

Abstract
Glucocorticoids (GCs) exert key metabolic influences on skeletal muscle. GCs increase protein degradation and decrease protein synthesis. The released amino acids are mobilized from skeletal muscle to liver, where they serve as substrates for hepatic gluconeogenesis. This metabolic response is critical for mammals’ survival under stressful conditions, such as fasting and starvation. GCs suppress insulin-stimulated glucose uptake and utilization and glycogen synthesis, and play a permissive role for catecholamine-induced glycogenolysis, thus preserving the level of circulating glucose, the major energy source for the brain. However, chronic or excess exposure of GCs can induce muscle atrophy and insulin resistance. GCs convey their signal mainly through the intracellular glucocorticoid receptor (GR). While GR can act through different mechanisms, one of its major actions is to regulate the transcription of its primary target genes through genomic glucocorticoid response elements (GREs) by directly binding to DNA or tethering onto other DNA-binding transcription factors. These GR primary targets trigger physiological and pathological responses of GCs. Much progress has been made to understand how GCs regulate protein and glucose metabolism. In this review, we will discuss how GR primary target genes confer metabolic functions of GCs, and the mechanisms governing the transcriptional regulation of these targets. Comprehending these processes not only contributes to the fundamental understanding of mammalian physiology, but also will provide invaluable insight for improved GC therapeutics.
Keywords: Glucocorticoids, Glucocorticoid receptor, Insulin, Skeletal muscle, Glucose metabolism, Protein metabolism
1. Introduction
Glucocorticoids (GCs) are steroid hormones secreted from the adrenal cortex to regulate whole-body metabolic homeostasis. The three major endogenous GC hormones are cortisol (hydrocortisone), corticosterone, and cortisone. In humans, the principal endogenous GC is cortisol, while in rodents it is corticosterone. Cortisone is inactive until converted to cortisol or corticosterone by 11β hydroxysteroid dehydrogenase type 1 (11β-HSD1) (Seckl and Walker, 2001; Tomlinson et al., 2004). On the other hand, 11β-HSD type 2 (11β-HSD2) converts active cortisol to inactive cortisone (Krozowski et al., 1999). GCs convey their signals mainly through an intracellular glucocorticoid receptor (GR). Cortisol has equal binding affinity for the mineralocorticoid receptor (MR) and GR; therefore, aldosterone-selective tissues, such as kidney, express 11β-HSD2 to inactivate cortisol. Thus, the levels of 11β-HSD1 and 11β-HSD2 in tissues help determine the tissue concentrations of active GCs, thereby modulating these effects. Notably, in some tissues, such as hippocampus and macrophages, GCs act via both GR and MR under normal physiological conditions, in which MR significantly influences cellular responses to GCs.
GCs exert specific metabolic influence on different tissues. Skeletal muscle accounts for approximately 40% of body mass and is a major GC target tissue. Based on myosin heavy-chain isoform expression profile, skeletal muscle fibers are classified into type I, type IIa, type IIx and type IIb. Type I myofibers are also known as slow-twitch fibers, and type II, fast-twitch fibers. Slow-twitch myofibers are slow to fatigue, are rich in mitochondria, and have long contraction times. Fast-twitch myofibers fatigue rapidly and display quick contractions. Type I muscles appear red in color due to the presence of oxygen-binding protein, myoglobin, while type II appear pale. On the basis of the degree of oxidative phosphorylation, type I and IIa fibers exert oxidative metabolism, while type IIx and IIb mainly use glycolytic metabolism to generate ATP. Interestingly, GCs appear to impact type II muscle fibers much more than type I (Dekhuijzen et al., 1995; Falduto et al., 1990; Fournier et al., 2003), though the mechanism of such fiber-specific GC response is unclear.
In skeletal muscle, GCs mainly regulate protein and glucose metabolism (Fig. 1). Under stressful conditions, such as fasting and starvation, circulating GC levels are increased, which in turn decreases the rate of protein synthesis and increases proteolysis to generate amino acids to serve as precursors for hepatic gluconeogenesis. The resulting glucose can then be used by the brain as fuel. Under pathophysiological conditions, having excess endogenous (Cushing’s Syndrome) or exogenous (prolonged medical treatment) sustained GC-mediated protein degradation can lead to skeletal muscle atrophy and muscle weakness. Moreover, GCs preserve plasma glucose through inhibiting glucose uptake and utilization in skeletal muscle, and play a permissive role in epinephrine-induced glycogenolysis. This adaptive course maintains adequate circulating glucose to fuel the brain during stress. However, this course becomes maladaptive upon chronic or excess exposure to GCs. Notably, reducing the level of available bioactive GCs has been shown to improve insulin sensitivity in animal models. The approach of inhibiting 11β-HSD1, thus reducing the level of available bioactive GCs in tissues, is currently under clinical trials for treating type 2 diabetes (Hollis and Huber, 2011; Rosenstock et al., 2010).
Fig. 1.

Metabolic influences of glucocorticoids (GCs) in skeletal muscle to regulate glucose homeostasis. (1) GCs inhibit insulin-stimulated glucose uptake. (2) GCs decrease protein synthesis and increase proteolysis to release amino acids for hepatic gluconeogensis. (3) GCs downregulate glucose utilization by inhibiting glycolysis. GCs also suppress glycogen synthesis, and act with catecholamine to upregulate glycogenolysis.
Upon binding to GCs, cytosolic GR enters the nucleus and associates with specific genomic sequences called glucocorticoid response elements (GREs). Direct binding of GR to GRE, or negative GRE (nGRE) (Surjit et al., 2011), leads to the recruitment of transcription cofactors to activate, or repress, the transcriptional rate of nearby genes, respectively. Other modes of GR action include tethering (GR binding to other transcription regulators) and squelching (GR binding to and taking away transcription regulator from DNA), which often lead to transcription repression. These genes, defined as GR primary target genes, in turn trigger biological responses of GCs. The goal of this review is to discuss the current understanding of mechanisms governing GC-regulated glucose and protein metabolism, with a main focus on potential GR primary target genes identified in skeletal muscle and mediating the metabolic functions of GCs.
2. The regulation of glucose metabolism by glucocorticoids
Skeletal muscle is one the major tissues accountable for glucose homeostasis in mammals. Approximately 80% of glucose utilization occurs in skeletal muscle (DeFronzo and Tripathy, 2009; Ferrannini et al., 1988). Skeletal muscle also serves as a reservoir for glycogen storage. GCs inhibit glucose uptake and utilization and glycogen synthesis, and play a permissive role for catecholamine-stimulated glycogen breakdown in skeletal muscle (Fig. 1). These actions counteract those of insulin, which promotes glucose utilization and glycogen synthesis. Mice treated with GCs have reduced insulin-stimulated glucose uptake, caused by attenuated insulin-induced GLUT4 translocation to the cell membrane in myotubes (Dimitriadis et al., 1997; Morgan et al., 2009; Weinstein et al., 1998). Furthermore, insulin signaling and glycogen synthase activity are suppressed by GCs (Coderre et al., 1991, 1992; Morgan et al., 2009). These metabolic effects are due to, at least in part, the direct effect of GCs on myotubes, as GC treatment of cultured myotubes inhibits insulin-stimulated glucose utilization (Gathercole et al., 2007; Morgan et al., 2009).
One major mechanism by which GCs regulate glucose metabolism is to inhibit insulin signaling (Morgan et al., 2009; Pivonello et al., 2010; Schakman et al., 2008). Insulin binds to the cell-surface insulin receptor (IR), a tyrosine kinase that autophosphorylates and phosphorylates the insulin receptor substrate (IRS) (Lee and White, 2004). Tyrosine-phosphorylated IRS associates with IR and activates downstream signaling pathways (Lee and White, 2004). Mice treated with GCs have reduced levels of tyrosine-phosphorylated IR and total IRS-1 proteins in skeletal muscle (Morgan et al., 2009). The activities of phosphoinositide-3-kinase (PI3K) and Akt, two key signaling molecules downstream of IR and IRS-1, are also decreased (Giorgino et al., 1993; Morgan et al., 2009; Saad et al., 1993). Moreover, the phosphorylation of serine 307 of IRS-1 (pSer307-IRS-1) is increased upon GC treatment (Morgan et al., 2009). This phosphorylation disrupts the association between IR and IRS-1, thus reducing the insulin response (Draznin, 2006; Gual et al., 2005). However, recent studies showed that mice harboring IRS-1 serine 307 mutated to alanine had reduced insulin sensitivity (Copps et al., 2010; Copps and White, 2012). It suggested that serine 307 phosphorylation positively regulates insulin sensitivity in vivo. Downstream of Akt, glycogen synthase kinase-3 (Gsk3) phosphorylates and inhibits glycogen synthase (Cohen and Goedert, 2004; Rayasam et al., 2009), an enzyme involved in converting glucose to glycogen. While Akt phosphorylates and reduces the activity of Gsk3, GCs, on the other hand, decrease the phosphorylation status of Gsk3 (Buren et al., 2008; Ruzzin et al., 2005).
