

Summary
Activation of Toll-like receptor (TLR) signaling and related pathways by microbial products drives inflammatory responses, host defense pathways and adaptive immunity. The cost of excessive inflammation is cell and tissue damage, an underlying cause of many acute and chronic diseases. Coincident with activation of TLR signaling, a plethora of anti-inflammatory pathways and mechanisms begin to modulate inflammation until tissue repair is complete. Whereas most studies have focused on the signaling components immediately downstream of the TLRs, this review summarizes the different levels of anti-inflammatory pathways that have evolved to abate TLR signaling and how they are integrated to prevent cell and tissue destruction.
The inflammatory response must be constantly constrained to prevent molecular, cellular and organ damage. The consequences of unregulated inflammation are associated with, or directly underpin, a substantial fraction of diseases that plague us, including autoimmune and metabolic diseases, infectious diseases caused by large macroparasites to viruses, chronic neurological diseases, malignancy and life-threatening acute responses to pathogen products such as sepsis and shock. Correspondingly, a proportionate percentage of the modern pharmacopoeia is devoted to blocking inflammation, from widely used drugs such as aspirin and the non-steroidal anti-inflammatory medication to humanized anti-cytokine antibodies.
Given that the inflammatory response is essential to constrain infection, recruit and activate lymphocytes and then promote wound healing and repair, how are these processes regulated such that horror autotoxicus is mitigated and organ systems return to homeostasis? The integration of inflammatory inhibition and homeostasis is especially important in large animals that must live for decades to successfully reproduce and raise the next generation. It should not be surprising therefore that elaborate mechanisms to regulate inflammation have co-evolved with pro-inflammatory pathways, that non-resolving or chronic inflammation is linked to the chronic maladies of aging and that older organisms are especially sensitive to inflammatory perturbation1.
Before the discovery of ‘innate’ detection systems such as the Toll-like receptors (TLRs), Nod-like receptors (NLRs), C-type lectin receptors (CLRs) and the diverse nucleic acid detectors, 'negative' pathways had been recognized to mediate multiple layers of the inflammatory response. Subsequent to the notion that innate receptors have preeminent roles in pathogen detection and the initiation of inflammation, a massive literature has established additional layers of regulatory control over inflammation. In this overview, we cannot cover the primary literature on the fine details of each molecule attributed to have negative regulatory influence on inflammation. Instead, we first propose that modulation of inflammation involves inter-connected layers or strata that begins with the anatomy of mammals and extends to the precise control of the metabolic state of inflammatory cells. From these regulatory strata, we will focus on three interrelated pathways whose mechanistic details are emerging and represent new strategies to manipulate and interrupt excessive responses initiated by TLR signaling and related pathways that activate inflammation. As other recent reviews have covered aspects of inflammatory modulation including TLR signaling components and post-transcriptional pathways2–7 we will focus on the wider context of inflammatory regulation.
Discovery of anti-inflammatory pathways
Three types of investigation have uncovered negative regulators of inflammation. First, observation of unanticipated phenotypes in humans and mutant mice with inflammatory disease have been the starting point for the discovery of numerous key pathways including the interleukin 10 (IL-10) and TGF-β pathways8–10. Second, standard experimental models of inflammation including cecal ligation and puncture, endotoxin challenge, bleomycin-mediated lung injury, graft versus host disease, airway challenges of mice with allergens and TLR agonists and infection models that include acute and chronic inflammation-associated damage have led to the definition of many anti-inflammatory pathways. Third, in vitro systems to measure the TLR- and NLR-activated pathways on primary cultures of macrophages and dendritic cells from knockout and transgenic mice have been instrumental in pinpointing where a given regulatory factor fits into a signaling pathway. Coupled with complementary approaches such as forward genetics and biochemical reconstitution experiments, many anti-inflammatory factors been discovered and their activities defined to differing extents (Table 1). Nevertheless, caveats apply with these approaches that center on the model systems used and their context. First, many types of experiments in mice cannot be applied to humans. Murine models of inflammation can amplify the preeminence of pathways that are subject to compensatory or redundant effects in people. For example, MyD88-deficient humans have a narrow range of infection phenotypes even in adulthood, while MyD88-deficient mice have broad phenotypes consistent with MyD88’s key role in all TLR signaling pathways other than TLR311. These observations are best reconciled by considering that humans are not exposed to the same experimental pressure as would be found in a procedure such as cecal ligation and puncture12. Future studies on humans, mice and new model organisms such as the pig13,14, will likely refine our understanding of the existing anti-inflammatory pathways and uncover new regulatory layers.
Table 1.
Representative gene deletions that result in enhanced inflammation are shown in this table. Numerous other gene deletions have been linked to negative effects on inflammatory responses. Murine loss-of-function alleles were the primary criteria for inclusion.
| STRATA | REPRESENTATIVE EXAMPLES | EXAMPLE GENE DISRUPTION LEADS TO EXCESSIVE INFLAMMATION* |
|---|---|---|
|
1. Anatomical barriers and Inter-organ communication systems |
•Skin | |
| •Peristaltic flow of the intestines | ||
| •Vagus nerve-mediated inhibition of splenic macrophage activation35 |
||
| •Sentinel innate B cells that suppress endotoxoc shock33 | ||
| •Cytokine-mediated hematopoiesis | ||
|
2. Mucosal surfaces and innate wound responses |
•Mucous-mediated inhibition of TLR signaling | MUC1 (Muc1)32 |
| •Extracellular matrix pathways | CD44 (Cd44)95 | |
| •Complement activation | C5aR (C5ar1)96 | |
| •Coagulation cascade | Anti-thrombin III (Serpinc1)97 | |
|
3. PAMP sequestration and repair receptor modification |
•PAMP sequestration (soluble TLRs, PAMP binding factors) | sTLR2, sTLR45 |
| •Receptor competition | SIGIRR (Sigirr)98 | |
| •Signaling cross-talk | CD11b (Itgam)99 | |
| •Signal abatement at the membrane | RP105 (Cd180)100, DAP12 (Tyrobp)101 | |
| 4. PAMP signaling | •Sentinel inhibitors (can also be induced/regulated) | IRAK-M (Irak3)26, TAK1 (Map3k7)102, CYLD (Cyld)103 , Tollip (Tollip)104, TRAF3 (Traf3)105,106 |
| •Inhibitors (regulated) | A20 (Tnfaip3)27, ABIN1 (Tnip1)28,29,107, IκBα (Nfkbia)25 |
|
| •Phosphatases | DUSP1 (Dusp1)90,108,109 | |
| •Kinases | PI3Kp85 (Pik3rl)110,111, MSK1 (Msk1)112, MSK2 (Msk2)112 |
|
| 5. Transcription | Transcription factors | STAT3 (Stat3)76, IRF4 (Irf4)48, NFIL3 (Nfil3)87,88, ATF3 (Atf3)62 |
| Chromatin modification | BCL6 (Bcl6) | |
|
6. Post-transcriptional mRNA regulation |
