

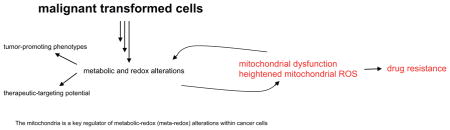
Abstract
Under physiological conditions, a well-coordinated and balanced redox system exists to ensure that reactive oxygen species (ROS) are appropriately utilized to accomplish specific functions, such as signaling and protein regulation. The influence of ROS within malignant cells, whether for good or bad may depend on several factors, such as tumor and tissue type, disease stage, treatment strategy, as well as duration, specificity and levels of ROS. What then are the known roles of ROS in cancer? Firstly, ROS significantly impacts cancer phenotypes. Secondly, the oxidative ROS property responsible for killing cancer cells, also impact secondary signaling networks. Thirdly, a strong correlation exist between ROS and genetic instability which may promote mutations. Finally, emerging observations suggest a role for mitochondrial ROS in cancer drug resistance, with implications for therapy. The mitochondria is a key regulator of metabolic-redox (meta-redox) alterations within cancer cells. Like a double-edged sword, mitochondrial ROS perturbations in cancer therapy may be beneficial or detrimental. However, harnessing ROS-specific cancer-targeting benefits remain a major challenge.
Keywords: Mitochondrial ROS, Oxidative stress, Cancer drug resistance, Metabolic alterations
Graphical abstract
1. Introduction
Introduction of improved anti-cancer drugs over the last couple of decades have been aimed at effective ablation of tumor growth or progression while providing minimal side-effects. New-generation, target-specific drugs, such as tyrosine kinase inhibitors (e.g. gefitinin, erlotinib) and monoclonal antibodies (e.g. trastuzumab) have joined the lists of other established cancer therapies (chemo, and radiation-based treatments) in the fight against cancer. While combination strategies are now widely used and accepted, the overall outcomes are variable. Together, these anti-cancer agents suffer a major and common challenge, unresponsiveness of tumors to previously effective drugs. As would be expected, several variables and factors contribute to the loss of response, which may reflect survival-adaptations employed by cancer cells. A major aspect of such adaptations will usually involve metabolic alterations designed to support and maintain highly active processes undertaken by cancer cells, such as proliferation, angiogenesis and metastasis. Metabolism is an intrinsic cellular process utilized by “normal” non-cancer cells, as well as disease tissues in order to accomplish energy-dependent processes. Whether by design or default arrangement, the mitochondria is the “powerhouse” of cellular metabolic functions under patho-physiological conditions. As a dynamic organelle, the mitochondria modulates its functions to reflect prevailing changes, such as starvation or oxygen deficiency (hypoxia). Furthermore, response to extrinsic factors, such as drug treatments inadvertently trigger mitochondrial adaptations that impact its functions.
Various redox systems at play within biological systems, and their essential but often conflicting functions in physiology and disease have been reported [1–4]. ROS is widely implicated in cancer initiation, progression and survival phenotypes [4,5]. Although further research questions are required to delineate the relationship between redox signaling and cancer, this review article approaches the subject from a perspective designed to provide unique and fresh insight on direct links between mitochondrial ROS and cancer drug resistance, with broader implications for therapy. While ROS-mediated mechanisms of action represent a major cancer-targeting strategy, emerging data demonstrate that chronic and abnormally high ROS levels may instigate or accentuate cancer phenotypes, including drug resistance [2,6].
