly, apoptotic death and caspase-3 activation were absent in hace1–/– MEFs challenged with TNF plus cycloheximide (CHX) (Figure 2D; Figure S1F), an inhibitor of the NF-κB-mediated survival pathway (Tsuchida et al., 1995). Importantly, re-expression of WT HACE1, but not E3 ligase-dead C876S HACE1, restored TNF/ActD- as well as TNF/CHX-induced cell death in hace1–/– MEFs (Figures 2E and 2F). SMAC mimetic drugs such as BV6 induce intrinsic apoptosis by blocking the inhibitor of apoptosis (IAP) proteins downstream of TNFR1 (Vercammen et al., 1998, Vucic et al., 2011). We therefore used this model as an additional trigger of TNF-induced apoptotic cell death. Baseline cIAP1 and XIAP expression levels before and after TNF stimulation were comparable in control and hace1–/– MEFs (Figure S1G). As expected, we observed TNF/BV6-induced cell death in control MEFs; by contrast, hace1–/– MEFs again were resistant to apoptosis induced by TNF and BV6 (Figure S1H). These results show that TNFR1-induced apoptosis depends on HACE1.
To test whether HACE1 also regulates TNFR1-mediated apoptosis in vivo, we first challenged control and hace1–/– mice with recombinant murine TNF. Hace1–/– mice were completely resistant to TNF-induced death and systemic inflammation, as determined by the liver injury markers ALT and AST (Figures 3A–3C). To extend these data to additional in vivo models, we next challenged mice with lipopolysaccharide (LPS)/D-Gal, which induces liver damage and death in WT mice, but not in TNFR1 mutant mice (Pfeffer et al., 1993). Hace1 knockout mice were, to a large extent albeit not completely, resistant to LPS/D-Gal-induced liver damage and apoptotic death (Figures 3D–3F). These in vivo data indicate that hace1 deficiency renders mice resistant to apoptotic liver damage and death downstream of TNFR1.
Figure 3.

Hace1–/– Mice Are Protected from TNFR1-Induced Lethality In Vivo
(A–C) Hace1–/– mice are protected from liver damage in response to a high-dose (450 μg/kg) TNF challenge. (A) Kaplan-Meier survival curves are shown for hace1+/+ and hace1–/– mice after TNF injection (n = 6 mice per group, p < 0.05). (B) Serum AST and ALT levels served as markers for liver damage in littermate hace1+/+ and hace1–/– mice following in vivo challenge with high-dose TNF. Data from pooled sera (n = 4 mice per group) are shown. (C) H&E staining of representative histological liver sections 9 hr after the TNF challenge is shown.
(D–F) Hace1–/– mice are protected from death in response to LPS/D-Gal (LPS 10 μg/kg and D-Gal 1 g/kg mouse, i.p.). (D) Kaplan-Meier survival curves of hace1+/+ and hace1–/– littermates injected intravenously (i.v.) with a lethal dose of LPS/D-Gal (n = 10 mice per group, p < 0.01). (E) H&E staining and active caspase-3 immunodetection on liver sections 7 hr after LPS/D-Gal injections are shown. (F) Serum AST and ALT levels served as markers for liver damage in littermate hace1+/+ and hace1–/– mice following in vivo challenge with a lethal dose of LPS + D-Gal. Data from pooled sera (n = 5 mice per group) are shown.
HACE1 Is a Gatekeeper of Apoptotic versus Necroptotic Cell Fate
In the condition where both NF-κB and caspase activation are either genetically or pharmacologically blocked, TNF stimulation may result in a non-apoptotic form of cell death termed necroptosis (Cho et al., 2009, Christofferson and Yuan, 2010, Declercq et al., 2009, Galluzzi and Kroemer, 2008). To determine whether loss of HACE1 also interferes with TNF-induced necroptosis, WT and hace1–/– MEFs were pre-treated with the pan-caspase inhibitor Z-Val-Ala-Asp-fluoromethyl ketone (Z-VAD) and then stimulated with TNF/ActD to induce cell death. We observed that both WT and hace1–/– MEFs are comparably sensitive to necroptosis triggered by TNF/ActD/Z-VAD (Figure 4A). These findings also were confirmed using TNF/CHX/Z-VAD as an alternative necroptotic drug combination (Figure 4A). Of note, treatment with Z-VAD alone or in combination with either ActD or TNF did not induce cell death in control or hace1–/– MEFs (Figures S1D and S2A).
Figure 4.

Hace1–/– Mutant Cells Undergo Necroptotis
(A) Hace1+/+ and hace1–/– MEFs were treated with TNF (10 ng/ml), Z-VAD (5 μg/ml), and Nec-1 (50 nM) plus either ActD (1 μg/ml) or CHX (10 μg/ml) using the indicated combinations. Cell viability was determined in triplicates (mean values ± SD).
(B) Hace1+/+, hace1–/–, and hace1–/–ripk3–/– MEFs were treated with TNF (10 ng/ml), Z-VAD (5 μg/ml), Nec-1 (50 nM), and ActD (1 μg/ml). Cell viability was determined in quadruplicates (mean values ± SD). Results are representative of at least three independent experiments.
See also Figure S2.
Two kinases, RIP1 and RIP3, mediate necroptotic cell fate downstream of TNFR1 (Christofferson and Yuan, 2010, Declercq et al., 2009). Pre-treatment with the specific RIP1 kinase inhibitor necrostatin 1 (Nec-1) (Degterev et al., 2008) reverted necroptotic cell death in both control and hace1–/– MEFs (Figure 4A). We next generated hace1 ripk3 double-knockout (hace1–/– ripk3–/–) MEFs to genetically determine the contribution of RIP3 on cell death in hace1–/– MEFs. These double-mutant cells exhibited growth curves comparable to control MEFs (Figure S2B). Importantly, whereas hace1–/– MEFs were sensitive to necroptosis, hace1–/– ripk3–/– MEFs were resistant to necroptotic death (Figure 4B). Thus, HACE1 is required for TNF-induced NF-κB activation and apoptosis induction, but it is dispensable for the necroptotic response following TNFR1 stimulation.