mRNA destabilization | TTP (Zfp36)7,49, HuR (Elav11), AUF1 (Hnrnpd), TIA-1 (Tia1)113 |
| MicroRNAs | mIR-146a (Mir146a)50 | |
| Translation | ||
|
7. Processing and secretion of inflammatory modifiers |
||
|
8. Expression of decoy receptors and receptor antagonists |
IL-1 receptor modifiers IL-13 decoy receptor |
IL-1Ra (I11rn), IL-1 decoy receptor (Il1r2)114,115 |
| IL-13 decoy receptor (I113r2)93 | ||
| 9. Anti-inflammatory cytokines | Sentinel/homeostatis | TGF-β (Tgfb1)8 |
| Inducible | Type I IFNs (Ifnb)116 | |
| Both sentinel and inducible | IL-10 (Il10)9 | |
| 10. Metabolic regulators | Global homeostatic mechanisms | |
| Autophagy | Atg16L1 (Atg1611)117 |
A second caveat concerns the interpretation of links between molecules that have multiple functions and whose disruption causes excessive inflammation. Tracing the connections between pathways has proven difficult when the starting point is a whole animal experiment. An informative example is the whole organism disruption of SOCS1, an inducible inhibitor of the type I and II interferon (IFN) receptors15. Deletion of Socs1 leads to death a few weeks after birth, a phenotype that can be rescued by crossing to mice into Ifng−/− or Rag1−/− backgrounds. However, Socs1−/− Ifng−/− mice (as well as juvenile Socs1−/− mice) are extraordinarily sensitive to systemic endotoxin administration16,17. Several models have been proposed to account for this phenotype, including absence of regulation of TLR signaling by direct binding of SOCS1 to TIRAP, IRAK1 and NF-κB, and excessive IFN-α/IFN-β signaling in the Socs1−/− Ifng−/− mice16–20. Collectively these data illustrate that for a protein like SOCS1 that has multiple effects in multiple cell types and contexts, probing the inflammatory strata to assign a relative hierarchy of negative regulation is complex.
Inflammation has a clock
In vivo, inhibition of inflammatory pathways occurs across a time frame that extends from seconds to years in wound healing and tissue repair, or is continuously ongoing in chronic inflammation. For productive immunity to pathogens to emerge, the initial inflammatory insult needs to be sufficient to trigger a response beyond the homeostatic anti-inflammatory threshold21. For example, in the gut, IL-10 constitutively dampens TLR and NLR signaling from the gut flora to maintain normal intestinal functions. Infection with a pathogen like Shigella flexneri, that invades the mucosal layers, triggers a response that exceeds the homeostatic threshold21, and causes massive inflammation22. The signals to repair the gut likely begin once the infection is ‘controlled’ such that bacteria may still be present but are no longer proliferating. In the gut, repair mechanisms to restore the epithelia and mucosa must work very quickly as all animals need to acquire nutrients to survive. By contrast, the wound healing and tissue restoration process for bones, deep tissue injuries and muscle takes months to years to restore tissue strength. Regardless of the time frame of tissue repair, negative regulation of inflammation has to be continuously engaged. Thus, productive regulatory pathways are induced proportionally to the inflammatory insult and are themselves subject to additional layers of regulation23. An informative example of the latter is the production of IL-10 which is essential to inhibit inflammation at multiple layers, but can also promote an immune environment permissive for multiple pathogens24. Obviously, lack of engagement of counter regulation at the correct time and place underlies a plethora of inflammatory diseases touched upon here.
Cell intrinsic and extrinsic anti-inflammation strata
It is plausible to consider that for every pro-inflammatory pathway activated by the microbial and cell damage sensing systems, there are at least as many anti-inflammatory pathways. Anti-inflammatory pathways can be crudely divided into cell intrinsic and extrinsic mechanisms, many of which have been uncovered by unexpected outcomes of the detailed examination of genetically-modified mice that manifest inflammatory diseases. Examples of cell intrinsic pathways include co-regulated inhibitors of TLR signaling such as IκBα, IRAK-M, A20 and ABIN1 (Table 1). Deletion of each of these factors in mice leads to complex inflammatory diseases because of failure to attenuate inflammatory signal after it has been initiated by a microbial product or pro-inflammatory cytokine such as TNF 25–29. Cell extrinsic mechanisms include anti-inflammatory cytokines such as IL-10 and TGF-β, as well as a myriad of factors that function to block inflammation through the sequential ‘repair’ process outlined above. Another way to define anti-inflammatory pathways is to break down each step from inflammatory initiation to resolution. These strata constitute an integrated system to mitigate the negative effects of inflammation through the span of an inflammatory response. Table 1 summarizes these strata as a 'snapshot' of the breadth of inflammatory modulation and provides select examples of gene disruption experiments in the mouse that lead to excessive inflammation.
Strata 1 and 2: anatomy and mucosa
Many papers concerned with inflammation begin with a statement concerning the ‘first line’ functions of the innate immune response. However, the first line of defense against pathogens is mammalian anatomy (Stratum 1) and its associated mucosal system (Stratum 2). The lungs, gut and skin receive constant stimulation by commensals and pathogens30. However, it is rare that inflammatory responses are ever sufficiently troublesome to send someone to the doctor. Our barrier systems have evolved to mechanically repel or constrain microorganisms that could trigger inflammation. A key example is the mucous lining of the gut where both the viscosity and forward motion ensures that only a fraction of the gut flora encounters the underlying immune cell-rich mucosa. Similarly, the lungs are bathed in surfactant to restrain colonization by microorganisms; an effect complemented by the cough reflex to continuously propel mucus and debris from the lungs upward. The effects of disruption of the lung mucosal lining and failure of coughing are key elements of cystic fibrosis and bacterial overgrowth observed in the terminally ill. Similarly, defects in intestinal mucus production or flow are associated with dramatic inflammatory responses31,32. The gut, liver, skin, spleen and lungs are also endowed with sentinel immune cells programmed not to overreact to pathogens or their products30,33,34. The spleen is also the target of the neural reflex anti-inflammatory pathway35. Collectively, the anatomy of mammals contributes the bulk of 'defense' against pathogen challenge and initiation of inflammation.
Stratum 3: PAMP sequestration
Removal of microbial products including cell wall components before they ever interact with TLRs and NLRs is a key innate mode of inflammatory regulation. When considered with anatomical restriction of TLR ligand exposure via strata 1 and 2, sequestration of PAMPs operates continuously. The complexity of the pathways in strata 3 has been reviewed5. Two other ‘innate’ pathways that interface with anatomy, mucous membranes and the circulatory system are the complement and coagulation pathways. The effect of these two pathways on pro-and anti-inflammatory TLR signaling modulation is an emerging area of investigation, along with modulation of the extracellular matrix, and has been discussed in other reviews36–38.