2. Cancer drug resistance: definitions, readouts and phenotypes
Beyond the loss of response to a particular drug or treatment regimen, a single definition for drug resistance is non-existent due to the often confounding processes associated with resistance. In the absence of well-defined drug resistance properties, researchers are locked in a “game” devoid of established rules. Paradoxically, the heterogeneity of cancer cells make any given set of rules limited, and tumor-specific. The wide variety of drugs, mechanisms of action, as well as off-target effects contribute further to the complexity of deciphering drug resistance. It is important to note that ablation of a targeted signaling pathway by specific anti-cancer agents may not necessarily imply absence of resistance. Cancer cells can and do evolve in a dynamic manner, utilizing various and/or multiple alternative survival mechanisms. For example, EGFR activation (the primary gefitinib target) was effectively abrogated following chronic, long-term treatments in lung cancer cell lines. However, prolonged gefitinib treatments correlated with defective cell cycle, mitochondrial dysfunction, increased ROS and epithelial-mesenchymal transition (EMT) [6]. What then is the readout for drug resistance? What are the established, standard genetic markers, phenotypes or morphology that correlate with resistance? EMT, which is associated with upregulation of mesenchymal markers (such as, vimentin and fascin) and downregulation of epithelial markers (such as, E-cadherin and keratin) is a widely accepted phenotype of drug resistant cells [6–11]. During EMT, non-motile parental cells, gain motility with associated expression of targets typically required during early developmental stages [6,7]. EMT ties in nicely with metastatic potential of cancer cells, which is a major contributor to decreased drug efficacy and ultimately resistance. Additional drug-resistant markers may include certain microRNAs (miRNAs), as well as cancer stem cells [12–14]. However, cautious interpretation is required as the absence of designated markers or phenotypes may not necessarily translate into absence of resistant cancer cells. To this end, it would be important to continue identifying processes and markers that strongly correlate with the loss of response to therapies, in addition to the factors that drive malignant transformations. Expansion, characterization and validation of drug resistant markers and/or phenotypes will enable early determination or onset of drug resistance, allowing better intervention strategies to counter the transition.
3. Reactive oxygen species (ROS)
ROS has been aptly described as a heterogeneous group of diatomic oxygen from free and non-free radical species [1]. Enzyme-catalyzed reactions are major contributors of endogenous ROS, with mitochondrial metabolism being central to the process [15,16]. ROS can be interconverted from one form to another, e.g. superoxide (O2−) to hydrogen peroxide (H2O2). Hydrogen peroxide can in turn lead to different oxidizing derivatives, such as hydroxyl radicals [1]. Hence, a cooperative or positive feed-forward mechanism can be set in motion by the action of a single ROS, with resultant exaggerated effects. Furthermore, superoxide can form the reactive nitrogen specie (RNS), peroxynitrite (ONOO−) following reaction with nitric oxide (NO). As would be expected, such a heterogeneous group of oxygen-derived free radicals possess intrinsically diverse properties that define specific reactivity and functions, either directly or as second messengers in cellular signaling networks [1]. Unique affinity for a particular substrate or pathway may vary, and ROS levels in response to cellular perturbations are unpredictable. Hence, the factors that determine ROS abundance, stability, functions, regulation and perturbation are intricate and complex, with several inputs and modifications at multiple points. ROS are highly reactive and promote oxidative stress, which may result in tissue damage. Depending on context, direct or indirect effects of ROS on the immune system may further act to support disease progression. Although the focus of this review is on cancer, free radicals contribute to other disease conditions, including cardiovascular damage, aging, arthritis, and neurodegenerative disorders [15,17–19].
4. Factors that impact ROS balance and functions
ROS arise from physiological cellular processes, such as respiration. The mitochondria being at the center of ATP synthesis remains perhaps the most important source of cellular ROS. Therefore, tight regulation of metabolic reactions is essential to healthy ROS balance. Even within non-cancer cells, physiological processes, such as apoptosis, autophagy and proliferation are likely to involve heightened metabolic activity, which may in turn propagate oxidative stress in the absence of a robust antioxidant balancing system. Sustained oxidative damage may trigger genetic instability and malignant transformations [1,17]. For example, H2O2 is membrane permeable, and can penetrate membrane barriers of various organelles to potentially instigate or accentuate genetic aberrations [1]. ROS attenuates cysteine-containing proteins, which affect protein structure, functions and interactions [1,2].
