

Abstract
Background
Leiomodin proteins, Lmod1, Lmod2 and Lmod3, are key regulators of the thin filament length in muscles. While Lmod1 is specifically expressed in smooth muscles, both Lmod2 and Lmod3 are expressed in striated muscles including both cardiac and skeletal muscles. We and others have previously shown that Lmod3 mainly function in skeletal muscles and the mutant mice display disorganized sarcomere. Lmod2 protein has been found to act as an actin filament nucleator in both cell-free assays and in cultured rat and chicken cardiomyocytes.
Results
To better understand the function of Lmod2 in vivo, we have identified and characterized a piggyBac (PB) insertional mouse mutant. Our analysis revealed that the PB transposon inserts in the first exon of the Lmod2 gene and severely disrupts its expression. We found that Lmod2PB/PB mice exhibit typical dilated cardiomyopathy (DCM) with ventricular arrhythmias and postnatal lethality. Electron microscope reveals that the Lmod2PB/PB hearts carry disordered sarcomere, disarrayed thin filaments, and distorted intercalated discs (ICDs). Those ICDs display not only decreased convolutions, but also reduced electron-dense staining, indicating less ICDs component proteins in Lmod2PB/PB hearts. Consistent with the phenotype, the expression of the ICD component genes, β-catenin and Connexin43, are down-regulated.
Conclusions
Taken together, our data reveal that Lmod2 is required in heart thin filaments for integrity of sarcomere and ICD and deficient mice exhibit DCM with ventricular arrhythmias and postnatal lethality. The Lmod2PB/PB mutant offers a valuable resource for interrogation of pathogenesis and development of therapeutics for DCM.
Keywords: Dilated cardiomyopathy, Leiomodin2 mutant, Sarcomere, Intercalated discs
Background
The Leiomodin proteins, Lmod1, Lmod2 and Lmod3, are a subgroup of the Tropomodulin (Tmod) protein family and are regulators of the thin filament length in muscles [1–3]. While Lmod1 is specifically expressed in smooth muscles, both Lmod2 and Lmod3 are expressed in striated muscles including both cardiac and skeletal muscles [4–6]. We and others have previously shown that Lmod3 mainly functions in skeletal muscles [6–8] and Lmod3 mutants exhibit muscle atrophy in fast fibers [6]. The mutant mice display disorganized sarcomere and the presence of nemaline bodies in skeletal muscles, a hallmark of the disease nemaline myopathy (NM), consistent with the finding that LMOD3 is mutated in the NM patients [7].
Lmod2 protein has been found to act as an actin filament nucleator in both cell-free assays and in cultured rat and chicken cardiomyocytes [2, 5]. Overexpression of Lmod2 results in the elongated thin filaments and knockdown exhibited disrupted sarcomere assembly in cultured cardiomyocytes [2, 5]. Furthermore, it has been shown that Lmod2 is an antagonist of Tmod1 in cardiomyocytes [2, 5]. Knockout mice of Tmod1 are embryonic lethal due to cardiac defects, and overexpression of Tmod1 in the heart causes myofibril disorganization and dilated cardiomyopathy (DCM) [9–13]. However, the physiological function of Lmod2 remains unknown. We hypothesized that the phenotype of loss of Lmod2 in mice might mimic that of the overexpression of its antagonist Tmod1, and the mutant mice are high likely to carry DCM.
Dilated cardiomyopathy is a common form of cardiomyopathies and the third most common inheritable heart disease [14]. DCM is diagnosed as dilated left ventricular accompanied by systolic dysfunction, less than 50 % ejection fraction (EF) [15]. These symptoms may be accompanied by other complications, including arrhythmias, coagulation, and congestive heart failure (CHF), which often lead to lethality [16, 17]. It has been estimated that DCM causes at least half of heart failures and less than 50 % of the DCM patients survive beyond year 5 after the diagnosis [16, 18, 19].
To better understand the function of Lmod2 in vivo, we have identified a piggyBac (PB) insertional mutant that disrupts Lmod2 expression and carried out phenotypic characterization of this mutant. We show here that Lmod2 is crucial for postnatal survival and essential for cardiac function. Lmod2 deficient mice display DCM with disrupted sarcomeres and intercalated discs (ICDs) including the expression of ICD genes, which offer a new mouse model for this deadly disease.
Results
Generation of Lmod2-deficient mice
Our group has shown that the PB transposon is highly active in mice and human cells and could be used to rapidly generate a large collection of insertional mouse mutants in a cost-effective manner [20]. One of the PB mutants that we generated has an insertion in the Lmod2 gene (Lmod2PB/+ in FVB/N background) [21]. This PB transposon is inserted in the non-coding region of the first exon of the Lmod2 transcriptional unit (Fig. 1a) and significantly down regulated the expression of the gene as revealed by quantitative RT-PCR (less than 5 and 10 % in homozygous mutant males and females in comparison to wild-type control Fig. 1b).