Stratum 4: signaling down-regulation
A myriad of cytoplasmic proteins work together to regulate cell autonomous TLR signaling3,39. These proteins fall into diverse structural and functional groups including sentinel proteins that rapidly inhibit signaling (IRAK-M, A20, ABIN1, IκBα, Tollip, DAP12), proteins that are further induced at the gene expression level to reinforce signaling down-regulation (A20, IκBα, DUSP family phosphatases), kinases that mediate downstream inhibitory pathways (MSK1, MSK2) and signaling proteins that initiate the production of cytokines that act through autocrine and paracrine pathways to block signaling (TRAF3). Other proteins such as the alanine-rich myristoylated protein MARCKS as one example of many, have been discovered to inhibit TLR signaling through unknown mechanisms40. The integration of each of these proteins into greater signaling networks is central to each TLR-responsive cell's decision to terminate or perpetuate an inflammatory response.
Stratum 5: transcriptional regulation
After the integration of initial TLR signaling pathways in the cytoplasm, a large cohort of positively and negatively acting transcription factors control the thousands of genes regulated by TLR signaling41. Negative regulation of TLR-mediated transcription can be described by at least four phenomena: specific negative feedback mechanisms to suppress the activities of factors that activate inflammatory gene transcription, such as NF-κB42; acquired resistance to transcriptional activation following chronic exposure to stimuli such as LPS and TNF43, usually referred to as endotoxin tolerance44; gene-specific effects of steroid hormone receptors and their co-factors45,46, and the autocrine-paracrine effects of IL-10, which signals to suppress many TLR-activated genes47. The effects of these pathways are discussed in more depth below. In addition to these four mechanisms, constitutively expressed transcription factors and chromatin proteins have been demonstrated to have negative effects of TLR-regulated gene expression in macrophages, although the relationship between these factors and tolerance, steroid hormone repression, IL-10 signaling and other physiological processes that are related to inflammatory diseases and the resolution of normal inflammatory responses remains unclear48.
Stratum 6: Post-transcription and translation
Post-transcriptional regulation and translation of mRNAs encoding inflammatory mediators is essential for inflammatory control7. Multiple RNA binding proteins such as TTP and HuR, and miRNAs including miR-155 are involved in fine-tuning the post-transcription inflammatory response49–51. Although the identity of several components of the post-transcriptional signaling network has been uncovered, further work is needed to pinpoint how proteins and miRNAs with broad mRNA substrate specificity locate their targets, suppress transcription/translation, and are themselves regulated.
Stratum 7: Processing and secretion
Inflammatory stress is coupled to the endoplasmic reticulum (ER) stress response. The ER stress response inhibits the processing and secretion of many proteins, presumably as a means to conserve resources during stress. However, the ER stress response is regulated by TLR signaling such that many secreted inflammatory cytokines and chemokines are allowed to escape the ER52,53. This remarkable process is required for the overall inflammatory response, but how is it regulated? Conceivably, numerous anti-inflammatory pathways could converge on the ER to suppress the production of multiple pro-inflammatory mediators that so far remain unknown.
Strata 8,9: decoys, antagonists and hijacked cytokines
These inhibitory pathways are discussed in more detail below.
Stratum 10:metabolic regulation of inflammation
A final stratum for control of inflammation is the impact of metabolic states on immunity. The metabolic control of inflammation encompasses a large variety of linked processes including translation control, metabolic stress responses54, autophagy55, and forms of programmed cell death. Emerging information on metabolism and the immune response is discussed in depth by another review in this issue (Ref. to add- Green review, this issue).
An example of the linkages between the strata of inflammatory control is the production, secretion, and bioavailability of IL-1β, which is negatively regulated at the level of at least eight different check points (Fig. 1).
Figure 1.
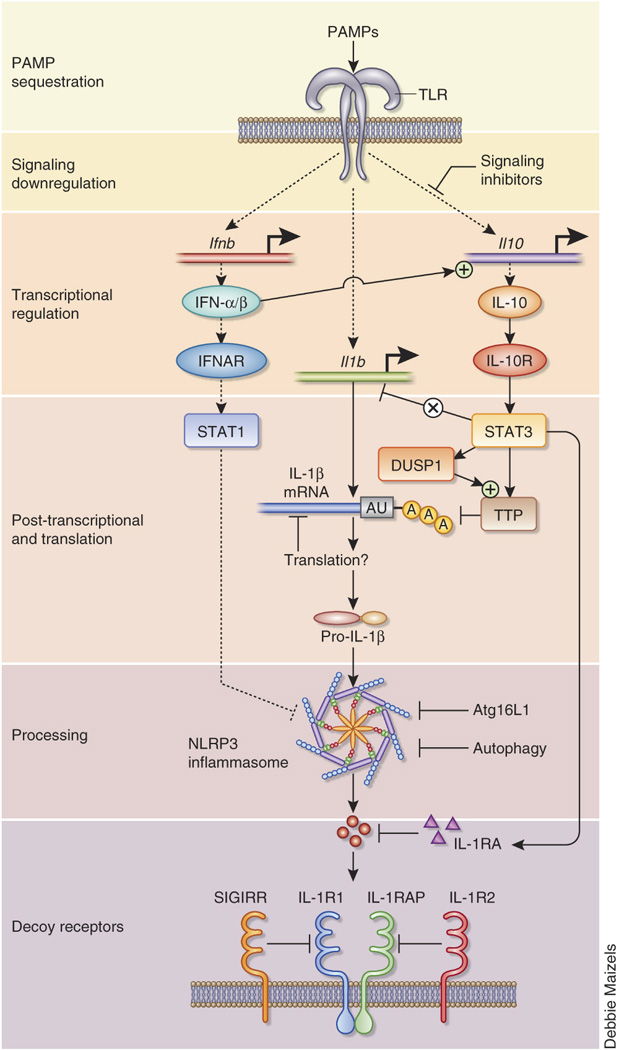
Negative regulation of IL-1β production, signaling and bioavailability as an example of multi-tiered anti-inflammatory integration. IL-1β is inhibited by at least eight interrelated mechanisms including the initial counter-regulation of TLR signaling, sustained TTP activity via DUSP1, transcription by IL-10, mRNA processing by TTP and related mRNA binding proteins (regulated by IL-10), the type I IFN-mediated inhibition of the NLRP3 inflammasome116, autophagy regulation by Atg16L1, autophagy-mediated destruction of inflammasomes118 and then the signaling and bioavailability of IL-1β itself. Several levels of IL-1β regulation are discussed in detail in the text.