ROS balance is typically mediated by antioxidant enzymes, such as, catalase, superoxide dismutase and glutathione. Mutation(s) of antioxidant genes may therefore compromise or contribute to the loss of ROS regulation with deleterious consequences to cell function and processes. In our study, attenuated protein expression of catalase was observed in gefitinib resistant clones, which correlated with enhanced ROS levels [6]. The unique functions of a cell determine energy requirements, and hence metabolic activity. For example, cardiac muscle cells are inherently more active and contain ~40% more mitochondria than skeletal muscle cells. Cellular energy demands may also fluctuate depending on rest (e.g. sleep) or activity (e.g. exercise). Therefore, the mitochondria must constantly adapt to ensure it closely matches the dynamic cellular energy requirements. Such alternating metabolic conditions may be context-dependent, especially in disease states, and directly influence ROS production. In summary, ROS is not a homogenous event that occurs across cells with uniformity. The net ROS levels of a healthy or diseased tissue is subject to various factors, with no certainty at predicting signaling events or outcomes.
5. The mitochondria: an age-old organelle with evolving functions
The mitochondria is a versatile organelle with self-replication ability (semi-autonomous). While metabolism may represent the most described and well-known function, the mitochondria plays other important roles, including haem protein, calcium ion buffering and regulation of biosynthetic intermediates in lipid and protein production [2,16]. The mitochondria is usually passed down from maternal line, hence mitochondria-specific mutations in disease conditions (such as, Leber’s hereditary optical neuropathy, and mitochondrial encephalomyopathy) are likely to be inherited by all the offsprings. Mitochondrial DNA within a cell may be homogenous (homoplasmic) or vary (heteroplasmic), and it is likely that mitochondrial functions, mutations, as well as ROS production reflect the variabilities unique to each cell type or population. It is conceivable that metabolic-intensive cells may be greatly impacted by mitochondrial defects relative to quiescent, low-level metabolic cells.
6. Mitochondrial redox signaling
Tasked with meeting cellular energy requirements, the mitochondria relies on a build-up of proton gradient across the inner mitochondrial membrane which link oxidation of NADH and FADH2 to the phosphorylation of ADP. This gradient potential and transfer of high-energy electrons to oxygen forms water and generates ATP. The entire process involve redox reactions that continuously generate and consume high-energy coenzymes, such as NAD+, NADP+ and FAD+ during glycolysis and Krebs cycle. While physiological redox reactions are beneficial and provide several important signaling ques integral to cell functions, perturbation of mitochondrial enzymes, or certain transcription factors may impact redox balance. For example, HIF activation due to mitochondrial ROS signaling has been reported, with several target genes under HIF regulation (or other transcription factors) becoming activated or silenced [2,16]. A tumor’s ability to resist therapy may not be unconnected with such aberrant proto-oncogenes that become switched-on, or inhibition of tumor suppressor genes. Unsurprisingly, a link between AMP-activated protein kinase (AMPK) and oxidative stress via NADPH has been reported [2,20,21]. AMPK is an important energy sensor in disease and physiology, and linked to the NF-kB transcription factor, as well as major signaling networks, such as mTOR and TSC [22]. Given its prominent role in cellular energy regulation, AMPK is likely involved with mitochondrial ROS, especially under conditions of cancer-cell metabolic alterations. Additional targets, including p53, EGFR and p38-MAPK have been linked to ROS signaling [2,23–25]. The confounding roles of mitochondrial ROS may be due to varying factors, such as duration of exposure, ROS levels, disease type, stage or dominant signaling network.