Fig. 1.
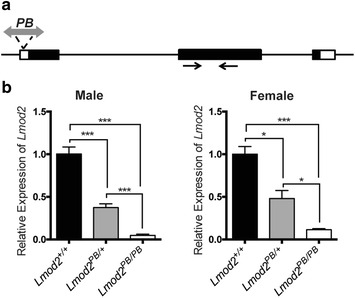
PB transposon disrupts Lmod2 expression. a Schematic representation of the Lmod2 transcription unit and the position of PB insertion. Coding and untranslated region are depicted as black and white boxes, respectively. b Quantitative RT-PCR analysis of Lmod2 mRNA from heart of 3-week-old male and female mice with indicated genotypes, n = 3. Arrows PCR primers
Tmod1 is an antagonist of Lmod2 at the pointed end of the thin filaments in cardiac muscle [5]. It has been suggested that the expression of Tmod1 could be affected by Lmod2 [5]. We therefore analyzed the transcriptional level of Tmod1 by quantitative PCR and found that the mRNA level of Tmod1 remains the same in 25 days old Lmod2PB/PB hearts compared to controls (Fig. 2a).
Fig. 2.
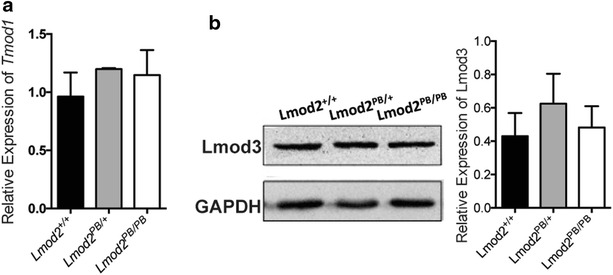
Tmod1 and Lmod3 Expression in Lmod2 mutant mice. a mRNA level of Tmod1 is not changed in Lmod2PB/PB hearts compared with wild-type control. b Lmod3 protein level is also not changed in Lmod2PB/PB mice as shown here in western blotting and statistics
Lmod3, another member of the Leiomodin family, also expresses in heart together with Lmod2. It might be up regulated to functionally compensate the down-regulation of Lmod2. However, Western analysis revealed that the Lmod3 protein level in the hearts of Lmod2PB/PB is unchanged compared to its controls (Fig. 2b). Together, these data indicate that the phenotypes observed below are likely caused by reduction of Lmod2 gene activity alone and not a compound effect disrupting other Tmod family members.
Lmod2PB/PB mice exhibit postnatal lethality
Lmod2PB/PB mice are born with the expected ratio as well as their Lmod2PB/+ and wild-type littermates. This indicates that Lmod2 is not essential for mouse embryonic development.
While Lmod2PB/PB mice are born alive with normal appearance and body weight, the mutant animals exhibit postnatal death around 3rd week of age and are all dead by 9th week (Fig. 3a). Furthermore, male mutant animals are also underweight after three weeks (Fig. 3b). Heterozygous Lmod2PB/+ animals have normal life spans and display no discernable phenotype including fertility. This result indicates that Lmod2 is crucial for postnatal survival.
Fig. 3.
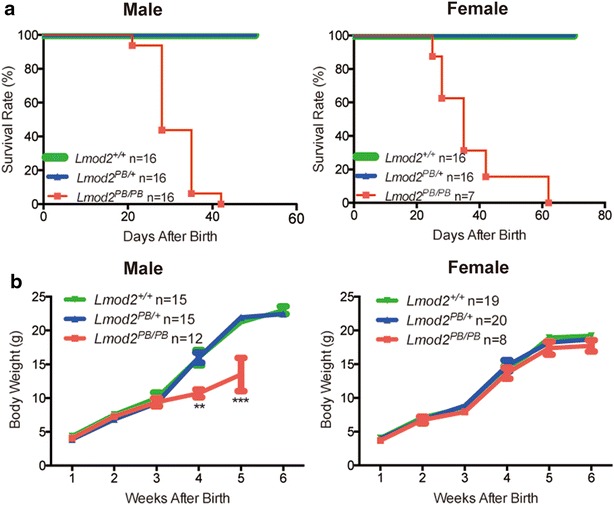
Lmod2PB/PB mice exhibit postnatal lethality and underweight in surviving males. Survival a and growth b curves of male and female Lmod2PB/PB mice compared with Lmod2PB/+ and Lmod2+/+ mice
Lmod2PB/PB hearts display dilated cardiomyopathy defects
The Lmod2PB/PB mice have no overt signs of distress until 1 to 2 days before death. These signs include curling up, less movement, and dyspnea. Given that Lmod2 is expressed in heart [4], these phenotypes suggest that the mutant animals may suffer from cardiac dysfunction.