In parallel with the PI3K/Akt signaling, another mechanism by which insulin promotes glucose uptake is to initiate the TC10 pathway (Leto and Saltiel, 2012). Insulin treatment triggers the phosphorylation of APS (adapter protein with Pleckstrin homology (PH) and Src homology 2 (SH2) domains), which then facilitates the phosphorylation of Cbl (Hu et al., 2003). Cbl-associated protein (CAP, also known as Sorbs1), as its name suggests, associates with Cbl (Baumann et al., 2000). This phosphorylated Cbl-CAP protein complex translocates to the lipid-raft microdomain at the plasma membrane, leading to the recruitment of Rho GTP-binding protein TC10 and the assembly of the exocyst complex, and finally the translocation of Glut4 to cell surface. Whether GCs can modulate this alternative insulin-initiated pathway is unknown. In contrast to insulin resistance induced by excess GCs, reducing GC level improves insulin sensitivity. Circulating GC levels are higher in obese ob/ob, db/db and lipotrophic A-ZIP/F-1 mice than normal mice, and adrenalectomy improved insulin-stimulated muscle glucose disposal in these mice (Haluzik et al., 2002; Ohshima et al., 1989). A-ZIP/F-1 mice expressed a dominant-negative protein, A-ZIP/F, under the control of the adipose-specific aP2 promoter, and had no WAT and markedly reduced brown adipose tissue (Moitra et al., 1998). This A-ZIP/F protein prevents DNA binding of basic leucine zipper (B-ZIP) transcription factor families, such as C/EBP and Jun. Kuo Kondo (KK) mice, derived from selective inbreeding for large body size, develop diabetes and demonstrate neurologic, renal, and retinal complications comparable to those observed in human diabetes (Ikeda, 1994; Reddi and Camerini-Davalos, 1988). Treating C57/B6 or KK mice with an 11β-HSD1-specific inhibitor decreased fasting blood glucose level and improved insulin sensitivity (Morgan et al., 2009). In skeletal muscle of 11β-HSD1 inhibitor-treated KK mice, pSer307-IRS-1 decreased and pThr308-Akt, a hallmark of activated Akt, increased (Morgan et al., 2009). While skeletal muscle 11β-HSD1 mRNA does not correlate with metabolic parameters (Nair et al., 2004), the activity of 11β-HSD1 in muscle biopsies of type 2 diabetic patients is lower than normal people (Jang et al., 2006, 2007; Whorwood et al., 2001). Also, in the skeletal muscle of diabetic patients, elevated GR mRNA expression correlates with the degree of insulin resistance, and treatments that improve insulin sensitivity normalize GR expression (Vestergaard et al., 2001; Whorwood et al., 2002). These studies indicate that GC signaling could be a target of therapeutic intervention against type 2 diabetes (Alamdari et al., 2010; Morton, 2010).
Overall, how GCs affect glucose metabolism in skeletal muscle is not entirely clear. To date, only a few GR primary target genes, outlined below, have been linked to GC-regulated glucose and glycogen metabolism in in vitro cultured cell models and in vivo animal models.
2.1. Pik3r1 (p85α)
A GR primary target gene contains GRE(s) near or in its genomic region, and its expression is regulated by GCs. Based on these criteria, a combination of chromatin immunoprecipitation sequencing (ChIPseq) and microarray was conducted to identify primary GR targets in muscle. It is important to note that ChIPseq gives ~300–800 bp DNA sequences (referred to as GR binding region, or GBR) likely containing GRE(s). We identified 173 genes whose expression was modulated by GC treatment and containing GBRs in mouse C2C12 myotubes (Kuo et al., 2012). Notably, whether these GBRs can confer GC response, thus contain GREs, requires further studies. Therefore, these 173 genes are referred to as “potential” GR primary targets. Interestingly, among these targets, nine genes have been shown to antagonize insulin action: Sorbs1, Grb10 (Holt et al., 2009), Cblb (discussed in a later section) (Nakao et al., 2009), Pid1 (Wu et al., 2011), Ddit4, Sesn1, Depdc6, Mknk2 and Pik3r1 (Fig. 2). Pik3r1 (a.k.a. p85α) encodes the regulatory subunit of PI3K. Pik3r1 plays a key role in conferring insulin action. However, excess Pik3r1 monomers can compete with Pik3r1 and PI3KC (catalytic subunit of PI3K, a.k.a. p110) heterodimers to interact with IRS-1 (Barbour et al., 2004; Draznin, 2006), resulting in decreased insulin response. Conversely, reducing the expression of Pik3r1 improves insulin sensitivity (Mauvais-Jarvis et al., 2002). It is important to note that elevated expression of PIK3R1 is found in patients with insulin resistance.
Fig. 2.
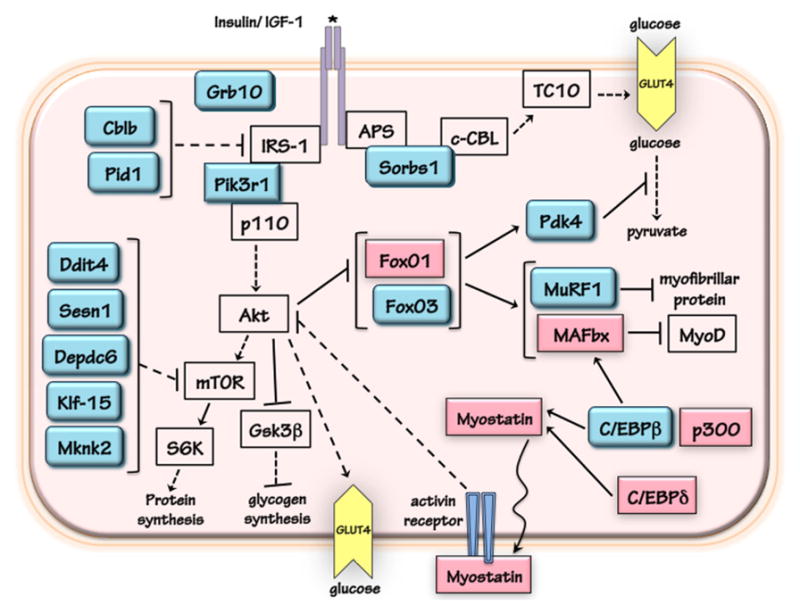
The regulation of protein and glucose metabolism by glucocorticoid receptor (GR) in skeletal muscle cells. The genes in teal are GR primary target genes, and the expression of genes in magenta are upregulated by glucocorticoids with GRE not yet defined, if any. Dashed line represents the involvement of multiple steps. Arrow shows activation while bar indicates inhibition.
Treating C2C12 myotubes with dexamethasone (Dex), a synthetic GC, reduces the total IRS-1 level, increases the pSer307-IRS-1 level and decreases the level of phosphorylation on serine 473 of Akt (pSer473-Akt), which is required to potentiate its kinase activity. These insulin resistance-associated phenotypes; however, are all compromised when Pik3r1 expression in C2C12 myotubes is reduced specifically by RNA interference (RNAi) (Kuo et al., 2012). Thus, Pik3r1 mediates, at least in part, GC-suppressed insulin signaling. The role of Pik3r1 in GC-induced insulin resistance in vivo is the subject of current research. CAP (Sorbs1), another potential GR primary target gene, is involved in the alternative insulin signaling pathway discussed above. It is essential to learn whether reducing the expressions of genes other than Pik3r1 that are capable of inhibiting insulin signaling compromises GC response.
2.2. Pyruvate dehydrogenase kinase 4 (Pdk4)
Pdk4 inhibits pyruvate dehydrogenase complex and leads to a reduced conversion of pyruvate to acetyl-CoA (Fig. 2) (Sugden and Holness, 2003). Thus, the induction of Pdk4 plays a critical role in GC-decreased glucose utilization (Sugden and Holness, 2003). Recent studies indicated that, in human primary myotubes, GC–repressed glycogen synthesis is diminished with reduced expression of PDK4 (Salehzadeh et al., 2009). GCs upregulate Pdk4 gene transcription in both liver and skeletal muscle, and GREs of human PDK4 and rat Pdk4 have been identified (Connaughton et al., 2010; Kwon et al., 2004). For human PDK4, the GRE is located between −824 and −809 (relative to transcription start site, TSS) (Kwon et al., 2004). Interestingly, three binding sites of the FoxO transcription factors, FoxO1 and FoxO3, are located near the GRE (from −738 to −731, −533 to −525, and −361 to −354, respectively). These FoxO binding sites could represent the insulin response element (IRE) that mediates the repressive effect of insulin on PDK4 gene transcription (Kwon et al., 2004). However, with no insulin present, these FoxO binding sites are required for the maximal induction of PDK4 gene transcription by GCs. This functional interaction between GR and FoxO proteins have also been reported in hepatic gluconeogenic genes, such as phosphoenolpyruvate carboxykinase (Pepck) (Hall et al., 2007) and glucose-6-phosphatase (G6Pase) (Vander Kooi et al., 2005), as well as in MuRF1 gene which is involved in GC-induced protein degradation (see below).
For rat Pdk4 gene, two GREs are identified between −6634 and −6377 relative to TSS (Connaughton et al., 2010). A FoxO binding site is found between −591 and −338. A mutation at FoxO binding decreases Dex-induced Pdk4 promoter-containing reporter gene activity. This FoxO binding site is also responsible for insulin repression on Pdk4 gene (Connaughton et al., 2010). Chromatin immunoprecipitation (ChIP) experiments show that Dex treatment increases GR occupancy at the GREs, whereas FoxO1 is already present at the FoxO binding site even without Dex treatment. Insulin treatment decreases GR recruitment to the GREs and decreases FoxO1 recruitment to the FoxO binding sites (Connaughton et al., 2010). Overall, although GREs of rat Pdk4 are located further away from its TSS, the players involved in GC and insulin response are similar to human PDK4 gene.
2.3. Additional potential GR primary targets that may confer glucocorticoid-regulated glucose metabolism
Several studies have shown that GCs increase the expression of FoxO1 and FoxO3 in skeletal muscle (Fig. 2) (Nishimura et al., 2008; Waddell et al., 2008). ChIPseq conducted in C2C12 myotubes and 3T3-L1 adipocytes found several GR binding regions (GBRs) in or near the FoxO3 genomic region, indicating that FoxO3 is a GR primary target gene (Kuo et al., 2012; Yu et al., 2010). No GBR was identified for FoxO1 gene. However, FoxO1 gene expression is induced by GC in mouse liver, and such induction is markedly reduced in mice expressing a GR mutant that has decreased DNA binding ability (Frijters et al., 2010). It is likely that FoxO1 and/or FoxO3 are involved in GC-regulated glucose oxidation, as demonstrated by FoxO1 playing an important role in the stimulation of Pdk4 gene transcription. Moreover, transgenic mice overexpressing FoxO1 gene showed impaired glycemic control (Kamei et al., 2004), suggesting that an elevated FoxO activity can cause insulin resistance. Nonetheless, the precise roles of FoxO1 and FoxO3 in GC-regulated glucose metabolism have not yet been examined.