7. Mitochondrial ROS and cancer drug resistance
Tumor heterogeneity dictates that while certain cancer types or cell sub-population benefit from ROS-based therapies, oxidative stress may potentially instigate untoward effects on other cells. Off-target ROS insults potentially contribute to genetic instability and mutations within healthy, as well as cancer cells. These dynamic sequence of events, and constant pressure for cell re-adjustments eventually promote the evolution of resilient, drug-resistant cells. While it is unlikely that mitochondrial ROS-mediated mechanisms are the sole contributor to cancer drug resistance, its prominent roles and modulation of metabolic events may be central to the process. Mitochondrial dysfunction and/or unrelated genetic aberrations may work in concert to facilitate maximum survival advantages, or operate independently to ensure that cancer cells possess alternative survival mechanisms. In our study, abnormal mitochondrial morphology and function, as well as heightened ROS levels correlated with gefitinib-resistant H1650 clones [6]. Given that EGFR is the primary target of gefitinib, we screened but failed to detect any new EGFR mutations following chronic drug treatments. However, specific downregulation of the mitochondrial enzyme, pyruvate dehydrogenase was evident within gefitinib resistant clones. Although further work is required, our observations suggest that chronic gefitinib treatment may adversely impact mitochondrial enzymes. Whether such aberrations contribute to or are a consequence of ROS is unclear. In essence ROS due to mitochondrial dysfunction, or mitochondrial dysfucntion from redox aberrations (e.g. hypoxia, cellular degradation or detoxification processes), may work in concert to promote cancer progression, even in the presence of drug targeting.
Additionally, ROS impact cancer stem cells. Usually, only a small sub-population of cancer cells develop resistance against therapies, similar to small proportion of cancer stem cells that determine cell differentiation property. Importantly, the expression of stem cell markers within drug resistant cancer cells [14] support the notion that the processes are interconnected, and is consistent with self-renewal potentials of cancer stem cells that may be responsible for drug resistant cancer cells. However, it would be interesting to test whether similar mechanisms are at play in the development of drug resistance and cancer stem cells. As a mutagen, ROS possess the ability to either block self-renewal, or stimulate the differentiation of stem cells [14]. Interconnected or parallel ROS signaling networks may influence cancer stem cells and cancer drug resistant cells. Therefore, ability to coordinately or independently modulate a repertoire of ROS signaling networks that impinge on cellular functions and processes is critical to regulation of ROS-mediated drug resistance.
8. Implications for cancer therapy: major roads lead to metabolism
Metabolic alterations represent a hallmark of cancer cells, and was described decades ago as the Warburg effect [26,27]. However, a salient aspect of such metabolic perturbations that may not have been so obvious at the time is the broader contribution to cancer phenotypes in the absence or presence of therapies [27]. Perhaps, alterations within other organelles or processes involved in redox balance would not be so grave compared to occurrence in the mitochondria. Inherent in the process of metabolism is cellular respiration and oxidative phosphorylation, with glucose as the primary source of ATP. Variations that integrate metabolic pathways, such as beta-oxidation of fatty acids, intermediates of amino acids and pentose phosphate pathway converge at the mitochondria, and likely involve ROS perturbations. Metabolic and redox alterations which are critical steps of malignant cell transformation make the mitochondria an attractive therapeutic target. Increased knowledge in the field of redox biology, its reactions, signaling networks and interplay in disease and physiology has enabled a better realization of potential benefits in disease targeting. However, ROS also presents grave danger with respect to cancer phenotypes and drug resistance. In addition to conventional cancer therapies that are heavily reliant on ROS-mediated mechanisms, recent observations indicate that targeted agents can potentially enhance cellular ROS levels [6]. These observations raise several concerns with respect to current cancer treatment modalities. Cancer therapies, whether directly dependent on ROS or other mechanisms should consider the implications and potential roles of ROS. The frequency, duration and dosage of anti-cancer agents may require modifications aimed at reducing untoward, off-target ROS consequences. ROS levels may provide information on cellular redox balance, as well as potential off-target perturbations during treatments. Devising accurate and robust means of monitoring ROS status before and during treatments may offer useful information that could influence appropriate interventions at an early and critical stage of disease transition into resistance. Drastic cellular ROS fluctuations should be matched with drug resistance phenotypes, such as EMT, cancer stem cell markers and mitochondrial dysfunction. Such information may enable doctors make informed judgements on whether to continue with a particular drug or change treatment.