We therefore conducted histological and physiological analyses of the Lmod2PB/PB mutant hearts from 25 days old mice before the animals display any signs of distress. Interestingly, all the analyzed Lmod2PB/PB mutants display enlarged ventricular lumens and thin ventricular walls, phenocopying the symptoms in human DCM patients (n=17, Fig. 4a). The histopathological defects were also confirmed by echocardiography analysis. The mutant hearts had dilated ventricular lumens (Fig. 4b) and thinner heart walls during both systolic and diastolic periods (Fig. 4c). Consistent with these, morphometric ratios (heart weight/body weight) were increased in these animals as well (Fig. 4d).
Fig. 4.
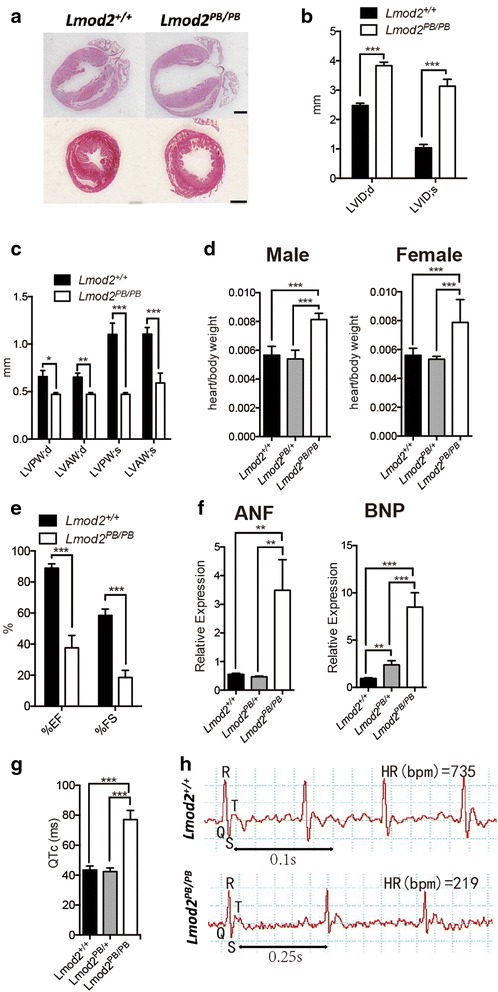
Lmod2PB/PB hearts display dilated cardiomyopathy defects. 25-day old Lmod2PB/PB mice were examined for cardiac morphology and functions. a Longitudinal (upper) and transverse (lower) H&E stain sections of paraffin-embedded hearts (Scale bar 1 mm). b, c, e Echocardiography analysis of Lmod2PB/PB hearts in comparison to wild-type control n = 5. b Higher LV end diastolic and systolic diameter. LVID left ventricular diameter. c Thinner LV anterior and posterior wall. LVPW left ventricular posterior wall; LVAW left ventricular anterior wall. d Morphometric ratios (heart/body weight) of both male and female Lmod2PB/PB mice are significantly increased. e Reduced ejection fraction and fractional shortening values. f Quantitative RT-PCR analysis of atrial natriuretic factor (ANF) and brain natriuretic peptide (BNP) revealed increased expression of the heart hypertrophy and heart failure biomarkers n = 3. LV left ventricle; d diastolic; s: systolic. g, h Electronic Cardiogram revealed lengthening QTc value of Lmod2PB/PB mice in comparison to controls
Echocardiography analysis also revealed that the mutant hearts displayed lower ejection fraction (EF) and fraction shortening (FS) values (Fig. 4e), characteristic DCM deficit. Furthermore, electrocardiography (ECG) analysis revealed lengthened corrected QT interval (QTc) in Lmod2PB/PB mice in comparison to controls (Fig. 4g, h). The lengthened QTc value means delayed electrical repolarization of the ventricles, indicating ventricular arrhythmia [22], complications that many DCM patients also develop.
The expression of atrial natriuretic factor (ANF) and brain natriuretic peptide (BNP) were examined in mutant animals before signs of heart failure [23]. We discovered that the Lmod2PB/PB mutant mice already exhibited a significant increase in gene expression for the heart hypertrophy and heart failure biomarkers (Fig. 4f). Finally, Masson’s Trichrome staining revealed that there was no observable increase in fibrosis in these animals (data not shown).