HACE1 Mediates TRAF2 Ubiquitylation within the TNFR1-Receptor Complex
To identify molecular targets of HACE1 that could explain impaired NF-κB activation and defective apoptosis downstream of TNFR1, we established a HACE1-dependent in vitro ubiquitylation assay using hemagglutinin (HA)-ubiquitin, the ubiquitin-activating E1 enzyme, and multiple E2 enzymes. The most efficient E2 for HACE1 E3 ligase activity was found to be Ubc13 (Figure S3A). Using recombinant HACE1, Ubc13, the E1 enzyme, and HA-ubiquitin, we then performed in vitro ubiquitylation assays on microchips spotted with >9,000 natively folded human proteins. Intriguingly, our microchip array data revealed multiple HACE1 ubiquitylation targets, many of which had been annotated previously to the TNFR1/NF-κB-signaling pathway (Figures S3B and S3C). Using in vitro ubiquitylation assays, we confirmed that HACE1 directly ubiquitylates all targets tested (not shown). Intriguingly, among the substrates we found TRAF2.
Previous reports indicated that TRAF2 plays a pivotal role in driving NF-κB and JNK activation and inhibiting necroptosis after stimulation with TNF. This function is modulated by K63-linked ubiquitylation of TRAF2 (Karl et al., 2014, Li et al., 2006, Li et al., 2009, Petersen et al., 2015, Yeh et al., 1997). To identify the nature of TRAF2 ubiquitylation mediated by HACE1, we performed an in vitro ubiquitylation assay. We found that HACE1 directly catalyzes K63-linked ubiquitylation of TRAF2 (Figure S4A). Importantly, HACE1 is essential for this particular modification: lack of HACE1 led to the abrogation of TNF-driven K63-linked ubiquitylation of TRAF2 (Figure S4B). We failed to detect TNF-induced K48-specific ubiquitylation of TRAF2 in control and hace1–/– MEFs (Figure S4C) as well as in in vitro ubiquitylation assays (not shown). Defective ubiquitylation was restored in hace1–/– MEFs reconstituted with WT HACE1, but not in the E3 ligase-defective HACE1 C876S mutant (Figure S4D). Similar to hace1–/– MEFs, RNAi-mediated downregulation of HACE1 in WT MEFs impaired ubiquitylation of TRAF2 following TNF stimulation (Figure S4E). These data indicate that HACE1 is a key regulator of K63-linked ubiquitylation of TRAF2.
Activation of TNFR1 induces rapid formation of a TRAF2-containing plasma membrane-bound signaling complex (complex I) (Micheau and Tschopp, 2003). Within complex I, K63 ubiquitylation of TRAF2 has been previously associated with the ability to activate NF-κB and to induce JNK signaling (Habelhah et al., 2004, Li et al., 2009, Liang et al., 2010, Xia and Chen, 2005). Endogenous HACE1 associated with TRAF2, an interaction that was markedly enhanced upon TNF stimulation (Figures S5A and S5B). Moreover, endogenous HACE1 co-immunoprecipitated with TNFR1 in WT MEFs, an interaction that appears to be constitutive (Figures S5C and S5D). Cell surface expression of TNFR1 in hace1–/– MEFs was comparable to that in control cells (Figure S5E). Immunoprecipitation of complex I using FLAG-tagged human TNF (Micheau and Tschopp, 2003) showed that all the essential components of complex I, such as IKKα, IKKγ, TRAF2, as well as RIP1, were recruited to the activated TNFR1 in both control and hace1–/– MEFs (Figure 5A; Figure S5C). RIP1 kinase appeared to be normally phosphorylated and ubiquitylated within complex I (Figure 5A; Figures S5F and S5G). Serial immunoprecipitation experiments, carried out to pull down TRAF2 from the TNF-activated complex I (Lu et al., 2013), showed a marked defect in K63 ubiquitylation of TRAF2 in hace1–/– versus control MEFs (Figure 5B; Figure S5H). Thus, loss of HACE1 has no apparent effect on the assembly of complex I; however, HACE1 is essential for the ubiquitylation of complex I-associated TRAF2.
Figure 5.

Complex I and Complex II Formation
(A) Hace1+/+ and hace1–/– MEFs were stimulated with FLAG-tagged human TNF (FLAG-hTNF, 50 μg/ml) for the indicated time points followed by an anti-FLAG immunoprecipitation to detect proteins associated with the activated TNFR1 (complex I). FLAG-immunoprecipitates (IPs) and input lysates were probed with antibodies to IKKα, RIP1 kinase, TRAF2, and TNFR1. RIP1 kinase ubiquitylation as well as phosphorylation (p-RIP1) are indicated.
(B) Serial immunoprecipitation of TRAF2 from the activated TNFR1 complex. Western blots were probed to detect K63-ubiquitylated TRAF2.
(C) Interactions of TRAF2 with FADD were determined in anti-TRAF2 IPs from TNF- (10 ng/ml) stimulated (+) and untreated (−) hace1+/+ and hace1–/– MEFs.
(D) RIP1/TRAF2 interactions in hace1+/+ and hace1–/– MEFs were determined by western blot on TRAF2 IPs from TNF-stimulated (+) and untreated (−) MEFs.