9. Conclusion
While ROS may represent a property of cancer cells, heightened levels may serve as an indicator of drug resistance [2,6]. Recent observations from our lab demonstrated that gefitinib resistance correlate with mitochondrial dysfunction and increased ROS in lung cancer cells [6]. Conventional anti-cancer strategies still in common use, such as chemo-and radio-therapy are essentially designed to destroy cancer cells via ROS-mediated mechanisms. Like a double-edge sword, ROS significantly influences cellular signaling networks with a potential for beneficial or deleterious outcomes [1,2,28–30]. Further complication exist with respect to the roles of antioxidant in cancer treatments. While antioxidants may minimize the effectiveness of ROS-mediated therapies, antioxidants potentially attenuate oxidative damage and the risk of malignant transformations. A contributory factor in divergent antioxidant outcomes is in part due to variations, such as disease models, antioxidant specificity, concentrations and treatment durations. Given heterogeneous nature of ROS, oxidizing strengths and signaling networks, it is conceivable that the effects of antioxidants will likely vary with respect to attenuation of ROS-mediated events. Perhaps, optimized and selective targeting of ROS-specific organelles with antioxidants, or intermittent ROS delivery may provide a balanced approach. The mitochondria represents a key regulator of meta-redox alterations within cancer cells and an attractive target.
Observations that heightened cellular ROS may instigate or accentuate cancer drug resistance present serious consequences for therapy. While initial ROS treatments may be effective against certain cancers, long-term exposure to abnormally high ROS levels may become counter-productive [2,6]. Chronic and heightened ROS exposure as the primary therapy, or indirectly from secondary effects of other anti-cancer drugs contribute to adaptation mechanisms employed by cancer cells in order to survive “unfavorable” micro-environments [2]. The heterogeneity of cancer cells varies within tissue types, disease stage and patients, further compounding the unpredictability of response to ROS perturbations. Off-target ROS insults may contribute to genetic instability and mutations within healthy cell populations. These dynamic sequence of events, and persistent adjustments by cancer cells eventually result in the evolution of resilient, drug-resistant clones. To this end, basal mitochondrial ROS range and redox status during treatments may provide important clues to guide appropriate intervention strategies. Although the ideal goal is to prevent drug resistance and effectively eliminate cancer cells, this may not be achievable due to the robust, dynamic nature of tumor cells. However, a realistic and achievable expectation is the identification of processes that can delay the onset of drug resistance, or enable early detection of resistant markers/phenotypes. Mitochondrial ROS may fulfill these roles.
Acknowledgments
The present paper has not been funded in anyway. The authors did not receive any funding related to the present paper.
Footnotes
Contributors
I.S Okon and M.-H. Zou conceived and drafted this paper. All authors participated in critical review of the report, and have approved the final version.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Kwee JK. A Paradoxical chemoresistance and tumor suppresive role of antioxidant in solid cancer cells: a strange case of Dr. Jekyll and Mr. Hyde. Biomed Res Int. 2014;2014:1–9. doi: 10.1155/2014/209845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen G, Wang F, Trachootham D, et al. Preferential killing of cancer cells with mitochondrial dysfunction by natural compounds. Mitochondrion. 2010;10(6):614–625. doi: 10.1016/j.mito.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J. Reactive oxygen species and drug resistance in cancer chemotherapy. Austin J Clin Pathol. 2014;1(14):1–8. [Google Scholar]
- 4.Poillet-Perez L, Despouy G, Delage-Mourroux R, et al. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015;4:184–192. doi: 10.1016/j.redox.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toyokuni S. Molecular mechanisms of oxidative stress-induced carcinogenesis: from epidemiology to oxygenomics. IUBMB Life. 2008;60(7):441–447. doi: 10.1002/iub.61. [DOI] [PubMed] [Google Scholar]