Lmod2-deficiency leads to sarcomere and ICD disorganization
We further characterized cardiac muscles of Lmod2PB/PB mice (25 days old) by paraffin section and transmission electron microscope (EM). We noticed that the area between the muscle fibers is larger in Lmod2PB/PB cardiac muscles than the wild-type control (Fig. 5a, b, i). EM analysis revealed altered sarcomeres with irregular Z-discs and unidentifiable M-lines in Lmod2PB/PB cardiac fibers (Fig. 5c–f). For those fibers with recognizable Z-discs, measuring the length of the sarcomeres revealed shorter sarcomeres than wild-type control (Fig. 5j). Moreover, the filaments in the Lmod2PB/PB cardiac fibers are also disarrayed in comparison to wild-type control (Fig. 5e, f). Finally, the ICDs in Lmod2PB/PB cardiac fibers are less convoluted and have reduced electron dense (Fig. 5g, h). The ICD morphological changes suggest that the mutant fibers might have less ICD proteins. Quantitative RT-PCR (QPCR) indeed identified reduced expression of two of major ICD genes, β-catenin and Connexin43 (Cx43), components of fascia adherens and gap junctions, respectively (Fig. 5k). Together, these data indicate that Lmod2 deficient cardiac fibers have disrupted sarcomeres and ICDs including the expression of ICD genes.
Fig. 5.
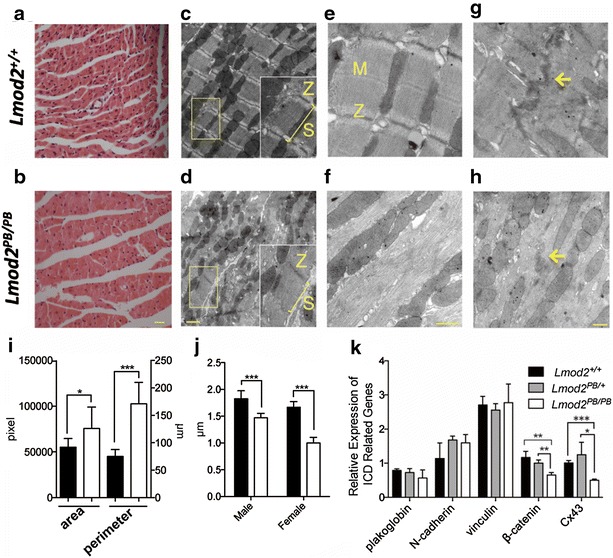
ICD and sarcomere defects of Lmod2PB/PB heart fibers and reduced expression of ICDs proteins. a, b Longitudinal sections of 25d paraffin-embedded hearts stained with H&E. c–h Representative electron micrographs of cardiac LV tissue from 25 days old Lmod2+/+ (c, e, g) and Lmod2PB/PB (e, f, h) mice. Sarcomeres (S) are shortened and disorganized in Lmod2PB/PB myocardium (c, d). Z-discs (Z) and M-lines (M) are disorganized and unrecognizable, respectively, in Lmod2PB/PB myocardium (e, f). Reduced convolution and electron dense of ICD (arrow) in Lmod2PB/PB myocardium (g, h). i Area and perimeter of the area between the muscle fibers are increased in Lmod2 PB/PB hearts compared with wild-type control n = 8. j Statistics of sarcomere length in Lmod2PB/PB myocardium n = 20. k Quantitative RT-PCR of major intercalated discs (ICD) genes n = 3. Scale bars 200 μm in b, 1 μm in d, 500 μm in f, h
Discussion
Using PB insertional mutants, we have studied two members of the Lmod family, Lmod2 and Lmod3, which express in the cardiac and skeletal muscles. We previously reported that Lmod3 deficiency lead to severe skeletal muscle weakness with atrophy specific to fast fibers [6]. EM analysis revealed disorganized sarcomeres in skeletal muscles of Lmod3 mutant mice.
Here we report the characterization of Lmod2-deficient mice caused by PB insertion in the first exon. Unlike Lmod3-deficient mice that are born small with skeletal muscle atrophy [6], Lmod2-deficient animals are born with normal appearance and body weight. Only male Lmod2-deficient mice exhibit underweight in the 4th week. On the other hand, Lmod2-deficient mice exhibit postnatal lethality and all mutant animals die from 3 to 9 weeks after birth. This postnatal lethality together with previous report of Lmod2 in cultured cardiomyocytes [2, 5] suggests a role of the gene in cardiac function.