Myostatin (a.k.a. growth differentiation factor 8, Gdf8) is a secreted protein and a member of the transforming growth factor β (TGFβ) family. Myostatin binds to activin type 2 receptor located on the membrane of muscle cells, and initiates a cellular signaling cascade that activates transcription factors smad2 and smad3, which in turn regulate the transcription of specific target genes. The function of myostatin in GC-regulated protein metabolism has been studied extensively and will be discussed in a later section. Recent studies suggest that myostatin could also participate in glucose homeostasis (Burgess et al., 2011; Zhang et al., 2011). However, the role of myostatin in GC-regulated glucose metabolism has not been reported.
2.4. Factors that modulate glucocorticoid effects on glucose metabolism
While GCs directly reduce insulin-stimulated glucose uptake and glycogen synthesis in skeletal muscle cells, GCs also affect glucose homeostasis by stimulating lipid mobilization from white adipose tissue to skeletal muscle. Upon GC treatment, increased intramuscular accumulation of triglyceride (TG) is observed (Gounarides et al., 2008). Also, the level of diacylglycerol (DAG), which can activate protein kinase c (PKC) thus causing insulin resistance (Samuel et al., 2010), is elevated (Gounarides et al., 2008). Treating mice with an 11β-HSD1 inhibitor reduces the expression of genes involved in lipogenesis and lipid metabolism in skeletal muscle (Morgan et al., 2009). Very likely, GCs positively regulate the expression of these genes. The activation of these genes involved in lipid metabolism by GCs has been well documented in liver and adipose tissues (Amatruda et al., 1983; Cai et al., 2011; Dolinsky et al., 2004; Gathercole et al., 2011; Yu et al., 2010). However, whether GCs can directly affect the transcription of these genes require more studies. Previous studies have shown that treating mice with an inhibitor of lipolysis in white adipose tissue decreased GC-induced insulin resistance (Tappy et al., 1994), seemingly by reducing ectopic lipid accumulation in both liver and skeletal muscle. It is important to note that the accumulation of lipid intermediates, such as DAG and ceramides, rather than neutral lipids, such as triglycerides, is linked to antagonize insulin signaling. In skeletal muscle, when beta oxidative capacity is high, concentrations of ceramide, acyl-CoA and DAG stay low. In contrast, under the condition of low beta oxidative capacity with lipid overload, concentrations of these lipid intermediates of lipid metabolism increase (Hulver et al., 2003). In human muscle, insulin resistance caused by elevated free fatty acid levels during euglycemic-hyperinsulinemic clamping was associated with increased DAG (Itani et al., 2002). Furthermore, elevated intracellular ceramide level in muscle is associated with GC-induced insulin resistance (Holland et al., 2007). In any case, whether GCs modulate the production of lipid intermediates, and how this may affect glucose homeostasis requires more mechanistic studies.
Interestingly, treating mice with anti-oxidants, such as vitamin C and E, attenuates GC-induced insulin resistance in skeletal muscle, liver and white adipose tissue (Williams et al., 2012). The improvement of glucose tolerance is independent of Akt phosphorylation; instead, AMP-activated kinase (AMPK) activity is elevated in skeletal muscle (Williams et al., 2012). GC treatment can repress AMPK activity in skeletal muscle (Christ-Crain et al., 2008; Kola et al., 2008; Lutzner et al., 2012) and increase the level of malonyl-CoA (Nakken et al., 2010), an intermediate of fatty acid synthesis that inhibits fatty acid oxidation. Anti-oxidants counteract such effects of GCs to improve chronic/excess GC-induced insulin resistance. Previous studies done with adipocytes indicated that long-term GC treatment increases the level of reactive oxygen species (ROS), which activates c-Jun N-terminal kinase (JNK) to suppress insulin signaling (Houstis et al., 2006). It has been shown that GC treatment increases ROS levels in L6 myotubes, which leads to an inhibition in protein synthesis, and an elevation in proteolysis and apoptosis (Orzechowski et al., 2003). However, whether oxidative stress could be one of the mechanisms by which GCs repress insulin action on glucose metabolism in skeletal muscle has not been reported.
3. The regulation of protein metabolism by glucocorticoids
It is well documented that the treatment with GCs induces a catabolic response in skeletal muscle. Treating animals with physiological or synthetic GCs causes a decrease in skeletal muscle size (Auclair et al., 1997; Baehr et al., 2011; Hu et al., 2009; Shimizu et al., 2011). In fact, induction of muscle atrophy by several pathological conditions, such as diabetes, metabolic acidosis and sepsis, all involves GCs. The muscle atrophy phenotype is caused by cell autonomous effects of GR both in vitro and in vivo. Mouse C2C12 and rat L6 myotubes treated with synthetic GCs, such as Dex, have markedly decreased cell diameters (Kuo et al., 2012; Menconi et al., 2008). Furthermore, using RNAi to reduce GR expression in C2C12 myotubes compromises GC-reduced protein degradation (Zhao et al., 2009). In muscle-specific GR knockout (MGRKO) mice, Dex-induced muscle atrophy was completely abolished (Watson et al., 2012). In these mice, muscle atrophy induced by nutritional deprivation, which elevates the circulating corticosterone, is also reduced though not completely blocked (Watson et al., 2012). Similarly, nutritional deprivation-induced muscle atrophy is inhibited after adrenalectomy (Almon and Dubois, 1988). These observations suggest that GR and its primary target genes play a pivotal role in GC-regulated protein metabolism. These are important studies given the previous suggestion that GCs effects on muscle were in part secondary to primary effects on the pituitary and liver from reduced GH and IGF1 secretion. Identifying the primary causative GR target genes responsible for muscle atrophy is important in the hunt for selective GR modulators that retain anti-inflammatory properties of GCs, but have less adverse effects. This issue is particularly relevant in the treatment of critically ill patients who often suffer from “ICU myopathy” (Puthucheary et al., 2010) and in the treatment of inflammatory disease of either muscle or the lungs where decreased muscle function results in decreased pulmonary function (loss of bellows).
3.1. MuRF1
About a decade ago, systematic gene expression analyses identified a list of genes that were upregulated during muscle wasting. These genes are called the atrogenes (Bodine et al., 2001). Well recognized atrogenes include two muscle-specific E3 ubiquitin ligases: muscle RING finer 1 (MuRF1, a.k.a. Trim63) and muscle atrophy F-box (MAFbx, a.k.a. Atrogin-1, Fbxo32) (Bodine et al., 2001; Sandri et al., 2004; Stitt et al., 2004) (Fig. 2). MuRF1 and Atrogin-1 gene expression are increased in most of the muscle wasting scenarios, including denervation, nutritional deprivation, unloading and exogenous GC treatment. For the mouse MuRF1 gene, the GRE is located between −210 and −196. Interestingly, a binding site for the FoxO family of transcription factors is located adjacent to the MuRF1 GRE (Waddell et al., 2008). Overexpression of GR and FoxO1 in C2C12 cells synergistically activates a reporter plasmid containing mouse MuRF1 promoter. Mutation of either the GRE or the FoxO binding site reduces the ability of GCs to induce the activity of the MuRF1 promoter reporter gene. ChIP experiments demonstrated that GR and FoxO1 occupy the genomic region containing the MuRF1 GRE. Insulin-like growth factor-1 (IGF-1), which inhibits GC-induced MuRF1 gene transcription, decreases the recruitment of FoxO1 but not GR to the MuRF1 GRE.
Mice lacking MuRF1 gene were generated to examine its role in GC-induced muscle atrophy in vivo (Baehr et al., 2011). Following 14 days of Dex treatment, wild type (WT) mice have markedly decreased tricep surae (TS) and tibialis anterior (TA) muscle weights. In MuRF1 null mice, Dex treatment still decreased the weight of TS and TA muscles; however, these decreases were significantly less than those of WT mice. Also, MuRF1 null mice showed a significant sparing of fiber cross-section area and tension output of gastrocnemius muscles after Dex treatment. These results clearly confirm the important role of MuRF1 in GC-induced muscle atrophy. Interestingly, while Dex-reduced protein synthesis was compromised in MuRF1 null mice, Dex-induced protein degradation was not affected in these mice, although the basal protein degradation rate is lower in MuRF1 null mice (Baehr et al., 2011). Previous studies have reported that MuRF1 targets several myofibrillar proteins for degradation (Clarke et al., 2007; Cohen et al., 2009; Polge et al., 2011), and these results suggest that MuRF1 targets more than myofibrillar proteins when conferring GC response. Interestingly, transgenic mice overexpressing MuRF1 in skeletal muscle have altered whole-body energy and glucose homeostasis (Hirner et al., 2008; Koyama et al., 2008). Eight-week old transgenic mice do not show a muscle atrophy phenotype. Their plasma insulin levels are two-fold higher than those of WT mice, and they have lower hepatic glycogen levels. Pyruvate dehydrogenase (Pdh) levels are lower in these mice, and yeast two-hybrid showed that MuRF1 interacts with Pdh, Pdk2, and Pdk4, key regulators of glycolysis (Hirner et al., 2008). Overall, MuRF1 could be a multi-functional protein that participates in both protein and glucose metabolism. It is important to note that nutritional deprivation-induced muscle atrophy is not spared in MuRF1 null mice (Baehr et al., 2011), suggesting either GC-independent pathways act during nutritional deprivation to mediate atrophy or other GR primary targets are involved in glucocortioid-regulated protein metabolism during nutritional deprivation.
3.2. MAFbx
MAFbx encodes an ubiquitin E3 ligase, and its GRE, if any, has not been identified. It is possible that MAFbx is not a primary but a secondary target gene of GR. MAFbx gene transcription is activated by transcription factors FoxO1, FoxO3 and Klf15 (Sandri et al., 2004; Shimizu et al., 2011; Stitt et al., 2004). These three transcription factors are potential GR primary target genes in skeletal muscle (see below). Following 14 days of Dex treatment, WT mice and mice lacking MAFbx gene show similar extent of muscle atrophy (Baehr et al., 2011). Surprisingly, there is no muscle sparing in MAFbx null mice under this experimental condition. While we cannot rule out the potential role of MAFbx in GC-regulated protein metabolism in other conditions, these results clearly indicate that if MAFbx has a role in GC-regulated protein metabolism, it must be distinct from that of MuRF1. MAFbx has been shown to target and degrade MyoD, myogenin, and Eif3f (Csibi et al., 2009; Jogo et al., 2009; Lagirand-Cantaloube et al., 2008; Tintignac et al., 2005), and degradation of any of these proteins reduces myotube diameters. Interestingly, treatment of GCs has been reported to increase N-terminal ubiquitination of MyoD (Sun et al., 2008).