(E) Hace1+/+ and hace1–/– MEFs were treated with TNF (10 ng/ml), ActD (1 μg/ml), Z-VAD (5 μg/ml), and Nec-1 (50 nM) for 90 min followed by immunoprecipitation of FADD to detect FADD-RIP1 kinase and FADD-caspase-8 interactions. Lysates were assayed for protein expression of RIP1, caspase-8 (C8), and FADD. Phosphorylated RIP1 kinase (p-RIP1) is indicated.
(F) Hace1+/+ and hace1–/– MEFs were stimulated (+) or untreated (−) with TNF (10 ng/ml), ActD (1 μg/ml), and Z-VAD (5 μg/ml). Lysates were immunoprecipitated with anti-RIP1 antibodies to detect RIP1/RIP3 kinase interactions.
(G) Hace1+/+ and hace1–/– MEFs were left untreated (−) or stimulated (+) with TNF (10 ng/ml), ActD (1 μg/ml), and Z-VAD (5 μg/ml). Western blots show levels of total and phosphorylated MLKL (p-MLKL) at the indicated time points.
See also Figures S3–S6.
Impaired Assembly of the Caspase-8 Activation Complex
Following initial formation of complex I, TNFR1 stimulation results in the assembly of a secondary cytoplasmic complex, termed complex II (Micheau and Tschopp, 2003), which drives caspase-8-dependent apoptotic cell death (Declercq et al., 2009, Kaiser et al., 2011, Oberst et al., 2011, Welz et al., 2011). To investigate whether the observed resistance of hace1–/– MEFs to TNF-induced apoptosis originates from defective complex II formation, we analyzed the assembly of the apoptotic complex II following TNF/ActD stimulation. In hace1–/– MEFs, the association of TRAF2 with FADD (Figure 5C), as well as its TNF stimulation-dependent association with RIP1 kinase (Figure 5D), was severely impaired. Further in vitro assays using recombinant proteins showed that TRAF2 and FADD interaction depends on ubiquitylation of TRAF2 (Figure S6A). Moreover, the recruitment of RIP1 kinase and caspase-8 to FADD also was severely affected (Figure 5E), demonstrating that loss of HACE1 expression results in defective assembly of the caspase-8-activating complex.
Loss of HACE1 does not impair necroptosis downstream of TNFR1 stimulation (Figure 4A). Necroptosis is mediated via the formation of complex II containing, among other proteins, FADD, caspase-8, and both RIP1 and RIP3 kinases. When caspase-8 is inactivated, RIP1 and RIP3 kinases together induce programmed necrosis (Cho et al., 2009, Christofferson and Yuan, 2010, Declercq et al., 2009, Galluzzi and Kroemer, 2008, Zhang et al., 2011). As reported previously (Wang et al., 2008), Z-VAD inhibition of caspase-8 in TNF/ActD-treated WT MEFs was associated with enhanced stability of the FADD/caspase-8/RIP1 kinase complex (Figure 5E; Figure S6B). Intriguingly, the association of RIP1 kinase and caspase-8 with FADD was found to be virtually absent in hace1–/– MEFs undergoing TNF/ActD/Z-VAD-triggered necroptosis (Figure 5E; Figure S6B). In both control and hace1–/– MEFs, RIP3 kinase binds to RIP1 kinase under necroptotic conditions (Figure 5F). This interaction is mirrored by the phosphorylation of the necroptotic downstream mediator MLKL (Figure 5G) (Vanden Berghe et al., 2014), indicating that induction of RIP1/RIP3-dependent necroptosis is not affected by the absence of HACE1. Interestingly, RIP1/RIP3 interactions can be detected, albeit at reduced levels, in hace1–/– MEFs even under apoptotic conditions (Figure 5F). However, we failed to detect any association between the RIP1/RIP3 kinases and caspase-8 under necroptotic conditions in hace1–/– MEFs treated with either TNF/ActD/Z-VAD or with TNF/BV6/Z-VAD (Figures S6B and S6C) (He et al., 2009). Importantly, we still observed RIP1 phosphorylation, a marker of RIP1 kinase activity and necroptotic pre-disposition (Declercq et al., 2009), in both control and hace1–/– MEFs treated with TNF/ActD/Z-VAD (Figure 5E; Figure S6B) or TNF/BV6/Z-VAD (Figure S6C). Thus, loss of HACE1 expression impairs the assembly of the canonical complex II without any apparent effect on the formation and activity of the necrosome formed by RIP1 and RIP3 kinases and phosphorylation of MLKL.
Hace1-Deficient Mice Are Hyper-susceptible to DSS-Induced Colitis
Our data so far showed that loss of HACE1 protects cells from TNF-induced apoptosis in vitro while leaving TNF-mediated RIP1/RIP3 kinase-dependent necroptosis intact. Since TNFR1, NF-κB, and RIP3 kinase recently have been implicated in intestinal homeostasis (Günther et al., 2011, Pasparakis, 2009, Welz et al., 2011), we tested whether HACE1 might play a role in gut inflammation. Naive hace1–/– mice showed normal body weight (Figure S7A), and morphological and flow cytometric analyses of the intestine of naive hace1–/– mice did not reveal any overt alteration compared to WT littermates (Figures S7B and S7C). However, when we challenged mice with dextran sodium sulfate (DSS), a chemical irritant that disrupts the intestinal epithelial barrier and induces colitis (De Robertis et al., 2011), we observed markedly increased intestinal inflammation in hace1–/– versus control hace1+/+ mice. Hace1–/– mice suffered a more pronounced weight loss than WT mice when treated with DSS (Figure 6A), and histopathological analysis revealed markedly more severe inflammation, tissue destruction, and epithelial cell death in hace1–/– mice compared to WT littermate controls (Figure 6B; Figure S7D). In addition, hace1–/– mice showed more severe diarrhea and increased intestinal bleeding (hemoccult test) compared to WT littermates (Figures 6C and 6D). Flow cytometric analysis of the immune system in the lamina propria revealed markedly increased infiltration of CD11b+ Gr-1+ Ly6G– inflammatory monocytes in the intestines of hace1-deficient mice after treatment with DSS (Figure 6E), confirming the increased severity of intestinal inflammation in hace1–/– compared to control WT mice.