We have therefore carried out a pathological analysis of the Lmod2 mutant heart. Our study shows that the Lmod2 mutant hearts have enlarged ventricular with systolic dysfunction reflected with EF value less than 50 %, features that are characteristic to DCM patients. Investigation with transmission electron microscopy reveals that the Lmod2PB/PB cardiac muscles exhibit disordered sarcomeres and ICDs. The morphologies of Z-discs, M-lines, and thin filaments in sarcomeres are all affected.
Intercalated disc is composed of highly organized fascia adherens, gap junctions and desmosomes, and glues cardiomyocytes together. Disruption of ICD would also lead to cardiomyopathy [24]. Recently, it has been shown that mutation in non-ICD component protein could also result in ICD structure abnormality and cardiomyopathy [25]. In the previous reported case, an increase in convolutions of ICD was reported in the cardiomyopathy patient [25]. Here we show that Lmod2PB/PB cardiomyocytes have a decrease in ICD convolution and cardiomyopathy. Disruption of ICD is also confirmed by reduction in electron density and expression of two of major ICD genes, β-catenin and Connexin43 (Cx43), components of fascia adherens and gap junctions. Therefore, Lmod2PB/PB mouse model represents a new ICD phenotype related to DCM. During preparation of our manuscript, we noticed that a report of Lmod2 knockout mouse characterization [26]. While DCM was observed in both models, there were differences in ultrastructure of cardiac muscles. In contrast to Lmod2PB/PB phenotypes, Lmod2 knockout model reported widen Z-discs and increased convolutions of ICDs [26]. The difference could be due to the C57BL/6J background for the Lmod2 knockout and FVB/N for Lmod2PB/PB.
The morphological defects in cardiac muscles proceed the symptom of heart failure in animals. Similarly, the expressions of the heart hypertrophy and heart failure biomarkers, atrial natriuretic factor (ANF) and brain natriuretic peptide (BNP), are elevated before the detection of heart failure. Together, these data show that Lmod2 deficiency leads to structure abnormality of cardiac muscles, which results in DCM, and that Lmod2PB/PB mice offer a new model for studying DCM mechanisms and developing therapeutics.
Conclusion
Taken together, our data reveal that Lmod2 is required in heart thin filaments for integrity of sarcomere and ICD and deficient mice exhibit DCM with ventricular arrhythmias and postnatal lethality. The Lmod2PB/PB mutant offers a valuable resource for interrogation of pathogenesis and development of therapeutics for DCM.
Methods
Mouse strains
The founder Lmod2PB/+ mouse was produced by a large-scale genome-wide insertional mutagenesis with the PB transposon in the FVB/N mice [20, 27].
Genotyping
Genomic DNA was isolated by a 56 °C overnight digestion of mouse toes in 200 μL lysis buffer (100 mM NaCl, 10 mM Tris pH 8.0, 25 mM EDTA, and 0.5 % SDS) plus fresh 0.1 mg/ml proteinase K. PCR was performed with primers within exon 1 of Lmod2 (forward: 5′-AGCTGTCGGCTTTCAATTTTTTTCC-3′; reverse: 5′-TGTCTTCCAGCTCCCTCTCAAG-3′, 247 bp product) and primers within the PB transposon (forward: 5′-CTGAGATGTCCTAAATGCACAGCG-3′ and reverse: 5′-TGTCTTCCAGCTCCCTCTCAAG-3′, 819 bp product). PCR conditions used were as the following: an initial denaturation step at 93 °C for 3 min, then followed by a 40 cycle of 93 °C for 30 s/57 °C for 30 s/65 °C for 2 min, and one more elongation at 65 °C for 10 min as the final step.
Reverse transcription and quantitative RT-PCR
Total RNA was isolated from 3-week-old mouse heart with Trizol (Invitrogen). The cDNA was synthesized then using the PrimeScriptTM RT reagent Kit (Takara). Quantitative RT-PCR was performed with AceQ qPCR SYBR Green Master Mix (Vazyme) according to the manufacturer’s instruction. All quantitative RT-PCR primers are listed in Table 1.
Table 1.