3.3. Myostatin
Myostatin negatively regulates muscle growth and differentiation (Walsh and Celeste, 2005). Myostain has been shown to inhibit Akt (Amirouche et al., 2009; Morissette et al., 2006; Trendelenburg et al., 2009), which is activated by two major protein-synthesis-promoting hormones, insulin and insulin-like growth factor-1 (IGF-1). GCs can increase the expression of myostatin (Ma et al., 2001, 2003) (Fig. 2). GCs also increase the activity of a reporter gene containing 3.3 kb of the human MYOSTATIN promoter. However, the exact location of the MYOSTATIN GRE is not yet identified, and the occupancy of GR on this promoter region has not been addressed (Ma et al., 2001). The MYOSTATIN promoter contains two binding sites for CCAAT/enhancer binding protein family of transcription factors (C/EBP). A MYOSTATIN promoter-embedded reporter is activated by the overexpression of CCAAT/enhancer binding protein α, β and δ (C/EBPα, β, and δ, respectively), with C/EBPδ overexpression demonstrating the strongest induction (Allen et al., 2010a,b). In C2C12 myotubes, GCs elevate the expression of C/EBPδ and C/EBPβ (Gonnella et al., 2011), and mutating C/EBP binding sites in MYOSTATIN promoter reporter compromised GC response, suggesting that myostatin could be a secondary GR target (Allen et al., 2010a,b). It is unclear whether C/EBPδ is a GR primary target gene, since no GRE has been identified. C/EBPβ is discussed in detail in a later section.
Naturally occurring myostatin nulls have been found in human and cattles, and their muscles are enlarged significantly (Allen et al., 2010a,b). Mice lacking the myostatin gene were created to elucidate its role in GC-regulated muscle atrophy. Ten-days of Dex treatment (1 mg/kg body weight, BW) caused muscle atrophy in WT mice and this was greatly attenuated in myostatin null mice (Gilson et al., 2007). When WT mice were treated with a higher dose of Dex (5 mg/kg BW) for 4 days, the expression of MuRF1, Atrogin-1 and Cathepsin L and chymoytpsin-like proteasomal activity were increased. However, the induction of these genes was reduced in myostatin null mice (Gilson et al., 2007). Therefore, myostatin likely participates in GC-induced protein degradation. Another study that measured the rate of phenylalanine tracer incorporation into myofibrillar proteins in myostatin null mice; however, showed that muscle hypertrophy seen in myostatin null mice is a result of increased protein synthesis, rather than decreased proteolysis, per muscle fiber (Welle et al., 2011). This phenotype could be due to myostatin’s ability to inhibit Akt, since activated Akt leads to protein synthesis and the phosphorylation of FoxO1 and FoxO3 to exclude them from the nucleus. Notably, myostatin expression is induced upon food deprivation and its function is required for food deprivation-induced muscle atrophy (Allen et al., 2010a,b).
3.4. Pik3r1 (p85α)
Overexpression of Pik3r1 in C2C12 myotubes resulted in a marked decrease in cell diameters (Kuo et al., 2012). In contrast, Dex-decreased myotube diameters and Dex-increased protein synthesis are compromised when reducing Pik3r1 expression in C2C12 myotubes (Kuo et al., 2012). These observations suggest that Pik3r1 is involved in GC-inhibited protein synthesis, leading to decreased myotube diameters (Fig. 2). Interestingly, Dex-induced expression of MuRF1, Atrogin-1 and FoxO3 are all compromised in Pik3r1 knockdown cells (Kuo et al., 2012), implying that Pik3r1 participates in Dex-induced protein degradation in vitro, though the precise role of Pik3r1 in GC-regulated protein metabolism needs to be confirmed in Pik3r1 null mice.
Administration of a physiological dose of GCs to adrenalectomized and acutely diabetic (induced by injecting strepzotocin, STZ) mice demonstrated decreased IRS-1-associated PI3K activity in muscle and resulted in progressive muscle atrophy (Hu et al., 2009). These responses are related to increased association of PI3K with GR proteins. In muscle-specific GRKO mice, acute diabetes minimally reduces IRS-1-associated PI3K activity in skeletal muscle, and these mice are resistant to muscle atrophy. However, when a physiological dose of GCs is given to muscle-specific insulin receptor knockout mice, muscle protein degradation is elevated (Hu et al., 2009). Fluorescence resonance energy transfer and an in vitro binding assay showed that hormone-bound GR interacts with Pik3r1 protein thus reducing the association of PI3K with IRS-1. In this case, GCs exert non-genomic action to regulate protein metabolism by antagonizing insulin/IGF-1 responses. The interaction between GR and Pik3r1 proteins has been reported in other cell types (Arancibia et al., 2011; Hafezi-Moghadam et al., 2002; Leis et al., 2004). Identifying GR mutants that do not interact with Pik3r1 or Pik3r1 mutants which do not associate with GR will help dissect the mechanism of GC-regulated protein metabolism.
Grb10 is another potential GR primary target capable of negatively regulating insulin action. Mice lacking Grb10 exhibited increased muscle weight and hypermuscularity due to increased myofiber size (Holt et al., 2012) compared to WT mice. The role of Grb10 in GC-regulated protein metabolism is unclear. It is likely that additional GR primary targets inhibiting insulin signaling also participate in GC-regulated protein metabolism.
3.5. C/EBPβ
In L6 myotubes, reducing C/EBPβ expression significantly compromises Dex-induced MuRF1 and MAFbx gene expression and attenuates Dex-decreased cell diameters though overexpressing C/EBPβ did not affect MuRF1 and MAFbx gene expression (Gonnella et al., 2011). Since the MuRF1 gene contains a GRE, C/EBPβ could cooperate with GR to induce MuRF1 gene expression. For MAFbx, C/EBPβ may act with FoxO1, FoxO3, and/or Klf15 to activate its expression. The C/EBPβ gene expression is induced by Dex in both L6 and C2C12 myotubes (Gonnella et al., 2011). ChIPseq identified a GR binding region (GBR) located at approximately 9.5 kb upstream from the C/EBPβ TSS (Kuo et al., 2012). Whether this GBR mediates GC-stimulated C/EBPβ gene transcription has not been tested.
C/EBPβ interacts with a histone acetyltransferase (HAT), p300, whose expression is increased by GCs in L6 myotubes and skeletal muscles of rat (Fig. 2) (Yang et al., 2005). p300 serves as a coactivator for C/EBPβ to activate genes involved in protein metabolism. In L6 myotubes, knocking down p300 with RNAi reduces Dex-decreased cell diameters and Dex-increased protein degradation (Yang et al., 2007). Overexpressing p300 lacking HAT activity (Yang et al., 2007) or p300 inhibitor Cited2 reduces these GC responses (Tobimatsu et al., 2009). It is important to note that p300 also acts as a coactivator for GR to stimulate the transcription of GR primary target genes (Amat et al., 2007; Shipp et al., 2010). This function of p300 may contribute to GC-induced muscle atrophy. Interestingly, inducing sepsis in rats increases p300 expression and decreases histone deacetylase HDAC3 and HDAC6 expression in skeletal muscle (Alamdari et al., 2010). These effects are attenuated when treating septic rats with RU486, a partial GR antagonist. Therefore, GCs are required for sepsis-induced p300 expression. Interestingly, in L6 myotubes, RU486 did not block sepsis-induced p300 expression, instead, it induces p300 expression, suggesting that different mechanisms are adapted by Dex to regulate p300 expression in vitro and in vivo.
3.6. FoxO1 and FoxO3
The roles of FoxO1 and FoxO3 in the regulation of protein metabolism have been described in many review articles (Gross et al., 2008; Schakman et al., 2008). Briefly, they activate genes involved in protein degradation, such as MuRF1 and MAFbx (Fig. 2). In addition, they stimulate genes encoding proteins that inhibit protein synthesis, such as 4EBP1. Furthermore, in myotubes, FoxO3 can activate the expression of genes involved in autophagy, such as Bnip3, Lc3 and Atg14, and trigger protein degradation through lysosomal and ubiquitin-proteosomal pathway (Mammucari et al., 2007; Masiero et al., 2009). In C2C12 myotubes, overexpressing a dominant negative form of FoxO3 protein blocked Dex-decreased cell diameters (Sandri et al., 2004) and Dex-induced MuRF1 and MAFbx expression (Sandri et al., 2004; Stitt et al., 2004). In another pathway, FoxO3 is required for GC-inducing insulin receptor substrate 2 (IRS-2), which in turn activates Mek and Erk kinases. Erk then phosphorylates Sp1, a transcription factor that upregulates the expression of ubiquitin C (Ubc), contributing to protein degradation (Zheng et al., 2010). Overall, FoxO proteins can confer GC response and act with GR to regulate specific gene transcription, such as MuRF-1.
3.7. Genes encoding proteins in the mTOR pathway
GCs reduce the activity of mammalian target of rapamycin (mTOR), a protein kinase involved in the activation of protein synthesis that is downstream of Akt and upstream of p70 S6 kinase (p70S6K) (Shah et al., 2000; Wang et al., 2006). Several potential GR primary targets identified from ChIPseq in C2C12 myotubes can inhibit mTOR signaling. These genes include Sestrin 1 (Sesn1) (Budanov and Karin, 2008), Depdc6 (a.k.a. deptor) (Peterson et al., 2009), Ddit4 (a.k.a. Redd1, Rtp801) (Brugarolas et al., 2004) and Mknk2 (Hu et al., 2012) (Fig. 2). Recent studies also suggest that Mknk2 associates with mTOR and inhibits the phosphorylation and activation of p70S6 kinase (Hu et al., 2012). GR binding regions of these genes can mediate GC response when inserted upstream of a TATA box in a reporter plasmid. The role of Sesn1, Depdc6 and Mknk2 in GC-suppressed protein synthesis has not been examined. In L6 myotubes, reducing the expression of Ddit4 significantly attenuates Dex-suppressed protein synthesis. Although several potential GR primary targets are identified as inhibitors of the insulin/IGF-1-PI3K-Akt-mTOR signaling, reducing the expression of either Pik3r1 or Ddit4 already showed compromised phenotypes on GC-reduced protein synthesis in vitro (Wang et al., 2006). These studies demonstrated that these potential GR primary targets likely do not play a redundant role. Instead, each might confer specific GC responses, and in vivo studies are required to confirm their roles. If deleting one of these genes could spare GC-induced muscle atrophy in vivo, it would have invaluable implications in GC therapeutics with reduced side effects.