Figure 6.

Deletion of RIP3 Kinase or TNFR1 Alleviates the Severe Colitis in Hace1–/– Mice
(A) Percentage body weight changes during 7-day administration of DSS (2.5%) in WT, hace1–/–, hace1–/–ripk3–/–, and hace1–/–tnfr1–/– mice are shown.
(B) Representative colonic histopathology (H&E staining) from WT, hace1–/–, hace1–/–ripk3–/–, and hace1–/–tnfr1–/– mice after 7 days of treatment with DSS. Lower histology panels depict magnifications of the selected areas.
(C and D) Diarrhea and blood scores of WT, hace1–/–, hace1–/–ripk3–/–, and hace1–/–tnfr1–/– mice treated with DSS for the indicated time periods. Data are shown as mean values ± SD (n = 5 mice per group). The p values are relative to DSS-treated hace1–/– versus all other genotypes (WT, hace1–/–ripk3–/–, and hace1–/–tnfr1–/–).
(E) Myeloid cell recruitment to the colonic lamina propria of DSS-treated mice. Neutrophils were defined as CD11b+ Gr-1+ Ly6G+, inflammatory monocytes as CD11b+ Gr-1+ Ly6G–, and dendritic cells (DCs) as CD11b+/– CD11clo/+ Gr-1–. All data in (B)–(E) are from mice at day 7 after DSS administration. Values show average ± SEM. Results are representative of more than three independent experiments.
(F) Body weight changes during 7-day administration of DSS (2.5%) in WT, ripk3–/–, and tnfr1–/– mice (n = 5 per group).
See also Figure S7.
Both NF-κB and TNFR1 pathways play fundamental roles in immunity. NF-κB acts downstream of surface receptors recognizing pro-inflammatory cytokines, including TNF itself, as well as pathogen-associated pattern receptors, which in turn leads to the expression of further inflammatory mediators (Bonizzi and Karin, 2004). On the other hand, NF-κB expression in epithelia such as the intestine plays a fundamental role in ensuring epithelial cell survival and in maintaining the structural integrity of mucosal surfaces (Pasparakis, 2009). Hace1 mRNA is expressed at comparable levels in a variety of immune cells as well as in intestinal epithelial cells (Figure S7F). To discern in which cells HACE1 is acting to prevent severe colitis, we generated bone marrow chimeric mice lacking HACE1 exclusively in the immune system by transferring hace1-deficient bone marrow cells in lethally irradiated hace1-sufficient host mice. As T cells in the intestine are largely radioresistant, we used lymphopenic rag2–/– mice as hosts (KO→rag2–/–) to prevent host-derived WT T cells to persist in the intestine, thus affecting the validity of the system. As a control group, we reconstituted irradiated rag2–/– mice with WT bone marrow (WT→rag2–/–). Treatment of these chimeric mice with DSS revealed that the absence or presence of HACE1 in the immune system had no apparent effect on the development of colitis: both groups showed comparable weight loss (Figure S7G) and similar recruitment of inflammatory cells to the intestinal lamina propria (Figure S7H). These results indicate that the loss of HACE1 in radiosensitive immune cells is not responsible for the increased susceptibility of hace1–/– mice to colitis.
Importantly, genetic inactivation of RIP3 kinase in hace1–/– ripk3–/– double-knockout mice and inactivation of TNFR1 in hace1–/– tnfr1–/– double-knockout mice completely rescued all parameters of the severe inflammatory phenotype in hace1–/– mice to levels observed in WT littermates, including weight loss, tissue damage, death of epithelial cells, recruitment of inflammatory cells, as well as diarrhea and intestinal bleeding (Figures 6A–6E; Figure S7D). Also, the expression of several pro-inflammatory mediators was upregulated in the colon of hace1–/– mice; in hace1–/– tnfr1–/– and hace1–/– ripk3–/– mice the levels of these pro-inflammatory factors were again restored to levels detected in WT controls (Figure S7E). The increased death of intestinal epithelial cells in DSS-treated hace1–/– mice is probably the consequence of exaggerated immune activation and inflammation resulting from the preferential induction of TNF-driven necroptosis instead of apoptosis in the absence of HACE1 (Pasparakis and Vandenabeele, 2015). Of note, while others reported increased severity of colitis in ripk3–/– (Moriwaki et al., 2014) and, to a lesser extent, in tnfr1–/– mice (Stillie and Stadnyk, 2009, Wang et al., 2012), in our hands single-knockout mice showed no change in weight loss compared to WT littermates (Figure 6F). The difference in ripk3–/– mice might be due to the time point of analysis: Moriwaki et al. (2014) reported a defect of ripk3–/– animals in the recovery from DSS, while we focused on the earlier phase of tissue destruction during which enhanced necroptosis is known to be detrimental (Günther et al., 2011). Together, our data show that the increased susceptibility of hace1-deficient mice to DSS-induced colitis depends on deregulated TNFR1 signaling and RIP3 kinase.