Quantitative RT-PCR primers
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Lmod2 | ACCTTATCCCGATTTGCTGAAG | ACCTTGAGCATGTCTGCAATG |
| GAPDH | TGTTCCTACCCCCAATGTGTCC | GGAGTTGCTGTTGAAGTCGCAG |
| Tmod1 | TGAGCTAGATGAACTAGACCCTG | CGGTCCTTAAATTCCTTCGCTTG |
| ANF | CATCACCCTGGGCTTCTTCCT | CATCACCCTGGGCTTCTTCCT |
| BNP | GCGGCATGGATCTCCTGAAGG | GCGGCATGGATCTCCTGAAGG |
| Plakoglobin | TGGCAACAGACATACACCTACG | GGTGGTAGTCTTCTTGAGTGTG |
| N-cadherin | AGCGCAGTCTTACCGAAGG | TCGCTGCTTTCATACTGAACTTT |
| Vinculin | GAGGCTGAACTGCTTCAATCA | CCAGATTTGACGAGGTGCCTA |
| β-catenin | ATGGAGCCGGACAGAAAAGC | CTTGCCACTCAGGGAAGGA |
| Cx43 | ACAGCGGTTGAGTCAGCTTG | GAGAGATGGGGAAGGACTTGT |
Western blotting
Hearts were dissected and lysed in RIPA buffer with Complete Protease inhibitors (Roche) and PMSF (Phenylmercury acetate) (Sigma) on ice. The BCA Assay (Themo) was applied to quantify total proteins extracted. The western blotting procedure was carried out according to standard protocol with a brief introduction here. Protein bands were separated by SDS/PAGE and transferred to nitrocellulose membranes (Immobilon-P). The transferred membrane was blotted first 5 % skim milk for 1 h. Blots were then incubated with a primary antibody and a secondary antibody consecutively to detect protein of interest. Primary antibodies used are the following: Lmod3 (1:1000; HPA036034, Sigma), GAPDH (1:3000; AC001, ABclonal). Secondary antibody used is the Anti-rabbit IgG antibody conjugated to PerCP-Cy5.5 (1:3000; sc-45101, Santa Cruz). Signal was finally visualized by enhanced chemiluminescence (34,080, Themo).
Echocardiography and ECG
Mice were anesthetized by inhalation of 1–2 % isoflurane delivered in a gas mixture with oxygen, medical-grade compressed air, and nitrogen. The anesthetized mice went through echocardiography with the Vevo 770 microultrasound system (VisualSonics Inc. Toronto, Canada). Both parasternal long-axis and short-axis views of the left ventricles were analyzed with a 707B transducer according to the manufacturer’s instruction. Data were recorded in M mode.
Conscious and unrestrained mice were used for ECG analysis with the ECGenie electrocardiography system (Mouse Specifics, Inc., Boston, MA) according to the manufacturer’s instruction. Before experimental tests, mice should be trained for 3–5 days to behave normally in this machine. Only data from continuous recordings of 20–30 ECG signals were included in the final analyses. A physiologic waveform analysis platform “eMOUSE” (Mouse Specifics, Inc.) was applied for data analysis.
Transmission electron microscopy
Samples for transmission electron microscopy were prepared using the standard procedure: hearts were excised, washed in PBS, fixed in 2.5 % glutaraldehyde in 0.1 M cacodylate buffer for more than 2 h, washed in 0.1 M cacodylate buffer for 3 × 15 min, post-fixed with 1 % potassium ferrocyanide reduced OsO4 on ice for 2–3 h, dehydrated through graded ethanol and acetone, and finally embedded in EMbed 812 resin. Ultrathin sections (60 nm) were analyzed on a Technai G2 Spirit BioTWIN electron microscope, and images were collected with a 4 × 4 digital camera (Optronics). Length of sarcomere was measured using ImageJ.
Histology
Hearts were excised, washed in PBS, fixed in 4 % paraformaldehyde solution (Sangon Biotech) overnight, dehydrated, and embedded in paraffin. Longitudinal or transverse sections (10 μm) were stained with a Masson Trichrome Stain Kit (Maixin Biotech) or according to the classical hematoxylin and eosin (H&E) staining protocol (Sigma), and finally mounted with DPX Mountant (Sigma). Light microscopy images were captured using a Nikon Eclipse microscope with a Nikon DS-Ri1 camera and/or a Zeiss Axio Zoom V16 stereomicroscope with an AxioCam MRc camera.
Statistics
Data were presented as mean ± SED in figures. Two-tailed unpaired Student’s t-test was used unless otherwise stated. The significance is indicated as the label of one or more * with the following categories: (1) * P < 0.05; (2) ** P < 0.01; 3) *** P < 0.005. Prism 6 (GraphPad Software) was used for plotting.
Authors’ contributions
SL, TX and LVS designed the project; SL, KM, HT, CC, and SS did the experiments and collected data; SL, KM, LT, SD, TL, XW, FL, ZZ, TX and LVS analyzed the data; SL, TX and LVS prepared the manuscript; TX and LVS supervised all aspects of the work. All authors read and approved the final manuscript.