Notably, the activation of mTOR signaling appears to antagonize GR activity (Shimizu et al., 2011). L6 myoblasts treated with branched-chain amino acids (BCAAs), which activates a GTPase called Rheb that positively regulates mTORC1, resulted in a reduced GC response of a reporter gene with tandem repeats of perfect palindromic GREs (AGAACAGGATGTTCT). Mice treated with BCAA have compromised Dex response on the induction of known GR primary targets. Intriguingly, the recruitment of GR to its cognate GRE of these target genes is also diminished, suggesting that mTOR signaling affects GR and DNA interaction. In addition, BCAA treatment in rats compromises Dex-inhibited mTOR activity and Dex-induced muscle atrophy. Overall, the balance between GCs and mTOR signaling can determine the catabolic and anabolic nature of skeletal muscle.
3.8. Klf-15
Branched-chain amino acid transaminase 2 (Bcat2) encodes a mitochondrial enzyme catalyzing the first reaction in the catabolism of BCAA to promote BCAA degradation and alanine production in skeletal muscle. In skeletal muscle, Bcat2 expression is activated by a transcription factor called Klf15, which is a GR primary target gene (Fig. 2) (Shimizu et al., 2011). With ChIP and reporter assay, two rat Klf15 GREs were identified in the intron region between +479 and +494, and between +1014 and +1028. In addition to activating Bcat2 transcription to reduce mTOR activity, Klf15 also elevates MuRF1 and MAFbx transcription, with Klf15 binding sites identified in rat Bcat2, MuRF1 and MAFbx. Adenoviral mediated overexpression of Klf15 in skeletal muscle ex vivo caused muscle atrophy, which confirmed the involvement of Klf15 in protein metabolism. Interestingly, Klf15 null mice have abnormal lipid and energy flux, excessive reliance on carbohydrate fuels, exaggerated muscle fatigue, and impaired endurance exercise capacity (Haldar et al., 2012). However, whether Dex-induced muscle atrophy is spared in Klf15 null mice has not been reported.
3.9. Cblb
Cblb encodes an ubiquitin E3 ligase that stimulates the ubiquitination and degradation of IRS-1 (Fig. 2). In C2C12 myotubes, overexpressing Cblb attenuates IGF-1’s ability to antagonize Dex-reduced myotube diameters and Dex-induced MAFbx gene expression, possibly due to Cblb’s role in degrading IRS-1 thus suppressing IGF-1 action (Nakao et al., 2009). However, it is unclear whether reducing the expression of Cblb would compromise GC effects on myotube diameters and atrogene expression. Unloading-induced muscle atrophy is protected in Cblb null mice (Nakao et al., 2009). Whether Cblb null mice are protected from GC-induced muscle atrophy has not been reported. Notably, overexpressing Cblb did not affect myotube diameters or the expression of MAFbx gene. These results indicate that, unlike Pik3r1 or Ddit4, the induction of Cblb alone is not sufficient to induce muscle atrophy.
3.10. Factors that modulate glucocorticoid effects on protein metabolism
In skeletal muscle, several signaling pathways are capable of suppressing the catabolic effects of GCs. It is not surprising that IGF-1 is one of them, as IGF-1 is the main anabolic signal in skeletal muscle. In fact, factors that modulate insulin/IGF-1 signaling pathway have potential to regulate the catabolic effects of GCs. For example, reducing the expression of Gsk3β in C2C12 myotubes compromised Dex-reduced myofibrillar protein levels and MAFbx gene expression (Verhees et al., 2011). Androgens are another category of anabolic hormones that antagonize GC-induced muscle atrophy. Testosterone suppresses Dex-induced expression of MuRF1, MAFbx, Pik3r1, Ddit4, 4Ebp1 and FoxO1 in vivo (Wu et al., 2010; Yin et al., 2009). Testosterone treatment alone reduces FoxO1 expression but exerts no effect on Pik3r1 expression (Wu et al., 2010). Testosterone deprivation by castration in rats resulted in an increased expression of MuRF1 and MAFbx in testosterone-sensitive fast-twitch pelvic levator ani muscle (Pires-Oliveira et al., 2010). Administration of testosterone in these castrated rats then reduces the expression of these two genes. Androgen receptor (AR) binds to androgen response element (ARE) to modulate the transcription of its target genes. The consensus DNA sequence of ARE and GRE are very similar. The observations of these two transcription factors (AR and GR) that appear to be able to recognize the same DNA sequence yet exhibit opposite effects on protein metabolism in the same tissue (skeletal muscle) have puzzled endocrinologists for years. Notably, most endogenous AREs and GREs are composite elements that require the participation of other cis-acting elements and their trans-acting factors to carry complete hormonal responses. However, AR and GR are structurally distinct and likely interact with overlapping but different sets of proteins. Thus, a GRE in a specific genomic context may not be responsive to AR, because AR cannot interact with DNA-binding transcription factors occupying the cis-acting elements adjacent to this GRE. It would be intriguing to learn whether cis-acting elements co-localize with AREs and GREs are indeed distinct in myotubes. This question can be answered with ChIPseq assays.
4. Future directions
In the last two decades, much progress has been made to decipher how GCs regulate glucose and protein metabolism. Animal studies have confirmed the role of GR primary target genes, such as MuRF1, and a potential GR secondary target gene, Myostatin, in GC-induced muscle atrophy. It is clear that additional GR primary targets are involved in GC-regulated protein metabolism, as neither MuRF1 nor Myostatin null mice are completely spared from effects of GCs. In fact, GC-induced protein degradation is not affected in MuRF1 null mice. In addition to identifying additional GR primary targets involved in protein metabolism and mediate GC response, it is important to dissect in more detail how these identified GR primary target genes, such as MuRF1, mediate the physiological changes induced by GCs. There is still a sizable gap in our understanding linking known changes in gene expression to observed changes in glucose metabolism caused by GCs. To our knowledge, no GR primary target in skeletal muscle has been shown to confer GC-induced insulin resistance in vivo. Last but not least, whether GC-altered lipid metabolism results in increased levels of certain lipid intermediates to cause insulin resistance is another crucial issue awaiting future studies.
Acknowledgments
This work is supported by the NIH (R01DK083591), the Muscular Dystrophy Association (186068), and the Hellman Funds of UC Berkeley. T.K. is supported by the Dissertation Award Fellowship from the University of California Tobacco-Related Diseases Research Program (18DT-0010).
References
- Alamdari N, Smith IJ, Aversa Z, Hasselgren PO. Sepsis and glucocorticoids upregulate p300 and downregulate HDAC6 expression and activity in skeletal muscle. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2010;299:R509–R520. doi: 10.1152/ajpregu.00858.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DL, Cleary AS, Hanson AM, Lindsay SF, Reed JM. CCAAT/enhancer binding protein-delta expression is increased in fast skeletal muscle by food deprivation and regulates myostatin transcription in vitro. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2010a;299:R1592–R1601. doi: 10.1152/ajpregu.00247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DL, Cleary AS, Lindsay SF, Loh AS, Reed JM. Myostatin expression is increased by food deprivation in a muscle-specific manner and contributes to muscle atrophy during prolonged food deprivation in mice. Journal of Applied Physiology. 2010b;109:692–701. doi: 10.1152/japplphysiol.00504.2010. [DOI] [PubMed] [Google Scholar]
- Almon RR, Dubois DC. Adrenalectomy eliminates both fiber-type differences and starvation effects on denervated muscle. American Journal of Physiology. 1988;255:E850–E856. doi: 10.1152/ajpendo.1988.255.6.E850. [DOI] [PubMed] [Google Scholar]
- Amat R, Solanes G, Giralt M, Villarroya F. SIRT1 is involved in glucocorticoid-mediated control of uncoupling protein-3 gene transcription. Journal of Biological Chemistry. 2007;282:34066–34076. doi: 10.1074/jbc.M707114200. [DOI] [PubMed] [Google Scholar]
- Amatruda JM, Danahy SA, Chang CL. The effects of glucocorticoids on insulin-stimulated lipogenesis in primary cultures of rat hepatocytes. Biochemical Journal. 1983;212:135–141. doi: 10.1042/bj2120135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amirouche A, Durieux AC, Banzet S, Koulmann N, Bonnefoy R, Mouret C, Bigard X, Peinnequin A, Freyssenet D. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009;150:286–294. doi: 10.1210/en.2008-0959. [DOI] [PubMed] [Google Scholar]
- Arancibia S, Benitez D, Nunez LE, Jewell CM, Langjahr P, Candia E, Zapata-Torres G, Cidlowski JA, Gonzalez MJ, Hermoso MA. Phosphatidylinositol 3-kinase interacts with the glucocorticoid receptor upon TLR2 activation. Journal of Cellular and Molecular Medicine. 2011;15:339–349. doi: 10.1111/j.1582-4934.2009.00958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auclair D, Garrel DR, Chaouki Zerouala A, Ferland LH. Activation of the ubiquitin pathway in rat skeletal muscle by catabolic doses of glucocorticoids. American Journal of Physiology. 1997;272:C1007–C1016. doi: 10.1152/ajpcell.1997.272.3.C1007. [DOI] [PubMed] [Google Scholar]