Hace1 Controls Colon Cancer via TNFR1 and RIP3 Kinase
We initially identified HACE1 as a tumor suppressor gene downregulated in multiple cancers (Anglesio et al., 2004, Zhang et al., 2007). Since we observed increased intestinal inflammation, we next explored whether hace1–/– mice are prone to develop colon cancer upon challenge with DSS and the genotoxic drug azoxymethane (AOM) (Tanaka et al., 2003). Since hace1–/– mice are particularly sensitive to DSS, we reduced the DSS treatment regimen to a level not sufficient to induce colon cancer in WT mice (De Robertis et al., 2011, Tanaka, 2012). As expected using this low DSS dose, we failed to detect any tumors in control HACE1-expressing mice (Figure 7A). However, in hace1–/– mice, we observed a markedly increased incidence of colon cancer as determined by colonoscopy or histopathological examination (Figures 7A and 7B). Colonic tumors in hace1–/– mice show features consistent with adenomatous polyps as well as nuclear atypia, multiple mitotic figures, areas of carcinoma in situ, and focal invasion (Figure 7B). Most importantly, hace1–/– ripk3–/– as well as hace1–/– tnfr1–/– double-knockout mice were similar to WT mice and did not show AOM/DSS-induced colonic tumors (Figures 7A and 7B), indicating that simultaneous genetic inactivation of TNFR1 and RIP3 kinase considerably reduced colon cancer formation secondary to hace1 deficiency. In conclusion, hace1 deficiency predisposes to colonic inflammation and carcinogenesis, which is markedly alleviated by genetic inactivation of TNFR1 and RIP3 kinase.
Figure 7.

Genetic Inactivation of ripk3 or tnfr1 Reverts the Sensitivity of hace1–/– Mice to DSS + AOM-Induced Colon Cancer
(A) Representative colonoscopies and incidence of colon cancer of hace1+/+, hace1–/–, hace1–/–ripk3–/–, and hace1–/–tnfr1–/– mice (n = 5 mice per cohort) after 7 weeks of treatment with AOM + DSS. Arrow indicates tumor.
(B) Colonic histopathology (H&E stainings) on tissues harvested 14 weeks after AOM-DSS challenge. Note the large adenoma in hace1–/– mice, whereas no aberrant hyperplasias or adenomas were observed in hace1+/+, hace1–/–ripk3–/–, and hace1–/–tnfr1–/– mice. Magnifications are ×10; inset magnification are ×40. Experiments were repeated two more times with similar cohort sizes and we always observed analogous results.
Discussion
Our results highlight HACE1 as a gatekeeper for TNFR1-mediated cell fates. Loss of HACE1 impairs TNFR1-mediated activation of NF-κB signaling and apoptosis in vitro and in vivo, but it does not inhibit TNFR1-dependent RIP1/RIP3-induced necroptosis. Moreover, our data show that enhanced inflammation and colon cancer in hace1 mutant mice can be reverted by genetic inactivation of RIP3 kinase and TNFR1.
We propose the following scenario for HACE1-regulated NF-κB activation downstream of TNFR1. Loss of HACE1 does not impair formation of complex I at TNFR1; however, NF-κB and JNK activation are markedly impaired in hace1–/– MEFs, a function dependent on its E3 ligase activity. Our results indicate that HACE1 expression is essential for K63 ubiquitylation of TRAF2 within complex I upon TNFR1 stimulation, which previously has been associated with the ability to activate the canonical NF-κB pathway and to induce JNK signaling (Habelhah et al., 2004, Liang et al., 2010, Xia and Chen, 2005). Although NF-κB signaling is impaired, hace1–/– MEFs do not undergo apoptosis in response to TNFR1 stimulation, which can be biochemically explained by the fact that hace1–/– cells fail to form pro-apoptotic complex II. Moreover, our results show that TRAF2 ubiquitylation is required for TRAF2 to associate with the key death molecule FADD, which in turn nucleates the caspase-8 apoptosis effector complex. However, besides TRAF2, we found multiple additional HACE1 ubiquitylation targets that previously have been linked to cell death and signaling pathways downstream of TNFR1, suggesting that HACE1 could regulate TNFR1-regulated cell fate at various interception points.
Our in vitro and in vivo data indicate that necroptosis in hace1 mutant cells is mediated via the RIP1/RIP3/MLKL pathway. Since we failed to detect formation of an intact necrosome, such death signals might progress independently of the canonical molecular necroptotic complex. Necroptosis in the context of hace1 deficiency appears to occur without direct caspase-8 interaction with RIP1/RIP3 kinases, raising the possibility that alternative RIP1/RIP3 kinase complexes might exist when HACE1 is inactivated. Of note, in our experimental system, induction of necroptosis in hace1 knockout MEFs still requires caspase inhibition via Z-VAD. Whether regulation of cFLIP (Oberst et al., 2011) could explain this remaining caspase dependency requires further exploration. Moreover, a feedback inhibition loop via RIP3 kinase activity has been reported recently in the control of caspase-8-mediated apoptosis (Newton et al., 2014), suggesting complex interplays between the apoptotic and necroptotic death effector machineries.
We initially identified HACE1 as a candidate tumor suppressor gene in a child with Wilms’ tumor (Anglesio et al., 2004, Zhang et al., 2007). In accord with its putative tumor suppressor function, we found HACE1 to be downregulated in multiple cancers (Hibi et al., 2008, Sakata et al., 2009). That HACE1 can indeed suppress cancer was shown in hace1 knockout mice that develop tumors of multiple tissues (Zhang et al., 2007). Since the TNFR1 and RIP1/RIP3 kinase pathway has been reported to induce oxidative stress and possibly alter cellular metabolism (Christofferson and Yuan, 2010, Declercq et al., 2009, Galluzzi and Kroemer, 2008), our results suggest that this activation pathway downstream of TNFR1 might not only trigger necroptosis but also could be involved in oncogenesis. Moreover, RIP1/RIP3 kinases downstream of TNFR1 have been implicated in a pro-inflammatory cascade in the intestine unleashed by SMAC mimetics or caspase-8 deficiency (Kang et al., 2013, Vince et al., 2012), driven by the release of necroptosis-mediated damage-associated molecular pattern molecules (DAMPs) (Duprez et al., 2011, Kaczmarek et al., 2013). Hace1 mutant mice indeed develop increased intestinal inflammation following epithelial injury and increased incidence of colon cancer. Importantly, genetic deletion of either RIP3 kinase or TNFR1 not only reversed the enhanced inflammation but also strongly reduced the development of colon cancer in hace1 single-mutant mice. These data demonstrate that deregulation of the TNFR1-mediated RIP3 kinase signaling is a key driving principle of inflammation and subsequent colon cancer development controlled by HACE1.