- 6.Okon IS, Coughlan KA, Zhang M, et al. Gefitinib-mediated ROS instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J Biol Chem. 2015;290(14):9101–9110. doi: 10.1074/jbc.M114.631580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119(6):1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69(14):5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sommers CL, Heckford SE, Skerker JM, et al. Loss of epithelial markers and acquisition of vimentin expression in adriamycin-and vinblastine-resistant human breast cancer cell lines. Cancer Res. 1992;52:5190–5197. [PubMed] [Google Scholar]
- 10.Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009;1796(2):75–90. doi: 10.1016/j.bbcan.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Thompson EW, Torri J, Sabol M, et al. Oncogene-induced basement membrane invasiveness in human mammary epithelial cells. Clin Exp Metastasis. 1994;12(3):181–194. doi: 10.1007/BF01753886. [DOI] [PubMed] [Google Scholar]
- 12.Ma J, Dong C, Ji C. MicroRNA and drug resistance. Cancer Gene Ther. 2010;17(8):523–531. doi: 10.1038/cgt.2010.18. [DOI] [PubMed] [Google Scholar]
- 13.Majumder S, Jacob ST. Emerging role of microRNAs in drug-resistant breast cancer. Gene Expr. 2011;15(3):141–151. doi: 10.3727/105221611x13176664479287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dayem AA, Choi HY, Kim JH, et al. Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers. 2010;2(2):859–884. doi: 10.3390/cancers2020859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valko M, Rhodes CJ, Moncol J, et al. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 16.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14(11):709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobliakov VA. Mechanisms of tumor promotion by reactive oxygen species. Biochemistry. 2010;75(6):675–685. doi: 10.1134/s0006297910060015. [DOI] [PubMed] [Google Scholar]
- 18.Jimenez-Del-Rio M, Velez-Pardo C. The bad, the good, and the ugly about oxidative stress. Oxid Med Cell Longev. 2012;2012:1–13. doi: 10.1155/2012/163913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oliveira BF, Nogueira-Machado JA, Chaves MM. The role of oxidative stress in the aging process. Sci World J. 2010;10:1121–1128. doi: 10.1100/tsw.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong Y, Zhang M, Liang B, et al. Reduction of amp-activated protein kinase alpha2 increases. endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121(6):792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang S, Zhang M, Liang B, et al. Ampkalpha2 deletion causes aberrant expression and activation.of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26s proteasomes. Cir Res. 2010;106(6):1117–1128. doi: 10.1161/CIRCRESAHA.109.212530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veelen WV, Korsse SE, Laar LVD, et al. The long and winding road to rational treatment of cancer associated with LKB1/AMPK/TSC/mTORC1 signaling. Oncogene. 2010;30(20):2289–2303. doi: 10.1038/onc.2010.630. [DOI] [PubMed] [Google Scholar]
- 23.Giannoni E, Fiaschi T, Ramponi G, et al. Redox regulation of anoikis resistance of metastatic prostate cancer cells: key role for Src and EGFR-mediated pro-survival signals. Oncogene. 2009;28(20):2074–2086. doi: 10.1038/onc.2009.77. [DOI] [PubMed] [Google Scholar]
- 24.Dolado I, Swat A, Ajenjo N, et al. p38a MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11(2):191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 25.Runchel C, Matsuzawa A, Ichijo H. Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxid Redox Signal. 2011;15(1):205–218. doi: 10.1089/ars.2010.3733. [DOI] [PubMed] [Google Scholar]
- 26.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 27.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gogvadze V. Targeting mitochondria in fighting cancer. Curr Pharm Des. 2011;17(36):4034–4046. doi: 10.2174/138161211798764933. [DOI] [PubMed] [Google Scholar]
- 29.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 30.Wen S, Zhu D, Huang P. Targeting cancer cell mitochondria as a therapeutic approach. Future Med Chem. 2013;5(1):53–67. doi: 10.4155/fmc.12.190. [DOI] [PMC free article] [PubMed] [Google Scholar]