Transcriptional repression of inflammation
As mentioned above, studies of inflammatory gene transcription have uncovered a number of transcription factors, chromatin proteins and other transcription-related mechanisms that contribute to the suppression of inflammation. One notable example is the rapid transcriptional activation of the gene encoding IkBα by most or all inflammatory stimuli. IκBα induction leads to the suppression of NF-κB activity, thereby attenuating the transcription of NF-κB-dependent genes unless the stimulus is capable of circumventing the suppression40. The variable consequences of this suppression mechanism are apparent from a comparison of macrophages stimulated with TNF and LPS56,57. The initial response to TNF is transient, due to the upregulation of IκBα, the rapid internalization of the TNF receptor and other feedback inhibitory mechanisms. In contrast, LPS stimulates two distinct NF-κB-inducing pathways with different kinetics, the MyD88 and TRIF pathways, thereby allowing sustained NF-κB activation and a transcriptional cascade that differs substantially from that activated by TNF, despite the upregulation of IκBα by both stimuli. Several other mechanisms that have the potential to suppress NF-κB activity have been described and have been summarized in recent reviews40. One challenge in the study of these suppression mechanisms is that their precise physiological relevance during the course of a normal inflammatory response and during abnormal responses associated with disease have been difficult to uncover and therefore remain poorly understood.
In addition to the various mechanisms involved in the broad suppression of NF-κB activity, the transcription of specific sets of inflammatory genes is limited by several other proteins and protein complexes. Two examples are the Mi-2-NuRD complex and Bcl-6, both of which are constitutively expressed in macrophages. Mi-2-NuRD is a multiprotein complex containing histone deacetylase and ATP-dependent nucleosome remodeling activities and has been primarily implicated in transcriptional repression58. Deletion of this complex in mouse macrophages leads to greatly enhanced expression of a subset of LPS-induced genes in a stimulus-dependent manner59. The genes that were sensitive to Mi-2-NuRD knockdown correspond to those that require nucleosome remodeling by another family of ATP-dependent nucleosome remodeling complexes, the SWI/SNF family, for their transcriptional activation. Many of these remodeling-dependent genes play critical roles in regulating inflammatory and adaptive immunity, such as Il12b, Il6 and Nos2.
Bcl-6 appears to be a similarly potent suppressor of a subset of inflammatory gene, as many LPS-induced genes were found to be activated at greatly enhanced levels in macrophages from Bcl-6-deficient mice60. Bcl-6 directly binds control regions for a large percentage of the affected genes, suggesting that it may suppress transcription of these genes by directly competing with transcriptional activators, possibly leading a repressive chromatin environment. One possibility that has not yet been explored is that Bcl-6 may recruit Mi-2-NuRD to target genes to limit inducible transcription. A careful comparison of the sets of genes suppressed by the two factors may provide insight into this possibility, as regulation of a common set of genes would suggest that the two factors act in concert. Careful delineation of the gene suppressed by these proteins, preferably by RNA sequencing, may also provide clues into the biological reason for the existence of these suppression mechanisms. Several other transcription factors, including IRF4 and ATF3, have also been implicated in the repression of inducible gene transcription48,61,62. Although it has been possible to document the importance of these repression mechanisms in the context of mutant mice, the manner in which they are integrated into a normal inflammatory response is unknown and it is not known whether they directly participate in pathways that promote disease.
As discussed above, several factors and mechanisms capable of suppressing or limiting inflammatory gene transcription have been described, but their contributions to normal and abnormal inflammatory responses remain to be elucidated. With this in mind, it is interesting to consider an independent line of investigation that originated with a biological observation that is likely to be of considerable importance, but for which the underlying mechanisms have remained incompletely understood for many years. Specifically, it has long been known that exposure to a potent inflammatory stimulus can lead to acquired resistance to inflammatory gene induction upon subsequent stimulation. This observation was first made with LPS as the stimulus and is referred to as LPS or endotoxin tolerance. However, TNF has similarly been shown to induce tolerance43.
Multiple molecular mechanisms appear to contribute to tolerance, ranging from mechanisms to suppress the transduction of an inflammatory signal to active repression of inflammatory genes through the assembly of repressive chromatin structures63–65. The existence of multiple mechanisms has made it difficult to determine the relative importance of each mechanism that has been described. A few notable studies have provided compelling evidence that changes in chromatin structure contribute to stable suppression of inducible transcription43,64,66,67. Repressive histone modifications and chromatin changes that may prevent remodeling by ATP-dependent nucleosome remodeling complexes have been suggested to contribute to resistance to transcriptional activation. Interestingly, only a subset of inducible genes was found to be susceptible to LPS tolerance, with tolerance observed at some genes that contribute to inflammation but not at genes that contribute to anti-microbial immunity44.
An attractive hypothesis is that the two groups of genes may exhibit distinguishing chromatin characteristics that confer resistance or sensitivity to tolerance. However, initial efforts to identify these distinguishing characteristics have been unsuccessful, as both sensitive and resistant genes were included within a group of genes found to be dependent on nucleosome remodeling for their activation44,59,68. Genes that are sensitive and resistant to tolerance induction were also found in a class of nucleosome remodeling-independent genes. The two classes of genes also cannot be distinguished on the basis of common histone modifications in unstimulated and stimulated cells. Therefore, much remains to be learned about LPS tolerance and the precise mechanisms by which chromatin structure and other events, including signal transduction, contribute to this process.
IL-10 signaling integrates multiple regulatory strata
Genetics teaches that IL-10 is the central anti-inflammatory cytokine that impinges on multiple anti-inflammatory strata. The effects of germline deletion in IL-10- or the IL-10 receptor-encoding genes produce extreme and often lethal inflammatory syndromes in both humans and mice9,69. In mice housed in normal or SPF conditions, the effects of IL-10 disruption are first observed in the gut, as noted above. By contrast, germ-free IL-10-deficient mice do not have colitis70, arguing that the intestinal flora drives the excessive inflammatory response. Furthermore, mice lacking MyD88 and IL-10 do not have colitis, providing conclusive evidence that excessive TLR and IL-1 receptor (IL-1R) signaling in the intestine must be continuously suppressed by IL-1071,72. Indeed, depending on the stimulus or infection, pathogenic inflammatory responses are observed in most models of acute and chronic inflammation in IL-10-deficient mice73. However, anti-inflammatory effects of IL-10 come at a cost because IL-10 also inhibits productive inflammatory responses against intracellular pathogens, especially Mycobacteria and Leishmania74,75. Thus, the IL-10 anti-inflammatory signal is a trade-off between deleterious and productive inflammatory responses. Another evolutionary curiosity of IL-10 concerns its non-redundant nature in mammals: if IL-10 is so important, why don’t we have multiple IL-10-like cytokines? While the answers to this question are speculative, one possibility is that only one IL-10-IL-10R system is required and if it does not function properly then early lethality from excessive inflammation is likely, removing the mutation from the gene pool (similarly, some other essential cytokines like EPO and G-CSF may have the same properties).