Acknowledgements
We thank Ms. Yanfeng Tan and Ms. He Tan in the Institute of Developmental Biology and Molecular Medicine, Fudan University, for technical support, and 973 and 863 working group, Dr. Yuan Zhuang, and other IDM members for scientific discussion.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All animal procedures applied in this article have been approved by the Animal Care and Use Committee of the Institute of Developmental Biology and Molecular Medicine, Fudan University, Shanghai, China.
Funding
This work was supported by the following grants: Chinese Key Projects for Basic Research (973) Grants 2013CB945301 and 2013CB945304; Chinese Hi-tech Research and Development Project of China (863) Grants 2012AA022401 and 2014AA021104; National Natural Science Foundation of China grant 81170789; and National Institutes of Health grant U01HG004073. TX is a Howard Hughes Medical Institute Investigator.
Abbreviations
- ANF
atrial natriuretic factor
- BNP
brain natriuretic peptide
- CHF
congestive heart failure
- DCM
dilated cardiomyopathy
- ECG
electrocardiography
- EF
ejection fraction
- EM
electron microscope
- FS
fraction shortening
- ICD
intercalated discs
- Lmod
leiomodin
- Lmod1
leiomodin1
- Lmod2
leiomodin2
- Lmod3
leiomodin3
- LV
left ventricle
- LVAW
left ventricular anterior wall
- LVID
left ventricular diameter
- LVPW
left ventricular posterior wall
- PB
piggyBac
- QTc
corrected QT interval
- Tmod1
tropomodulin1
Contributor Information
Shuang Li, Email: li_s13@fudan.edu.cn.
Kaiqi Mo, Email: kenny.mo@live.com.
Hong Tian, Email: 18917134339@163.com.
Chen Chu, Email: chickenchu@163.com.
Shuna Sun, Email: shunasun@163.com.
Lei Tian, Email: lei.tian.us@gmail.com.
Sheng Ding, Email: sheng.ding@yale.edu.
Tong-ruei Li, Email: litongruei@fudan.edu.cn.
Xiaohui Wu, Email: xiaohui_wu@fudan.edu.cn.
Fang Liu, Email: liufang@fudan.edu.cn.
Zhen Zhang, Email: zhenzhang@sjtu.edu.cn.
Tian Xu, Email: tian.xu@yale.edu.
Ling V. Sun, Email: lingsun@fudan.edu.cn
References
- 1.Conley CA, Fritz-Six KL, Almenar-Queralt A, Fowler VM. Leiomodins: larger members of the tropomodulin (Tmod) gene family. Genomics. 2001;73(2):127–139. doi: 10.1006/geno.2000.6501. [DOI] [PubMed] [Google Scholar]
- 2.Chereau D, Boczkowska M, Skwarek-Maruszewska A, Fujiwara I, Hayes DB, Rebowski G, et al. Leiomodin is an actin filament nucleator in muscle cells. Science. 2008;320(5873):239–243. doi: 10.1126/science.1155313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nworu CU, Kraft R, Schnurr DC, Gregorio CC, Krieg PA. Leiomodin 3 and tropomodulin 4 have overlapping functions during skeletal myofibrillogenesis. J Cell Sci. 2015;128(2):239–250. doi: 10.1242/jcs.152702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nanda V, Miano JM. Leiomodin 1, a new serum response factor-dependent target gene expressed preferentially in differentiated smooth muscle cells. J Biol Chem. 2012;287(4):2459–2467. doi: 10.1074/jbc.M111.302224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsukada T, Pappas CT, Moroz N, Antin PB, Kostyukova AS, Gregorio CC. Leiomodin-2 is an antagonist of tropomodulin-1 at the pointed end of the thin filaments in cardiac muscle. J Cell Sci. 2010;123(Pt 18):3136–3145. doi: 10.1242/jcs.071837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian L, Ding S, You Y, Li TR, Liu Y, Wu X, et al. Leiomodin-3-deficient mice display nemaline myopathy with fast-myofiber atrophy. Dis Model Mech. 2015;8(6):635–641. doi: 10.1242/dmm.019430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuen M, Sandaradura SA, Dowling JJ, Kostyukova AS, Moroz N, Quinlan KG, et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest. 2014;124(11):4693–4708. doi: 10.1172/JCI75199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cenik BK, Garg A, McAnally JR, Shelton JM, Richardson JA, Bassel-Duby R, et al. Severe myopathy in mice lacking the MEF2/SRF-dependent gene leiomodin-3. J Clin Invest. 2015;125(4):1569–1578. doi: 10.1172/JCI80115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu X, Chen J, Reedy MC, Vera C, Sung KL, Sung LA. E-Tmod capping of actin filaments at the slow-growing end is required to establish mouse embryonic circulation. Am J Physiol Heart Circ Physiol. 2003;284(5):H1827–H1838. doi: 10.1152/ajpheart.00947.2002. [DOI] [PubMed] [Google Scholar]