Experimental Procedures
Mice and Study Approval
The mice, hace1+/+, hace1–/–, hace1–/– tnfr1–/–, and hace1–/– ripk3–/– on a C57BL/6 background, were bred and maintained under specific pathogen-free conditions at the Institute of Molecular Biotechnology of the Austrian Academy of Sciences (IMBA) animal facility. For experiments, age- and sex-matched mice (8–12 weeks of age) were used. All animal experiments were approved by Austrian authorities, and mice were kept following the guidelines of Austrian animal laboratory law.
Cell Lines and Western Blots
MEFs derived from hace1+/+ and hace1–/– mice were prepared as described previously (Zhang et al., 2007). MEFs were seeded at 2 × 106 cells per 60-cm2 Petri dish and cultured in DMEM supplemented with 5% fetal calf serum (FCS) and 1% Pen-Strep (Sigma) in a 5% CO2 incubator. The next day, cells were treated with 10 ng/ml mouse TNF (3014, Immunotools), 1 μg/ml ActD-Mannitol (A-5156, Sigma), 5 μg/ml Z-VAD (Bachem), and 50 nM Nec-1 (N-9037, Sigma). In addition, cells were treated with 10 ng/ml mouse TNF (3014, Immunotools) and 10 μg/ml CHX (C1988, Sigma). Following the treatments, the cells were lysed in 1 ml ice-cold NP-40 buffer (10 mM Tris [pH 8.0], 150 mM NaCl, and 1% Nonidet P-40) supplemented with protease and phosphatase inhibitor tablets (11873580001 and 04906837001, Roche). The lysates were then centrifuged at 14,000 rpm for 10 min at 4°C. Immunoprecipitations were performed according to the manufacturer’s protocol.
In Vivo Lethality Experiments
For high-dose lethal TNF challenge, animals were injected via the tail vein with 450 μg/kg of purified endotoxin-free TNF (Immunotools) in 200 μl PBS (pH 6.8). For induction of hyper-acute shock, TNF was used at a dose of 5 μg/kg. LPS (L2880, Sigma) was administrated intraperitoneally (i.p.) at a concentration of 10 μg/kg. D-Gal (G0500, Sigma) was given to mice together with LPS at a concentration of 1 g/kg. Liver enzymes were assayed using the Cobas C111 system (Roche). For histology, livers were embedded in paraffin, sectioned, and stained with H&E.
Listeria Infections
Listeria monocytogenes (strain LO28) was prepared for infection as described previously (Stockinger et al., 2009). The bacterial suspension (500 μl) was injected into the peritoneum of 8- to 10-week-old male hace1+/+ and hace1–/– littermate mice. Survival of infected mice was monitored for 10 days. For determination of the bacterial load, mice were euthanized 96 hr after infection and spleens were homogenized in PBS. Serial dilutions of homogenates were plated on Oxford agar plates and colonies were counted after growth at 37°C for 24 hr.
Statistics
If not otherwise stated, all data are expressed as the means ± SD. Significance was assessed by Student’s two-sided t test unless otherwise stated in the figure legends. For multiple comparisons, we used a two-way ANOVA test with a Bonferroni post-test. Differences with p values <0.05 were considered as significant (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001).
Author Contributions
L.T. and R.N. designed, performed, and interpreted experiments. M.J.M.B., I.K., I.U., P.V., M.K., Y.R., L.M.F., G.W., T.P., R.S., S.R., R.W., and T.H. helped in performing experiments. E.K., D.D., and T.D. performed Listeria infections. M.J.M.B., M.D., N.T., G.S.-F., F.I., G.K., P.V., H.K., A.P., and P.H.S. provided reagents and insight. J.M.P. coordinated the project and wrote the paper with L.T.
Acknowledgments
We thank all members of our laboratories and Helen Pickersgill for critical reading and expert advice, and we thank Cecile Pickart and Marc Timmers for the bacterial ubiquitin E2-expressing plasmids. We are grateful to the members of the IMP-IMBA Biooptics facility for assistance in cell sorting. SMAC mimetic BV6 and RIP3 kinase mutant mice were generously obtained by Genentech. The work was supported by the Austrian Academy of Sciences and an advanced European Research Council (ERC) grant (to J.M.P.), the Helmsley Foundation (VEO-IBD, to J.M.P.), an Innovator award by Era of Hope (to J.M.P.), the Max Planck Society (to A.P.), WWTF LS05003 (to A.P.), DFG PI 917/1-1 (to A.P.), and by the Swiss National Science Foundation (PBEZP3_145993 to L.T.). M.J.M.B. has a tenure track position in the Multidisciplinary Research Program of Ghent University (GROUP-ID). G.K. is supported by the Ligue Nationale contre le Cancer, Agence Nationale pour la Recherche, European Commission (ArtForce), Institut National du Cancer (INCa), Cancéropôle Ile-de-France, Fondation Bettencourt-Schueller, LabEx Immuno-Oncology, and PACRI. P.V. is supported by European grants (FP7 EC RTD Integrated Project, Apo-Sys, FP7-200767; Euregional PACT II), Belgian grants (Interuniversity Attraction Poles, IAP 6/18), Flemish grants (Research Foundation Flanders, FWO G.0875.11 and FWO G.0973.11), Ghent University grants (MRP, GROUP-ID consortium), and grants from Flanders Institute for Biotechnology (VIB). P.V. also holds a Methusalem grant (BOF09/01M00709) from the Flemish Government. R.N. is currently an employee of AstraZeneca; however, he contributed to this study during his previous occupation at IMBA.