IL-10 signaling is dependent on STAT347,76 (Fig. 2). The use of STAT3 raises another problematic aspect of deciphering how IL-10 suppresses TLR transcription because STAT3 is activated by numerous cytokines77,78. In IL-10-responsive myeloid cells, IL-6 is also a potent activator of STAT3, yet IL-6 mediates none of the suppressive effects of IL-10. The underlying mechanism involved in this dichotomy is mediated by SOCS3 inhibition of the IL-10 ‘signal’ from the IL-6R, while the IL-10R does not bind SOCS379. Furthermore, any cytokine receptor can be engineered into an ‘IL-10R’ by ensuring STAT3 activation in the absence of SOCS3 inhibition80. Thus, the IL-10-mediated anti-inflammatory response is ‘generic’ in that it depends on a specific way of activating STAT3, independent of the receptor. The underlying mechanisms involved in the ‘IL-10 type’ of STAT3 activation in comparison to other STAT3-activating receptors remain unknown.
Figure 2.
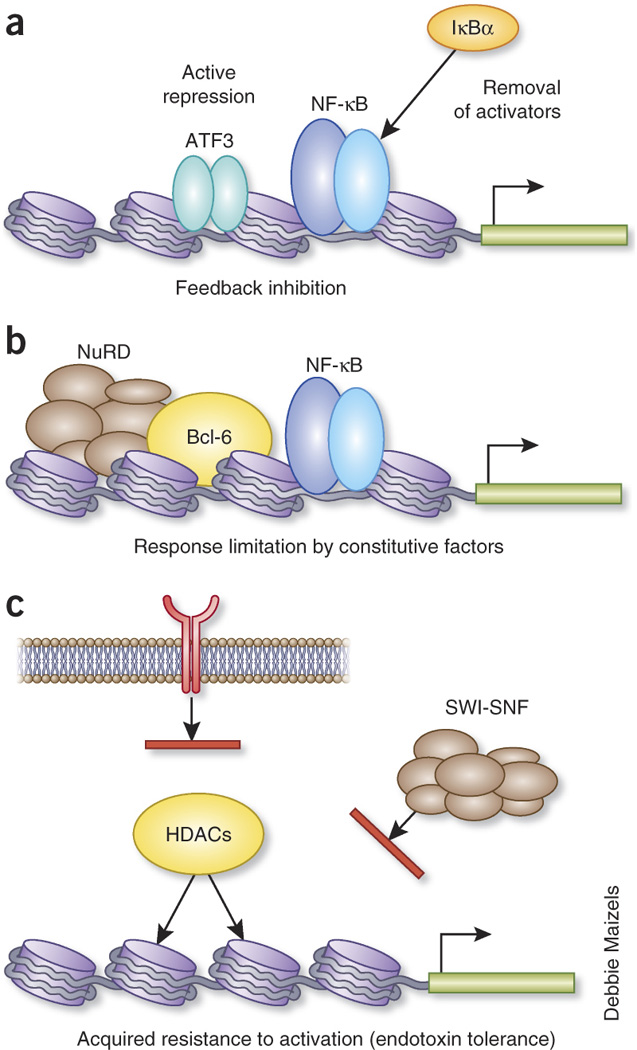
Three fundamental levels for transcriptional suppression of inflammation (Stratum 5). (a) Transcription factors induced during the inflammatory response, such as ATF-3 and IκBα, modulate feedback inhibition on the inducing gene. (b) Constitutively expressed factors, like Bcl-6 and NuRD, have sentinel effects on selected genes and limitation the transcriptional response. (c) Resistance to activation is acquired by suppressing TLR signaling or through creation of a repressive chromatin structure that may involve histone deacetylation and blockade of SWI/SNF-mediated activation. Secreted factors, such as IL-10, can promote transcriptional inhibition in different time frames: induced feedback mechanism (a) or a constitutively acting mechanism that limits the potency of activation (b), i.e. in the intestines, depending on context.
The effects of IL-10 (like LPS tolerance and steroid hormone inhibition) on transcription are gene specific: numerous TLR-regulated genes are unaffected by IL-10 (Nfkbia, Tnfaip3), while others show varying degrees of inhibition from complete (Il12b) to partial (Tnf)47. The underlying mechanisms involved in the selection of genes for inhibition remain unknown81,82. Two mechanisms are possible: a single master regulatory factor could mediate transcriptional repression or multiple factors could work together (Fig. 3). In the case of the former, no unique IL-10-regulated factor has been discovered that would be epistatic to STAT347,83,84. Instead, it seems likely that multiple factors suppress gene expression in a gene-specific way. To date, the best understood of these is NFIL3, a B-ZIP factor induced by IL-10 that regulates Il12b (encoding IL-12p40, the common subunit of IL-12 and IL-23) by binding to a distal enhancer ~10 kb upstream of the Il12b promoter85–87. NFIL3-deficient macrophages overproduce IL-12p40 and IL-12p70 in response to TLR stimulation87,88. However, while NFIL3 is necessary to regulate Il12b transcription, it is not sufficient because IL-10 retains residual inhibitory effects on Il12b transcription in Nfil3−/− macrophages87,88. Therefore, additional factors induced by IL-10 that are associated with transcription including Bcl-3, Sbno2, Etv3 and IkBNS may work together to suppress TLR-induced genes, along with factors that possibly regulate elongation on actively transcribed TLR-regulated genes82,87.
Figure 3.