- 10.Fritz-Six KL, Cox PR, Fischer RS, Xu B, Gregorio CC, Zoghbi HY, et al. Aberrant myofibril assembly in tropomodulin1 null mice leads to aborted heart development and embryonic lethality. J Cell Biol. 2003;163(5):1033–1044. doi: 10.1083/jcb.200308164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sussman MA, Welch S, Cambon N, Klevitsky R, Hewett TE, Price R, et al. Myofibril degeneration caused by tropomodulin overexpression leads to dilated cardiomyopathy in juvenile mice. J Clin Invest. 1998;101(1):51–61. doi: 10.1172/JCI1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKeown CR, Nowak RB, Moyer J, Sussman MA, Fowler VM. Tropomodulin1 is required in the heart but not the yolk sac for mouse embryonic development. Circ Res. 2008;103(11):1241–1248. doi: 10.1161/CIRCRESAHA.108.178749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ono Y, Schwach C, Antin PB, Gregorio CC. Disruption in the tropomodulin1 (Tmod1) gene compromises cardiomyocyte development in murine embryonic stem cells by arresting myofibril maturation. Dev Biol. 2005;282(2):336–348. doi: 10.1016/j.ydbio.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 14.Simpson S, Edwards J, Ferguson-Mignan TF, Cobb M, Mongan NP, Rutland CS. Genetics of human and canine dilated cardiomyopathy. Int J Genomics. 2015;2015:204823. doi: 10.1155/2015/204823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna CJ, Codd MB, McCann HA, Sugrue DD. Idiopathic dilated cardiomyopathy: familial prevalence and HLA distribution. Heart. 1997;77(6):549–552. doi: 10.1136/hrt.77.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grogan M, Redfield MM, Bailey KR, Reeder GS, Gersh BJ, Edwards WD, et al. Long-term outcome of patients with biopsy-proved myocarditis: comparison with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 1995;26(1):80–84. doi: 10.1016/0735-1097(95)00148-S. [DOI] [PubMed] [Google Scholar]
- 17.National Clinical Guideline Centre. Chronic heart failure: the management of chronic heart failure in adults in primary and secondary care. London: National Clinical Guideline Centre; 2010. Available from: http://guidance.nice.org.uk/CG108/Guidance/pdf/English.
- 18.Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010;12(11):655–667. doi: 10.1097/GIM.0b013e3181f2481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raju H, Alberg C, Sagoo GS, Burton H, Behr ER. Inherited cardiomyopathies. BMJ. 2011;343:d6966. doi: 10.1136/bmj.d6966. [DOI] [PubMed] [Google Scholar]
- 20.Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122(3):473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 21.PiggyBac Mutagenesis Information Center. http://www.idmshanghai.cn/PBmice/. Accessed 21 Nov 2012.
- 22.Towbin JA, Vatta M. Molecular biology and the prolonged QT syndromes. Am J Med. 2001;110(5):385–398. doi: 10.1016/S0002-9343(00)00715-4. [DOI] [PubMed] [Google Scholar]
- 23.Wang TJ, Larson MG, Levy D, Benjamin EJ, Leip EP, Omland T, et al. Plasma natriuretic peptide levels and the risk of cardiovascular events and death. N Engl J Med. 2004;350(7):655–663. doi: 10.1056/NEJMoa031994. [DOI] [PubMed] [Google Scholar]
- 24.Ehler E, Horowits R, Zuppinger C, Price RL, Perriard E, Leu M, et al. Alterations at the intercalated disk associated with the absence of muscle LIM protein. J Cell Biol. 2001;153(4):763–772. doi: 10.1083/jcb.153.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purevjav E, Arimura T, Augustin S, Huby AC, Takagi K, Nunoda S, et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet. 2012;21(9):2039–2053. doi: 10.1093/hmg/dds022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pappas CT, Mayfield RM, Henderson C, Jamilpour N, Cover C, Hernandez Z, et al. Knockout of Lmod2 results in shorter thin filaments followed by dilated cardiomyopathy and juvenile lethality. Proc Natl Acad Sci USA. 2015;112(44):13573–13578. doi: 10.1073/pnas.1508273112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun LV, Jin K, Liu Y, Yang W, Xie X, Ye L, et al. PBmice: an integrated database system of piggyBac (PB) insertional mutations and their characterizations in mice. Nucleic Acids Res. 2008;36 (Database issue):D729–D734. doi: 10.1093/nar/gkm790. [DOI] [PMC free article] [PubMed] [Google Scholar]