Published: May 5, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.04.032.
Supplemental Information
References
- Anglesio M.S., Evdokimova V., Melnyk N., Zhang L., Fernandez C.V., Grundy P.E., Leach S., Marra M.A., Brooks-Wilson A.R., Penninger J., Sorensen P.H. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum. Mol. Genet. 2004;13:2061–2074. doi: 10.1093/hmg/ddh215. [DOI] [PubMed] [Google Scholar]
- Beg A.A., Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Bergink S., Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009;458:461–467. doi: 10.1038/nature07963. [DOI] [PubMed] [Google Scholar]
- Bernassola F., Ciechanover A., Melino G. The ubiquitin proteasome system and its involvement in cell death pathways. Cell Death Differ. 2010;17:1–3. doi: 10.1038/cdd.2009.189. [DOI] [PubMed] [Google Scholar]
- Bhoj V.G., Chen Z.J. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- Bollrath J., Greten F.R. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10:1314–1319. doi: 10.1038/embor.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzi G., Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M., Chan F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofferson D.E., Yuan J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corn J.E., Vucic D. Ubiquitin in inflammation: the right linkage makes all the difference. Nat. Struct. Mol. Biol. 2014;21:297–300. doi: 10.1038/nsmb.2808. [DOI] [PubMed] [Google Scholar]
- Daugaard M., Nitsch R., Razaghi B., McDonald L., Jarrar A., Torrino S., Castillo-Lluva S., Rotblat B., Li L., Malliri A. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat. Commun. 2013;4:2180. doi: 10.1038/ncomms3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Robertis M., Massi E., Poeta M.L., Carotti S., Morini S., Cecchetelli L., Signori E., Fazio V.M. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declercq W., Vanden Berghe T., Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229–232. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Degterev A., Hitomi J., Germscheid M., Ch’en I.L., Korkina O., Teng X., Abbott D., Cuny G.D., Yuan C., Wagner G. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprez L., Takahashi N., Van Hauwermeiren F., Vandendriessche B., Goossens V., Vanden Berghe T., Declercq W., Libert C., Cauwels A., Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L., Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135:1161–1163. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Günther C., Martini E., Wittkopf N., Amann K., Weigmann B., Neumann H., Waldner M.J., Hedrick S.M., Tenzer S., Neurath M.F., Becker C. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habelhah H., Takahashi S., Cho S.G., Kadoya T., Watanabe T., Ronai Z. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-kappaB. EMBO J. 2004;23:322–332. doi: 10.1038/sj.emboj.7600044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S., Wang L., Miao L., Wang T., Du F., Zhao L., Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hibi K., Sakata M., Sakuraba K., Shirahata A., Goto T., Mizukami H., Saito M., Ishibashi K., Kigawa G., Nemoto H., Sanada Y. Aberrant methylation of the HACE1 gene is frequently detected in advanced colorectal cancer. Anticancer Res. 2008;28(3A):1581–1584. [PubMed] [Google Scholar]
- Hoeller D., Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- Kaczmarek A., Vandenabeele P., Krysko D.V. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Kaiser W.J., Upton J.W., Long A.B., Livingston-Rosanoff D., Daley-Bauer L.P., Hakem R., Caspary T., Mocarski E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T.B., Yang S.H., Toth B., Kovalenko A., Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Karl I., Jossberger-Werner M., Schmidt N., Horn S., Goebeler M., Leverkus M., Wajant H., Giner T. TRAF2 inhibits TRAIL- and CD95L-induced apoptosis and necroptosis. Cell Death Dis. 2014;5:e1444. doi: 10.1038/cddis.2014.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küçük C., Hu X., Iqbal J., Gaulard P., Klinkebiel D., Cornish A., Dave B.J., Chan W.C. HACE1 is a tumor suppressor gene candidate in natural killer cell neoplasms. Am. J. Pathol. 2013;182:49–55. doi: 10.1016/j.ajpath.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari S., Redouane Y., Lopez-Mosqueda J., Shiraishi R., Romanowska M., Lutzmayer S., Kuiper J., Martinez C., Dikic I., Pasparakis M., Ikeda F. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife. 2014;3:e03422. doi: 10.7554/eLife.03422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek W., Giermasz A., Kuc K., Wańkowicz A., Feleszko W., Gołab J., Zagozdzon R., Stokłosa T., Jakóbisiak M. Potentiation of the anti-tumor effect of actinomycin D by tumor necrosis factor alpha in mice: correlation between in vitro and in vivo results. Int. J. Cancer. 1996;66:374–379. doi: 10.1002/(SICI)1097-0215(19960503)66:3<374::AID-IJC18>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Li S., Wang L., Berman M.A., Zhang Y., Dorf M.E. RNAi screen in mouse astrocytes identifies phosphatases that regulate NF-kappaB signaling. Mol. Cell. 2006;24:497–509. doi: 10.1016/j.molcel.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Wang L., Dorf M.E. PKC phosphorylation of TRAF2 mediates IKKalpha/beta recruitment and K63-linked polyubiquitination. Mol. Cell. 2009;33:30–42. doi: 10.1016/j.molcel.2008.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J., Saad Y., Lei T., Wang J., Qi D., Yang Q., Kolattukudy P.E., Fu M. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J. Exp. Med. 2010;207:2959–2973. doi: 10.1084/jem.20092641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A., Bräsen J.H., Himmerkus N., Liu S., Huber T.B., Kunzendorf U., Krautwald S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81:751–761. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- Lu T.T., Onizawa M., Hammer G.E., Turer E.E., Yin Q., Damko E., Agelidis A., Shifrin N., Advincula R., Barrera J. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity. 2013;38:896–905. doi: 10.1016/j.immuni.