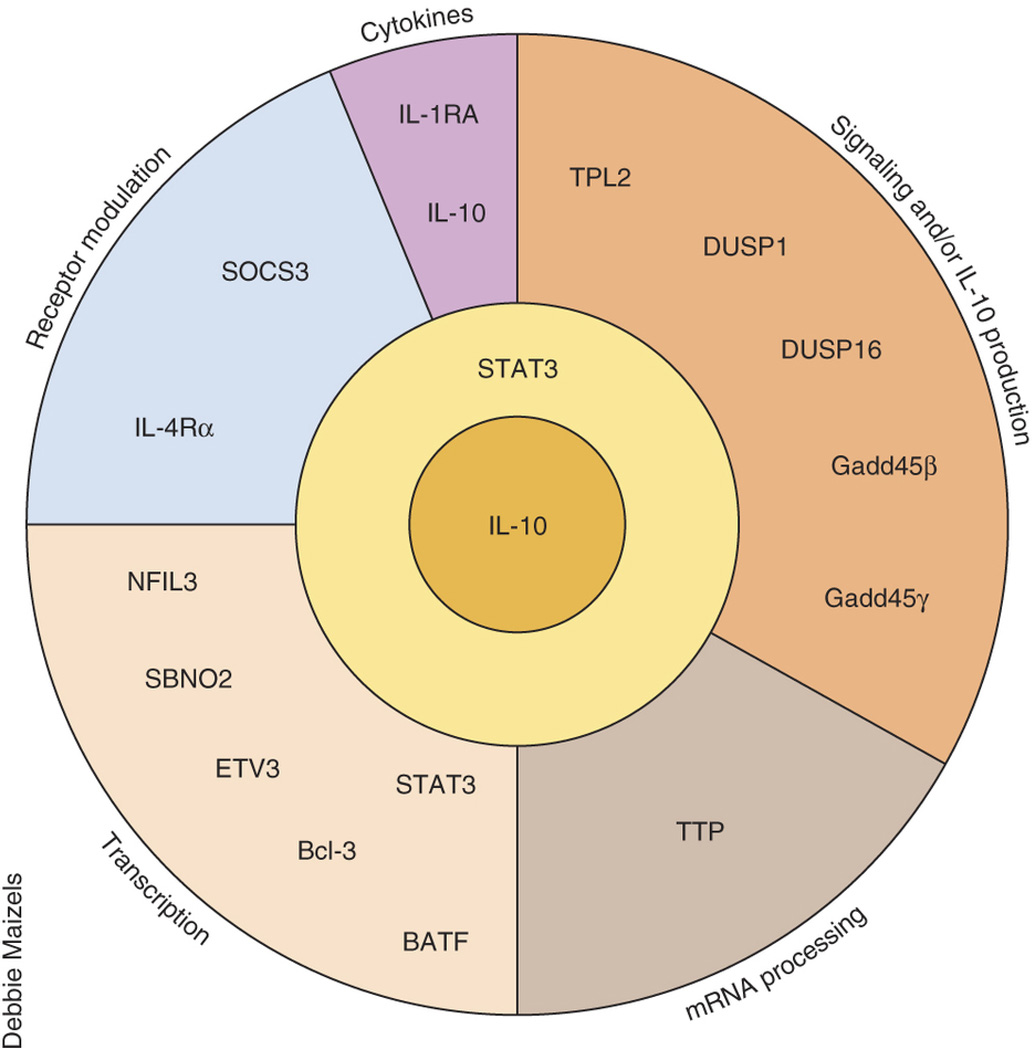
IL-10 regulates the production of downstream factors that control multiple strata of inflammation. Shown are a subset of known factors induced by IL-10 in a STAT3-dependent way and their known or speculated effects on inflammation. IL-10, via STAT3 also induces further IL-10 production in a self-reinforcing loop. The production of IL-10 by myeloid cells has been described in detail by Saraiva and O'Garra24, from which this diagram was inspired.
Although the majority of the inhibitory effects of IL-10 are focused on the level of transcriptional control, IL-10 also induces additional modifiers of inflammatory signaling that operate at other strata. For example, IL-10, via STAT3, increases the TLR-mediated expression of tristatraprolin (TTP, encoded by Zfp36) to enhance degradation of AU-rich 3' UTR target mRNAs targeted for degradation by TTP89. In the same time frame, IL-10 synergistically induces DUSP1, which can dephosphorylate p38 MAPK90. As p38-mediated phosphorylation of TTP is inactivating, DUSP1 maintains TTP activity, further enhancing the effects of IL-10 on mRNA stability (Fig. 1). Another target of IL-10 is the gene encoding the IL-1R antagonist, whose expression is highly induced by IL-10. Therefore, IL-10 regulates IL-1 signaling at the transcriptional, post-transcriptional and receptor levels in concert with other inhibitory processes (Fig. 1). A final example of the dichotomy of anti-inflammatory effects of IL-10 is the regulation of LPS-induced miRNAs. IL-10 is a potent transcription inhibitor of the pro-inflammatory miR-155 but not of miR-14691, which has cell-intrinsic anti-inflammatory effects50. How the IL-10 signaling pathway makes this discrimination remains unknown.
Decoys, antagonists and hijacked cytokines
Post-production removal of TLR-induced pro-inflammatory cytokines and chemokines coupled with receptor antagonism are key ‘downstream’ anti-inflammatory processes that are often overlooked in deciphering inflammatory control in vivo 92. Post-production removal of cytokines have been harnessed for successful therapies: soluble TNF receptors for rheumatoid arthritis and inflammatory bowel disease (etanercept), and the IL-1R antagonist for treatment of inflammatory cryopathies (anakinra). Soluble pro-inflammatory mediators are removed or inhibited by distinct mechanisms. For example, membrane-bound and soluble cytokine and chemokine receptors act as ‘sinks’ on a variety of different cell types to soak up pro-inflammatory mediators. Another type of inhibitory mechanism involves the active production of decoy receptors that inhibit a select group of cytokines, including IL-1, IL-13 and IL-22. Decoy receptors are non-signaling receptors that have an equal or higher affinity for their ligand than the signaling receptor. For example, the IL-13Rα2 decoy receptor binds IL-13 and has an essential role in blocking IL-13 and TH2-mediated inflammation93. A final type of inhibition involves the production of cytokine mimics that act as receptor antagonists. The best characterized example of inhibition of IL-1R signaling by the IL-1R antagonist encoded by Il1ra. The IL-1R antagonist competes with IL-1α and IL-1β for binding to the IL-1R, blocking signaling94. Why do so few cytokines have decoy receptors? While it is conceivable that IL-1, IL-13 and IL-22 have a high potential for inflammatory tissue destruction, other cytokines with known connections to pathogenic inflammation, including IFN-γ, IL-12 and IL-23, lack decoy receptors. Therefore, the decoy receptor system for IL-1, IL-13 and IL-22 likely has a fine-tuning role in inflammation that could be exploited therapeutically for other cytokines. A similar selectivity problem exists when considering the hijacking of cytokines by viruses. IL-10 has been hijacked at least three times (by Epstein-Barr virus, cytomegalovirus and Orf poxvirus) and IL-6 once (Kaposi's sarcoma-associated herpes virus. Yet most viruses that can fit additional genes into their genomes lack virokines, even though these molecules have powerful effects on host immune modulation. Like the cytokine decoy receptors, opportunities exist to engineer new drugs that can modulate acute and chronic inflammation with low toxicity (due to the selectivity for a given receptor).
Perspectives
In this brief overview we have attempted to emphasize that the pathways that restrain inflammation operate at many levels, and over broad time frames. Constitutive and inducible inflammation is regulated by a multitude of cell intrinsic and extrinsic mechanisms that are themselves regulated. Given that a large fraction of clinical medicine and health is concerned with inflammatory diseases, and that many of the most successful drugs target inflammation, it seems likely that new opportunities for disease mitigation can be developed by observing how the body naturally regulates inflammation. To achieve this goal, better tools and techniques are necessary to understand complex signaling pathways. Cell-specific deletions will be also required to assess molecular function in whole animal models of acute and chronic inflammation and these will need to be coupled to more sophisticated and realistic mouse models of inflammation, which will be essential for translation studies to humans. Finally comparative studies between animal models and human tissue samples and ex vivo primary cell cultures will be essential to pinpoint the key features inflammatory control relevant to humans.