- Baehr LM, Furlow JD, Bodine SC. Muscle sparing in muscle RING finger 1 null mice. response to synthetic glucocorticoids. Journal of Physiology. 2011;589:4759–4776. doi: 10.1113/jphysiol.2011.212845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour LA, Shao J, Qiao L, Leitner W, Anderson M, Friedman JE, Draznin B. Human placental growth hormone increases expression of the p85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscle. Endocrinology. 2004;145:1144–1150. doi: 10.1210/en.2003-1297. [DOI] [PubMed] [Google Scholar]
- Baumann CA, Ribon V, Kanzaki M, Thurmond DC, Mora S, Shigematsu S, Bickel PE, Pessin JE, Saltiel AR. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–207. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes & Development. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Karin M. P53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buren J, Lai YC, Lundgren M, Eriksson JW, Jensen J. Insulin action and signalling in fat and muscle from dexamethasone-treated rats. Archives of Biochemistry and Biophysics. 2008;474:91–101. doi: 10.1016/j.abb.2008.02.034. [DOI] [PubMed] [Google Scholar]
- Burgess K, Xu T, Brown R, Han B, Welle S. Effect of myostatin depletion on weight gain, hyperglycemia, and hepatic steatosis during five months of high-fat feeding in mice. PLoS ONE. 2011;6:e17090. doi: 10.1371/journal.pone.0017090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Song Z, Wang X, Jiao H, Lin H. Dexamethasone-induced hepatic lipogenesis is insulin dependent in chickens (Gallus gallus domesticus) Stress. 2011;14:273–281. doi: 10.3109/10253890.2010.543444. [DOI] [PubMed] [Google Scholar]
- Christ-Crain M, Kola B, Lolli F, Fekete C, Seboek D, Wittmann G, Feltrin D, Igreja SC, Ajodha S, Harvey-White J, Kunos G, Muller B, Pralong F, Aubert G, Arnaldi G, Giacchetti G, Boscaro M, Grossman AB, Korbonits M. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: a novel mechanism in Cushing’s syndrome. The FASEB Journal. 2008;22:1672–1683. doi: 10.1096/fj.07-094144. [DOI] [PubMed] [Google Scholar]
- Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ. The E3 ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metabolism. 2007;6:376–385. doi: 10.1016/j.cmet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Coderre L, Srivastava AK, Chiasson JL. Role of glucocorticoid in the regulation of glycogen metabolism in skeletal muscle. American Journal of Physiology. 1991;260:E927–E932. doi: 10.1152/ajpendo.1991.260.6.E927. [DOI] [PubMed] [Google Scholar]
- Coderre L, Srivastava AK, Chiasson JL. Effect of hypercorticism on regulation of skeletal muscle glycogen metabolism by insulin. American Journal of Physiology. 1992;262:E427–E433. doi: 10.1152/ajpendo.1992.262.4.E427. [DOI] [PubMed] [Google Scholar]
- Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nature Reviews Drug Discovery. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. Journal of Cell Biology. 2009;185:1083–1095. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connaughton S, Chowdhury F, Attia RR, Song S, Zhang Y, Elam MB, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Molecular and Cellular Endocrinology. 2010;315:159–167. doi: 10.1016/j.mce.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metabolism. 2010;11:84–92. doi: 10.1016/j.cmet.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012;55:2565–2582. doi: 10.1007/s00125-012-2644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csibi A, Leibovitch MP, Cornille K, Tintignac LA, Leibovitch SA. MAFbx/atrogin-1 controls the activity of the initiation factor eIF3-f in skeletal muscle atrophy by targeting multiple C-terminal lysines. Journal of Biological Chemistry. 2009;284:4413–4421. doi: 10.1074/jbc.M807641200. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(Suppl 2):S157–S163. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekhuijzen PN, Gayan-Ramirez G, Bisschop A, DeBock V, Dom R, Decramer M. Corticosteroid treatment and nutritional deprivation cause a different pattern of atrophy in rat diaphragm. Journal of Applied Physiology. 1995;78:629–637. doi: 10.1152/jappl.1995.78.2.629. [DOI] [PubMed] [Google Scholar]
- Dimitriadis G, Leighton B, Parry-Billings M, Sasson S, Young M, Krause U, Bevan S, Piva T, Wegener G, Newsholme EA. Effects of glucocorticoid excess on the sensitivity of glucose transport and metabolism to insulin in rat skeletal muscle. Biochemical Journal. 1997;321(Pt 3):707–712. doi: 10.1042/bj3210707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky VW, Douglas DN, Lehner R, Vance DE. Regulation of the enzymes of hepatic microsomal triacylglycerol lipolysis and re-esterification by the glucocorticoid dexamethasone. Biochemical Journal. 2004;378:967–974. doi: 10.1042/BJ20031320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draznin B. Molecular mechanisms of insulin resistance. serine phosphorylation of insulin receptor substrate-1 and increased expression of p85alpha: the two sides of a coin. Diabetes. 2006;55:2392–2397. doi: 10.2337/db06-0391. [DOI] [PubMed] [Google Scholar]
- Falduto MT, Czerwinski SM, Hickson RC. Glucocorticoid-induced muscle atrophy prevention by exercise in fast-twitch fibers. Journal of Applied Physiology. 1990;69:1058–1062. doi: 10.1152/jappl.1990.69.3.1058. [DOI] [PubMed] [Google Scholar]
- Ferrannini E, Simonson DC, Katz LD, Reichard G, Jr, Bevilacqua S, Barrett EJ, Olsson M, DeFronzo RA. The disposal of an oral glucose load in patients with non-insulin-dependent diabetes. Metabolism. 1988;37:79–85. doi: 10.1016/0026-0495(88)90033-9. [DOI] [PubMed] [Google Scholar]
- Fournier M, Huang ZS, Li H, Da X, Cercek B, Lewis MI. Insulin-like growth factor I prevents corticosteroid-induced diaphragm muscle atrophy in emphysematous hamsters. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2003;285:R34–R43. doi: 10.1152/ajpregu.00177.2002. [DOI] [PubMed] [Google Scholar]
- Frijters R, Fleuren W, Toonen EJ, Tuckermann JP, Reichardt HM, van der Maaden H, van Elsas A, van Lierop MJ, Dokter W, de Vlieg J, Alkema W. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genomics. 2010;11:359. doi: 10.1186/1471-2164-11-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gathercole LL, Bujalska IJ, Stewart PM, Tomlinson JW. Glucocorticoid modulation of insulin signaling in human subcutaneous adipose tissue. Journal of Clinical Endocrinology and Metabolism. 2007;92:4332–4339. doi: 10.1210/jc.2007-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gathercole LL, Morgan SA, Bujalska IJ, Hauton D, Stewart PM, Tomlinson JW. Regulation of lipogenesis by glucocorticoids and insulin in human adipose tissue. PLoS ONE. 2011;6:e26223. doi: 10.1371/journal.pone.0026223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D, Ketelslegers JM, Thissen JP. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology. 2007;148:452–460. doi: 10.1210/en.2006-0539. [DOI] [PubMed] [Google Scholar]
- Giorgino F, Almahfouz A, Goodyear LJ, Smith RJ. Glucocorticoid regulation of insulin receptor and substrate IRS-1 tyrosine phosphorylation in rat skeletal muscle in vivo. Journal of Clinical Investigation. 1993;91:2020–2030. doi: 10.1172/JCI116424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonnella P, Alamdari N, Tizio S, Aversa Z, Petkova V, Hasselgren PO. C/EBPbeta regulates dexamethasone-induced muscle cell atrophy and expression of atrogin-1 and MuRF1. Journal of Cellular Biochemistry. 2011;112:1737–1748. doi: 10.1002/jcb.23093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounarides JS, Korach-Andre M, Killary K, Argentieri G, Turner O, Laurent D. Effect of dexamethasone on glucose tolerance and fat metabolism in a diet-induced obesity mouse model. Endocrinology. 2008;149:758–766. doi: 10.1210/en.2007-1214. [DOI] [PubMed] [Google Scholar]
- Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh CM, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, Liao JK. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nature Medicine. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar SM, Jeyaraj D, Anand P, Zhu H, Lu Y, Prosdocimo DA, Eapen B, Kawanami D, Okutsu M, Brotto L, Fujioka H, Kerner J, Rosca MG, McGuinness OP, Snow RJ, Russell AP, Gerber AN, Bai X, Yan Z, Nosek TM, Brotto M, Hoppel CL, Jain MK. Kruppel-like factor 15 regulates skeletal muscle lipid flux and exercise adaptation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6739–6744. doi: 10.1073/pnas.1121060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RK, Wang XL, George L, Koch SR, Granner DK. Insulin represses phosphoenolpyruvate carboxykinase gene transcription by causing the rapid disruption of an active transcription complex: a potential epigenetic effect. Molecular Endocrinology. 2007;21:550–563. doi: 10.1210/me.2006-0307. [DOI] [PubMed] [Google Scholar]
- Haluzik M, Dietz KR, Kim JK, Marcus-Samuels B, Shulman GI, Gavrilova O, Reitman ML. Adrenalectomy improves diabetes in A-ZIP/F-1 lipoatrophic mice by increasing both liver and muscle insulin sensitivity. Diabetes. 2002;51:2113–2118. doi: 10.2337/diabetes.51.7.2113. [DOI] [PubMed] [Google Scholar]
- Hirner S, Krohne C, Schuster A, Hoffmann S, Witt S, Erber R, Sticht C, Gasch A, Labeit S, Labeit D. MuRF1-dependent regulation of systemic carbohydrate metabolism as revealed from transgenic mouse studies. Journal of Molecular Biology. 2008;379:666–677. doi: 10.1016/j.jmb.2008.03.049. [DOI] [PubMed] [Google Scholar]
- Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metabolism. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Hollis G, Huber R. 11Beta-hydroxysteroid dehydrogenase type 1 inhibition in type 2 diabetes mellitus. Diabetes, Obesity & Metabolism. 2011;13:1–6. doi: 10.1111/j.1463-1326.2010.01305.x. [DOI] [PubMed] [Google Scholar]