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O., Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Moriwaki K., Balaji S., McQuade T., Malhotra N., Kang J., Chan F.K. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014;41:567–578. doi: 10.1016/j.immuni.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay D., Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- Newton K., Dugger D.L., Wickliffe K.E., Kapoor N., de Almagro M.C., Vucic D., Komuves L., Ferrando R.E., French D.M., Webster J. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- Oberst A., Dillon C.P., Weinlich R., McCormick L.L., Fitzgerald P., Pop C., Hakem R., Salvesen G.S., Green D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolino M., Choidas A., Wallner S., Pranjic B., Uribesalgo I., Loeser S., Jamieson A.M., Langdon W.Y., Ikeda F., Fededa J.P. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507:508–512. doi: 10.1038/nature12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat. Rev. Immunol. 2009;9:778–788. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- Pasparakis M., Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- Petersen S.L., Chen T.T., Lawrence D.A., Marsters S.A., Gonzalvez F., Ashkenazi A. TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ. 2015;22:1846–1857. doi: 10.1038/cdd.2015.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer K., Matsuyama T., Kündig T.M., Wakeham A., Kishihara K., Shahinian A., Wiegmann K., Ohashi P.S., Krönke M., Mak T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- Rotblat B., Southwell A.L., Ehrnhoefer D.E., Skotte N.H., Metzler M., Franciosi S., Leprivier G., Somasekharan S.P., Barokas A., Deng Y. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. USA. 2014;111:3032–3037. doi: 10.1073/pnas.1314421111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata M., Kitamura Y.H., Sakuraba K., Goto T., Mizukami H., Saito M., Ishibashi K., Kigawa G., Nemoto H., Sanada Y., Hibi K. Methylation of HACE1 in gastric carcinoma. Anticancer Res. 2009;29:2231–2233. [PubMed] [Google Scholar]
- Sha W.C., Liou H.C., Tuomanen E.I., Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Stillie R., Stadnyk A.W. Role of TNF receptors, TNFR1 and TNFR2, in dextran sodium sulfate-induced colitis. Inflamm. Bowel Dis. 2009;15:1515–1525. doi: 10.1002/ibd.20951. [DOI] [PubMed] [Google Scholar]
- Stockinger S., Kastner R., Kernbauer E., Pilz A., Westermayer S., Reutterer B., Soulat D., Stengl G., Vogl C., Frenz T. Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes. PLoS Pathog. 2009;5:e1000355. doi: 10.1371/journal.ppat.1000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T. Development of an inflammation-associated colorectal cancer model and its application for research on carcinogenesis and chemoprevention. Int. J. Inflamm. 2012;2012:658786. doi: 10.1155/2012/658786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Kohno H., Suzuki R., Yamada Y., Sugie S., Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94:965–973. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D., Xiang Y., De Renzis S., Rink J., Zheng G., Zerial M., Wang Y. The ubiquitin ligase HACE1 regulates Golgi membrane dynamics during the cell cycle. Nat. Commun. 2011;2:501. doi: 10.1038/ncomms1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida H., Takeda Y., Takei H., Shinzawa H., Takahashi T., Sendo F. In vivo regulation of rat neutrophil apoptosis occurring spontaneously or induced with TNF-alpha or cycloheximide. J. Immunol. 1995;154:2403–2412. [PubMed] [Google Scholar]
- Van Antwerp D.J., Martin S.J., Kafri T., Green D.R., Verma I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Vercammen D., Beyaert R., Denecker G., Goossens V., Van Loo G., Declercq W., Grooten J., Fiers W., Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince J.E., Wong W.W., Gentle I., Lawlor K.E., Allam R., O’Reilly L., Mason K., Gross O., Ma S., Guarda G. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Vucic D., Dixit V.M., Wertz I.E. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 2011;12:439–452. doi: 10.1038/nrm3143. [DOI] [PubMed] [Google Scholar]
- Wang C.Y., Mayo M.W., Baldwin A.S., Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wang C.Y., Mayo M.W., Korneluk R.G., Goeddel D.V., Baldwin A.S., Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wang L., Du F., Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- Wang K., Han G., Dou Y., Wang Y., Liu G., Wang R., Xiao H., Li X., Hou C., Shen B. Opposite role of tumor necrosis factor receptors in dextran sulfate sodium-induced colitis in mice. PLoS ONE. 2012;7:e52924. doi: 10.1371/journal.pone.0052924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welz P.S., Wullaert A., Vlantis K., Kondylis V., Fernández-Majada V., Ermolaeva M., Kirsch P., Sterner-Kock A., van Loo G., Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- Xia Z.P., Chen Z.J. TRAF2: a double-edged sword? Sci. STKE. 2005;2005:pe7. doi: 10.1126/stke.2722005pe7. [DOI] [PubMed] [Google Scholar]
- Yeh W.C., Shahinian A., Speiser D., Kraunus J., Billia F., Wakeham A., de la Pompa J.L., Ferrick D., Hum B., Iscove N. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- Zhang L., Anglesio M.S., O’Sullivan M., Zhang F., Yang G., Sarao R., Mai P.N., Cronin S., Hara H., Melnyk N. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat. Med. 2007;13:1060–1069. doi: 10.1038/nm1621. [DOI] [PubMed] [Google Scholar]
- Zhang D.W., Shao J., Lin J., Zhang N., Lu B.J., Lin S.C., Dong M.Q., Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Zhang H., Zhou X., McQuade T., Li J., Chan F.K., Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–376. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Chen X., Sharma P., Moon M., Sheftel A.D., Dawood F., Nghiem M.P., Wu J., Li R.K., Gramolini A.O. HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat. Commun. 2014;5:3430. doi: 10.1038/ncomms4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.