Acknowledgments
We thank F. Kratochvill, P. Ward and G. Hajishengallis for informative comments of specific negative regulatory pathways. This work was supported by NIH grants R01 AI073868, R01 CA127279, R01 GM086372, CORE grant P30 CA21765, The Hartwell Foundation, and The American Lebanese Syrian Associated Charities.
References
- 1.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 2.Bezbradica JS, Medzhitov R. Integration of cytokine and heterologous receptor signaling pathways. Nat Immunol. 2009;10:333–339. doi: 10.1038/ni.1713. [DOI] [PubMed] [Google Scholar]
- 3.Ivashkiv LB. Cross-regulation of signaling by ITAM-associated receptors. Nat Immunol. 2009;10:340–347. doi: 10.1038/ni.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol. 2012;12:168–179. doi: 10.1038/nri3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 6.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 7.Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol. 2010;10:24–35. doi: 10.1038/nri2685. [DOI] [PubMed] [Google Scholar]
- 8.Christ M, et al. Immune dysregulation in TGF-beta 1-deficient mice. J Immunol. 1994;153:1936–1946. [PubMed] [Google Scholar]
- 9.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 10.Shull MM, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casanova JL, Abel L, Quintana-Murci L. Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu Rev Immunol. 2011;29:447–491. doi: 10.1146/annurev-immunol-030409-101335. [DOI] [PubMed] [Google Scholar]
- 12.Alcais A, Abel L, Casanova JL. Human genetics of infectious diseases: between proof of principle and paradigm. J Clin Invest. 2009;119:2506–2514. doi: 10.1172/JCI38111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapetanovic R, et al. Pig bone marrow-derived macrophages resemble human macrophages in their response to bacterial lipopolysaccharide. J Immunol. 2012;188:3382–3394. doi: 10.4049/jimmunol.1102649. [DOI] [PubMed] [Google Scholar]
- 14.Schroder K, et al. Conservation and divergence in Toll-like receptor 4-regulated gene expression in primary human versus mouse macrophages. Proc Natl Acad Sci U S A. 2012;109:E944–E953. doi: 10.1073/pnas.1110156109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- 16.Kinjyo I, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawa R, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 18.Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. 2004;279:54708–54715. doi: 10.1074/jbc.M410992200. [DOI] [PubMed] [Google Scholar]
- 19.Gingras S, Parganas E, de Pauw A, Ihle JN, Murray PJ. Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J Biol Chem. 2004;279:54702–54707. doi: 10.1074/jbc.M411043200. [DOI] [PubMed] [Google Scholar]
- 20.Mansell A, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 21.Blander JM, Sander LE. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol. 2012;12:215–225. doi: 10.1038/nri3167. [DOI] [PubMed] [Google Scholar]
- 22.Marteyn B, Gazi A, Sansonetti P. Shigella: A model of virulence regulation in vivo. Gut Microbes. 2012;3 doi: 10.4161/gmic.19325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev. 2008;226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 25.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi K, et al. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 27.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oshima S, et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature. 2009;457:906–909. doi: 10.1038/nature07575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou J, et al. A20-binding inhibitor of NF-kappaB (ABIN1) controls Toll-like receptor-mediated CCAAT/enhancer-binding protein beta activation and protects from inflammatory disease. Proc Natl Acad Sci U S A. 2011;108:E998–E1006. doi: 10.1073/pnas.1106232108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McAuley JL, et al. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J Clin Invest. 2007;117:2313–2324. doi: 10.1172/JCI26705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ueno K, et al. MUC1 mucin is a negative regulator of toll-like receptor signaling. Am J Respir Cell Mol Biol. 2008;38:263–268. doi: 10.1165/rcmb.2007-0336RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rauch PJ, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335:597–601. doi: 10.1126/science.1215173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. 2010;10:753–766. doi: 10.1038/nri2858. [DOI] [PubMed] [Google Scholar]
- 35.Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol. 2012;30:313–335. doi: 10.1146/annurev-immunol-020711-075015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. 2010;31:154–163. doi: 10.1016/j.it.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oikonomopoulou K, Ricklin D, Ward PA, Lambris JD. Interactions between coagulation and complement--their role in inflammation. Semin Immunopathol. 2012;34:151–165. doi: 10.1007/s00281-011-0280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 40.Mancek-Keber M, et al. MARCKS as a Negative Regulator of Lipopolysaccharide Signaling. J Immunol. 2012;188:3893–3902. doi: 10.4049/jimmunol.1003605. [DOI] [PubMed] [Google Scholar]
- 41.Smale ST. Selective transcription in response to an inflammatory stimulus. Cell. 2010;140:833–844. doi: 10.1016/j.cell.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruland J. Return to homeostasis: downregulation of NF-kappaB responses. Nat Immunol. 2011;12:709–714. doi: 10.1038/ni.2055. [DOI] [PubMed] [Google Scholar]
- 43.Park SH, Park-Min KH, Chen J, Hu X, Ivashkiv LB. Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat Immunol. 2011;12:607–615. doi: 10.1038/ni.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 45.Chinenov Y, et al. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1206059109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogawa S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 48.Negishi H, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A. 2005;102:15989–15994. doi: 10.1073/pnas.0508327102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kratochvill F, et al. Tristetraprolin-driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Mol Syst Biol. 2011;7:560. doi: 10.1038/msb.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- 51.Yiakouvaki A, et al. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J Clin Invest. 2012;122:48–61. doi: 10.1172/JCI45021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinon F, Glimcher LH. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr Opin Immunol. 2011;23:35–40. doi: 10.1016/j.coi.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woo CW, et al. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat Cell Biol. 2009;11:1473–1480. doi: 10.1038/ncb1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krawczyk CM, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Into T, Inomata M, Takayama E, Takigawa T. Autophagy in regulation of Toll-like receptor signaling. Cell Signal. 2012;24:1150–1162. doi: 10.1016/j.cellsig.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 56.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 57.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 58.Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26:5433–5438. doi: 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- 59.Ramirez-Carrozzi VR, et al. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006;20:282–296. doi: 10.1101/gad.1383206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barish GD, et al. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010;24:2760–2765. doi: 10.1101/gad.1998010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gilchrist M, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 62.Whitmore MM, et al. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol. 2007;179:3622–3630. doi: 10.4049/jimmunol.179.6.3622. [DOI] [PubMed] [Google Scholar]