- Holt LJ, Lyons RJ, Ryan AS, Beale SM, Ward A, Cooney GJ, Daly RJ. Dual ablation of Grb10 and Grb14 in mice reveals their combined role in regulation of insulin signaling and glucose homeostasis. Molecular Endocrinology. 2009;23:1406–1414. doi: 10.1210/me.2008-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Turner N, Mokbel N, Trefely S, Kanzleiter T, Kaplan W, Ormandy CJ, Daly RJ, Cooney GJ. Grb10 regulates the development of fiber number in skeletal muscle. The FASEB Journal. 2012;26:3658–3669. doi: 10.1096/fj.11-199349. [DOI] [PubMed] [Google Scholar]
- Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- Hu J, Liu J, Ghirlando R, Saltiel AR, Hubbard SR. Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Molecular Cell. 2003;12:1379–1389. doi: 10.1016/s1097-2765(03)00487-8. [DOI] [PubMed] [Google Scholar]
- Hu SI, Katz M, Chin S, Qi X, Cruz J, Ibebunjo C, Zhao S, Chen A, Glass DJ. MNK2 inhibits eIF4G activation through a pathway involving serine-arginine-rich protein kinase in skeletal muscle. Science Signalling. 2012;5:ra14. doi: 10.1126/scisignal.2002466. [DOI] [PubMed] [Google Scholar]
- Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. Journal of Clinical Investigation. 2009;119:3059–3069. doi: 10.1172/JCI38770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, Pories WJ, MacDonald KG, Cline GW, Shulman GI, Dohm GL, Houmard JA. Skeletal muscle lipid metabolism with obesity. American Journal of Physiology, Endocrinology and Metabolism. 2003;284:E741–E747. doi: 10.1152/ajpendo.00514.2002. [DOI] [PubMed] [Google Scholar]
- Ikeda H. KK mouse. Diabetes Research and Clinical Practice. 1994;24(Suppl):S313–S316. doi: 10.1016/0168-8227(94)90268-2. [DOI] [PubMed] [Google Scholar]
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- Jang C, Obeyesekere VR, Dilley RJ, Alford FP, Inder WJ. 11Beta hydroxysteroid dehydrogenase type 1 is expressed and is biologically active in human skeletal muscle. Clinical Endocrinology (Oxf) 2006;65:800–805. doi: 10.1111/j.1365-2265.2006.02669.x. [DOI] [PubMed] [Google Scholar]
- Jang C, Obeyesekere VR, Dilley RJ, Krozowski Z, Inder WJ, Alford FP. Altered activity of 11beta-hydroxysteroid dehydrogenase types 1 and 2 in skeletal muscle confers metabolic protection in subjects with type 2 diabetes. Journal of Clinical Endocrinology and Metabolism. 2007;92:3314–3320. doi: 10.1210/jc.2006-2729. [DOI] [PubMed] [Google Scholar]
- Jogo M, Shiraishi S, Tamura TA. Identification of MAFbx as a myogenin-engaged F-box protein in SCF ubiquitin ligase. FEBS Letters. 2009;583:2715–2719. doi: 10.1016/j.febslet.2009.07.033. [DOI] [PubMed] [Google Scholar]
- Kamei Y, Miura S, Suzuki M, Kai Y, Mizukami J, Taniguchi T, Mochida K, Hata T, Matsuda J, Aburatani H, Nishino I, Ezaki O. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. Journal of Biological Chemistry. 2004;279:41114–41123. doi: 10.1074/jbc.M400674200. [DOI] [PubMed] [Google Scholar]
- Kola B, Christ-Crain M, Lolli F, Arnaldi G, Giacchetti G, Boscaro M, Grossman AB, Korbonits M. Changes in adenosine 5′-monophosphate-activated protein kinase as a mechanism of visceral obesity in Cushing’s syndrome. Journal of Clinical Endocrinology and Metabolism. 2008;93:4969–4973. doi: 10.1210/jc.2008-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, Chiba T, Doi N, Kitamura F, Tanaka K, Abe K, Witt SH, Rybin V, Gasch A, Franz T, Labeit S, Sorimachi H. Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. Journal of Molecular Biology. 2008;376:1224–1236. doi: 10.1016/j.jmb.2007.11.049. [DOI] [PubMed] [Google Scholar]
- Krozowski Z, Li KX, Koyama K, Smith RE, Obeyesekere VR, Stein-Oakley A, Sasano H, Coulter C, Cole T, Sheppard KE. The type I and type II 11beta-hydroxysteroid dehydrogenase enzymes. Journal of Steroid Biochemistry and Molecular Biology. 1999;69:391–401. doi: 10.1016/s0960-0760(99)00074-6. [DOI] [PubMed] [Google Scholar]
- Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2012 doi: 10.1073/pnas.1111334109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HS, Huang B, Unterman TG, Harris RA. Protein kinase B-alpha inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes. 2004;53:899–910. doi: 10.2337/diabetes.53.4.899. [DOI] [PubMed] [Google Scholar]
- Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, Segura CT, Leibovitch SA. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO Journal. 2008;27:1266–1276. doi: 10.1038/emboj.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, White MF. Insulin receptor substrate proteins and diabetes. Archives of Pharmacal Research. 2004;27:361–370. doi: 10.1007/BF02980074. [DOI] [PubMed] [Google Scholar]
- Leis H, Page A, Ramirez A, Bravo A, Segrelles C, Paramio J, Barettino D, Jorcano JL, Perez P. glucocorticoid receptor counteracts tumorigenic activity of Akt in skin through interference with the phosphatidylinositol 3-kinase signaling pathway. Molecular Endocrinology. 2004;18:303–311. doi: 10.1210/me.2003-0350. [DOI] [PubMed] [Google Scholar]
- Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nature Reviews Molecular Cell Biology. 2012;13:383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- Lutzner N, Kalbacher H, Krones-Herzig A, Rosl F. FOXO3 is a glucocorticoid receptor target and regulates LKB1 and its own expression based on cellular AMP levels via a positive autoregulatory loop. PLoS ONE. 2012;7:e42166. doi: 10.1371/journal.pone.0042166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Mallidis C, Artaza J, Taylor W, Gonzalez-Cadavid N, Bhasin S. Characterization of 5′-regulatory region of human myostatin gene: regulation by dexamethasone in vitro. American Journal of Physiology, Endocrinology and Metabolism. 2001;281:E1128–E1136. doi: 10.1152/ajpendo.2001.281.6.E1128. [DOI] [PubMed] [Google Scholar]
- Ma K, Mallidis C, Bhasin S, Mahabadi V, Artaza J, Gonzalez-Cadavid N, Arias J, Salehian B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. American Journal of Physiology, Endocrinology and Metabolism. 2003;285:E363–E371. doi: 10.1152/ajpendo.00487.2002. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metabolism. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metabolism. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Mauvais-Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ, Iannacone M, Accili D, Cantley LC, Kahn CR. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. Journal of Clinical Investigation. 2002;109:141–149. doi: 10.1172/JCI13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menconi M, Gonnella P, Petkova V, Lecker S, Hasselgren PO. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. Journal of Cellular Biochemistry. 2008;105:353–364. doi: 10.1002/jcb.21833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, Feigenbaum L, Lee E, Aoyama T, Eckhaus M, Reitman ML, Vinson C. Life without white fat: a transgenic mouse. Genes & Development. 1998;12:3168–3181. doi: 10.1101/gad.12.20.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SA, Sherlock M, Gathercole LL, Lavery GG, Lenaghan C, Bujalska IJ, Laber D, Yu A, Convey G, Mayers R, Hegyi K, Sethi JK, Stewart PM, Smith DM, Tomlinson JW. 11Beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes. 2009;58:2506–2515. doi: 10.2337/db09-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morissette MR, Cook SA, Foo S, McKoy G, Ashida N, Novikov M, Scherrer-Crosbie M, Li L, Matsui T, Brooks G, Rosenzweig A. Myostatin regulates cardiomyocyte growth through modulation of Akt signaling. Circulation Research. 2006;99:15–24. doi: 10.1161/01.RES.0000231290.45676.d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton NM. Obesity and corticosteroids: 11beta-hydroxysteroid type 1 as a cause and therapeutic target in metabolic disease. Molecular and Cellular Endocrinology. 2010;316:154–164. doi: 10.1016/j.mce.2009.09.024. [DOI] [PubMed] [Google Scholar]
- Nair S, Lee YH, Lindsay RS, Walker BR, Tataranni PA, Bogardus C, Baier LJ, Permana PA. 11Beta-hydroxysteroid dehydrogenase Type 1: genetic polymorphisms are associated with Type 2 diabetes in Pima Indians independently of obesity and expression in adipocyte and muscle. Diabetologia. 2004;47:1088–1095. doi: 10.1007/s00125-004-1407-6. [DOI] [PubMed] [Google Scholar]
- Nakao R, Hirasaka K, Goto J, Ishidoh K, Yamada C, Ohno A, Okumura Y, Nonaka I, Yasutomo K, Baldwin KM, Kominami E, Higashibata A, Nagano K, Tanaka K, Yasui N, Mills EM, Takeda S, Nikawa T. Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Molecular and Cellular Biology. 2009;29:4798–4811. doi: 10.1128/MCB.01347-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakken GN, Jacobs DL, Thomson DM, Fillmore N, Winder WW. Effects of excess corticosterone on LKB1 and AMPK signaling in rat skeletal muscle. Journal of Applied Physiology. 2010;108:298–305. doi: 10.1152/japplphysiol.00906.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Mikura M, Hirasaka K, Okumura Y, Nikawa T, Kawano Y, Nakayama M, Ikeda M. Effects of dimethyl sulphoxide and dexamethasone on mRNA expression of myogenesis- and muscle proteolytic system-related genes in mouse myoblastic C2C12 cells. Journal of Biochemistry. 2008;144:717–724. doi: 10.1093/jb/mvn126. [DOI] [PubMed] [Google Scholar]
- Ohshima K, Shargill NS, Chan TM, Bray GA. Effects of dexamethasone on glucose transport by skeletal muscles of obese (ob/ob) mice. International Journal of Obesity. 1989;13:155–163. [PubMed] [Google Scholar]
- Orzechowski A, Jank M, Gajkowska B, Sadkowski T, Godlewski MM, Ostaszewski P. Delineation of signalling pathway leading to antioxidant-dependent inhibition of dexamethasone-mediated muscle cell death. Journal of Muscle Research and Cell Motility. 2003;24:33–53. doi: 10.1023/a:1024887431768. [DOI] [PubMed] [Google Scholar]