- 63.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 64.Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clin Immunol. 2009;130:7–15. doi: 10.1016/j.clim.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ivashkiv LB. Inflammatory signaling in macrophages: transitions from acute to tolerant and alternative activation states. Eur J Immunol. 2011;41:2477–2481. doi: 10.1002/eji.201141783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J, Ivashkiv LB. IFN-gamma abrogates endotoxin tolerance by facilitating Toll-like receptor-induced chromatin remodeling. Proc Natl Acad Sci U S A. 2010;107:19438–19443. doi: 10.1073/pnas.1007816107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem. 2011;286:9856–9864. doi: 10.1074/jbc.M110.196790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramirez-Carrozzi VR, et al. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 2009;138:114–128. doi: 10.1016/j.cell.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Glocker EO, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sellon RK, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006;25:319–329. doi: 10.1016/j.immuni.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 72.Biswas A, et al. Negative regulation of Toll-like receptor signaling plays an essential role in homeostasis of the intestine. Eur J Immunol. 2011;41:182–194. doi: 10.1002/eji.201040479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cyktor JC, Turner J. Interleukin-10 and immunity against prokaryotic and eukaryotic intracellular pathogens. Infect Immun. 2011;79:2964–2973. doi: 10.1128/IAI.00047-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4 + CD25 + regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 75.Redford PS, Murray PJ, O'Garra A. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol. 2011;4:261–270. doi: 10.1038/mi.2011.7. [DOI] [PubMed] [Google Scholar]
- 76.Takeda K, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 77.Delgoffe GM, Murray PJ, Vignali DA. Interpreting mixed signals: the cell's cytokine conundrum. Curr Opin Immunol. 2011;23:632–638. doi: 10.1016/j.coi.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O'Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 80.El Kasmi KC, et al. General nature of the STAT3-activated anti-inflammatory response. J Immunol. 2006;177:7880–7888. doi: 10.4049/jimmunol.177.11.7880. [DOI] [PubMed] [Google Scholar]
- 81.Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci U S A. 2005;102:8686–8691. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smallie T, et al. IL-10 inhibits transcription elongation of the human TNF gene in primary macrophages. J Exp Med. 2010;207:2081–2088. doi: 10.1084/jem.20100414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.El Kasmi KC, et al. Cutting edge: A transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J Immunol. 2007;179:7215–7219. doi: 10.4049/jimmunol.179.11.7215. [DOI] [PubMed] [Google Scholar]
- 84.Hutchins AP, Poulain S, Miranda-Saavedra D. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012;119:e110–e119. doi: 10.1182/blood-2011-09-381483. [DOI] [PubMed] [Google Scholar]
- 85.Zhou L, Nazarian AA, Smale ST. Interleukin-10 inhibits interleukin-12 p40 gene transcription by targeting a late event in the activation pathway. Mol Cell Biol. 2004;24:2385–2396. doi: 10.1128/MCB.24.6.2385-2396.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou L, et al. An inducible enhancer required for Il12b promoter activity in an insulated chromatin environment. Mol Cell Biol. 2007;27:2698–2712. doi: 10.1128/MCB.00788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smith AM, et al. A distal enhancer in Il12b is the target of transcriptional repression by the STAT3 pathway and requires the basic leucine zipper (B-ZIP) protein NFIL3. J Biol Chem. 2011;286:23582–23590. doi: 10.1074/jbc.M111.249235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kobayashi T, et al. NFIL3 is a regulator of IL-12 p40 in macrophages and mucosal immunity. J Immunol. 2011;186:4649–4655. doi: 10.4049/jimmunol.1003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schaljo B, et al. Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J Immunol. 2009;183:1197–1206. doi: 10.4049/jimmunol.0803883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hammer M, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med. 2006;203:15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McCoy CE, et al. IL-10 inhibits miR-155 induction by toll-like receptors. J Biol Chem. 2010;285:20492–20498. doi: 10.1074/jbc.M110.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dower SK. Cytokines, virokines and the evolution of immunity. Nat Immunol. 2000;1:367–368. doi: 10.1038/80799. [DOI] [PubMed] [Google Scholar]
- 93.Wilson MS, et al. IL-13Ralpha2 and IL-10 coordinately suppress airway inflammation, airway-hyperreactivity, and fibrosis in mice. J Clin Invest. 2007;117:2941–2951. doi: 10.1172/JCI31546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- 95.Liang J, et al. CD44 is a negative regulator of acute pulmonary inflammation and lipopolysaccharide-TLR signaling in mouse macrophages. J Immunol. 2007;178:2469–2475. doi: 10.4049/jimmunol.178.4.2469. [DOI] [PubMed] [Google Scholar]
- 96.Liang S, et al. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. 2011;186:869–877. doi: 10.4049/jimmunol.1003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yanada M, et al. Impact of antithrombin deficiency in thrombogenesis: lipopolysaccharide and stress-induced thrombus formation in heterozygous antithrombin-deficient mice. Blood. 2002;99:2455–2458. doi: 10.1182/blood.v99.7.2455. [DOI] [PubMed] [Google Scholar]
- 98.Wald D, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–927. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 99.Han C, et al. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. 2010;11:734–742. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- 100.Divanovic S, et al. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hamerman JA, Tchao NK, Lowell CA, Lanier LL. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol. 2005;6:579–586. doi: 10.1038/ni1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ajibade AA, et al. TAK1 negatively regulates NF-kappaB and p38 MAP kinase activation in Gr-1 + CD11b + neutrophils. Immunity. 2012;36:43–54. doi: 10.1016/j.immuni.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Courtois G. Tumor suppressor CYLD: negative regulation of NF-kappaB signaling and more. Cell Mol Life Sci. 2008;65:1123–1132. doi: 10.1007/s00018-007-7465-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277:7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 105.Hacker H, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 106.Oganesyan G, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 107.Ashida H, et al. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat Cell Biol. 2010;12:66–73. doi: 10.1038/ncb2006. sup pp 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhao Q, et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203:131–140. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chi H, et al. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci U S A. 2006;103:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Conley ME, et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85alpha subunit of PI3K. J Exp Med. 2012;209:463–470. doi: 10.1084/jem.20112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fukao T, et al. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3:875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 112.Ananieva O, et al. The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat Immunol. 2008;9:1028–1036. doi: 10.1038/ni.1644. [DOI] [PubMed] [Google Scholar]
- 113.Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc Natl Acad Sci U S A. 2004;101:2011–2016. doi: 10.1073/pnas.0400148101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Colotta F, et al. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science. 1993;261:472–475. doi: 10.1126/science.8332913. [DOI] [PubMed] [Google Scholar]
- 115.Colotta F, et al. Regulated expression and release of the IL-1 decoy receptor in human mononuclear phagocytes. J Immunol. 1996;156:2534–2541. [PubMed] [Google Scholar]
- 116.Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 118.Shi CS, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]