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires-Oliveira M, Maragno AL, Parreiras-e-Silva LT, Chiavegatti T, Gomes MD, Godinho RO. Testosterone represses ubiquitin ligases atrogin-1 and Murf-1 expression in an androgen-sensitive rat skeletal muscle in vivo. Journal of Applied Physiology. 2010;108:266–273. doi: 10.1152/japplphysiol.00490.2009. [DOI] [PubMed] [Google Scholar]
- Pivonello R, De Leo M, Vitale P, Cozzolino A, Simeoli C, De Martino MC, Lombardi G, Colao A. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology. 2010;92(Suppl 1):77–81. doi: 10.1159/000314319. [DOI] [PubMed] [Google Scholar]
- Polge C, Heng AE, Jarzaguet M, Ventadour S, Claustre A, Combaret L, Bechet D, Matondo M, Uttenweiler-Joseph S, Monsarrat B, Attaix D, Taillandier D. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. The FASEB Journal. 2011;25:3790–3802. doi: 10.1096/fj.11-180968. [DOI] [PubMed] [Google Scholar]
- Puthucheary Z, Montgomery H, Moxham J, Harridge S, Hart N. Structure to function: muscle failure in critically ill patients. Journal of Physiology. 2010;588:4641–4648. doi: 10.1113/jphysiol.2010.197632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase kinase 3: more than a namesake. British Journal of Pharmacology. 2009;156:885–898. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddi AS, Camerini-Davalos RA. Hereditary diabetes in the KK mouse: an overview. Advances in Experimental Medicine and Biology. 1988;246:7–15. doi: 10.1007/978-1-4684-5616-5_2. [DOI] [PubMed] [Google Scholar]
- Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, Yao W, Hollis G, Flores R, Levy R, Williams WV, Seckl JR, Huber R. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care. 2010;33:1516–1522. doi: 10.2337/dc09-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzin J, Wagman AS, Jensen J. Glucocorticoid-induced insulin resistance in skeletal muscles: defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor. Diabetologia. 2005;48:2119–2130. doi: 10.1007/s00125-005-1886-0. [DOI] [PubMed] [Google Scholar]
- Saad MJ, Folli F, Kahn JA, Kahn CR. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. Journal of Clinical Investigation. 1993;92:2065–2072. doi: 10.1172/JCI116803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehzadeh F, Al-Khalili L, Kulkarni SS, Wang M, Lonnqvist F, Krook A. Glucocorticoid-mediated effects on metabolism are reversed by targeting 11 beta hydroxysteroid dehydrogenase type 1 in human skeletal muscle. Diabetes/Metabolism Research and Reviews. 2009;25:250–258. doi: 10.1002/dmrr.944. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance. Unravelling the mechanism. Lancet. 2010;375:2267–2277. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schakman O, Gilson H, Thissen JP. Mechanisms of glucocorticoid-induced myopathy. Journal of Endocrinology. 2008;197:1–10. doi: 10.1677/JOE-07-0606. [DOI] [PubMed] [Google Scholar]
- Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- Shah OJ, Kimball SR, Jefferson LS. Among translational effectors, p70S6k is uniquely sensitive to inhibition by glucocorticoids. Biochemical Journal. 2000;347:389–397. doi: 10.1042/0264-6021:3470389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda S, Nakae J, Tagata Y, Nishitani S, Takehana K, Sano M, Fukuda K, Suematsu M, Morimoto C, Tanaka H. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metabolism. 2011;13:170–182. doi: 10.1016/j.cmet.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Shipp LE, Lee JV, Yu CY, Pufall M, Zhang P, Scott DK, Wang JC. Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS ONE. 2010;5:e13754. doi: 10.1371/journal.pone.0013754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Molecular Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. American Journal of Physiology, Endocrinology and Metabolism. 2003;284:E855–E862. doi: 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- Sun L, Trausch-Azar JS, Muglia LJ, Schwartz AL. Glucocorticoids differentially regulate degradation of MyoD and Id1 by N-terminal ubiquitination to promote muscle protein catabolism. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3339–3344. doi: 10.1073/pnas.0800165105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P. Widespread negative response elements mediate direct repression by agonist- liganded glucocorticoid receptor. Cell. 2011;145:224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- Tappy L, Randin D, Vollenweider P, Vollenweider L, Paquot N, Scherrer U, Schneiter P, Nicod P, Jequier E. Mechanisms of dexamethasone-induced insulin resistance in healthy humans. Journal of Clinical Endocrinology and Metabolism. 1994;79:1063–1069. doi: 10.1210/jcem.79.4.7962275. [DOI] [PubMed] [Google Scholar]
- Tintignac LA, Lagirand J, Batonnet S, Sirri V, Leibovitch MP, Leibovitch SA. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. Journal of Biological Chemistry. 2005;280:2847–2856. doi: 10.1074/jbc.M411346200. [DOI] [PubMed] [Google Scholar]
- Tobimatsu K, Noguchi T, Hosooka T, Sakai M, Inagaki K, Matsuki Y, Hiramatsu R, Kasuga M. Overexpression of the transcriptional coregulator Cited2 protects against glucocorticoid-induced atrophy of C2C12 myotubes. Biochemical and Biophysical Research Communications. 2009;378:399–403. doi: 10.1016/j.bbrc.2008.11.062. [DOI] [PubMed] [Google Scholar]
- Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocrine Reviews. 2004;25:831–866. doi: 10.1210/er.2003-0031. [DOI] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. American Journal of Physiology. Cell Physiology. 2009;296:C1258–C1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- Vander Kooi BT, Onuma H, Oeser JK, Svitek CA, Allen SR, Vander Kooi CW, Chazin WJ, O’Brien RM. The glucose-6-phosphatase catalytic subunit gene promoter contains both positive and negative glucocorticoid response elements. Molecular Endocrinology. 2005;19:3001–3022. doi: 10.1210/me.2004-0497. [DOI] [PubMed] [Google Scholar]
- Verhees KJ, Schols AM, Kelders MC, Op den Kamp CM, van der Velden JL, Langen RC. Glycogen synthase kinase-3beta is required for the induction of skeletal muscle atrophy. American Journal of Physiology. Cell Physiology. 2011;301:C995–C1007. doi: 10.1152/ajpcell.00520.2010. [DOI] [PubMed] [Google Scholar]
- Vestergaard H, Bratholm P, Christensen NJ. Increments in insulin sensitivity during intensive treatment are closely correlated with decrements in glucocorticoid receptor mRNA in skeletal muscle from patients with Type II diabetes. Clinical Science (Lond) 2001;101:533–540. doi: 10.1042/cs1010533. [DOI] [PubMed] [Google Scholar]
- Waddell DS, Baehr LM, van den Brandt J, Johnsen SA, Reichardt HM, Furlow JD, Bodine SC. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. American Journal of Physiology, Endocrinology and Metabolism. 2008;295:E785–E797. doi: 10.1152/ajpendo.00646.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh FS, Celeste AJ. Myostatin: a modulator of skeletal-muscle stem cells. Biochemical Society Transactions. 2005;33:1513–1517. doi: 10.1042/BST0331513. [DOI] [PubMed] [Google Scholar]
- Wang H, Kubica N, Ellisen LW, Jefferson LS, Kimball SR. Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. Journal of Biological Chemistry. 2006;281:39128–39134. doi: 10.1074/jbc.M610023200. [DOI] [PubMed] [Google Scholar]
- Watson ML, Baehr LM, Reichardt HM, Tuckermann JP, Bodine SC, Furlow JD. A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. American Journal of Physiology, Endocrinology and Metabolism. 2012;302:E1210–E1220. doi: 10.1152/ajpendo.00512.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein SP, Wilson CM, Pritsker A, Cushman SW. Dexamethasone inhibits insulin-stimulated recruitment of GLUT4 to the cell surface in rat skeletal muscle. Metabolism. 1998;47:3–6. doi: 10.1016/s0026-0495(98)90184-6. [DOI] [PubMed] [Google Scholar]
- Welle S, Mehta S, Burgess K. Effect of postdevelopmental myostatin depletion on myofibrillar protein metabolism. American Journal of Physiology, Endocrinology and Metabolism. 2011;300:E993–E1001. doi: 10.1152/ajpendo.00509.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorwood CB, Donovan SJ, Flanagan D, Phillips DI, Byrne CD. Increased glucocorticoid receptor expression in human skeletal muscle cells may contribute to the pathogenesis of the metabolic syndrome. Diabetes. 2002;51:1066–1075. doi: 10.2337/diabetes.51.4.1066. [DOI] [PubMed] [Google Scholar]
- Whorwood CB, Donovan SJ, Wood PJ, Phillips DI. Regulation of glucocorticoid receptor alpha and beta isoforms and type I 11beta-hydroxysteroid dehydrogenase expression in human skeletal muscle cells: a key role in the pathogenesis of insulin resistance? Journal of Clinical Endocrinology and Metabolism. 2001;86:2296–2308. doi: 10.1210/jcem.86.5.7503. [DOI] [PubMed] [Google Scholar]
- Williams DB, Wan Z, Frier BC, Bell RC, Field CJ, Wright DC. Dietary supplementation with vitamin E and C attenuates dexamethasone-induced glucose intolerance in rats. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2012;302:R49–R58. doi: 10.1152/ajpregu.00304.2011. [DOI] [PubMed] [Google Scholar]
- Wu WL, Gan WH, Tong ML, Li XL, Dai JZ, Zhang CM, Guo XR. Overexpression of NYGGF4 (PID1) inhibits glucose transport in skeletal myotubes by blocking the IRS1/PI3K/AKT insulin pathway. Molecular Genetics and Metabolism. 2011;102:374–377. doi: 10.1016/j.ymgme.2010.11.165. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhao W, Zhao J, Zhang Y, Qin W, Pan J, Bauman WA, Blitzer RD, Cardozo C. REDD1 is a major target of testosterone action in preventing dexamethasone-induced muscle loss. Endocrinology. 2010;151:1050–1059. doi: 10.1210/en.2009-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Menconi MJ, Wei W, Petkova V, Hasselgren PO. Dexamethasone upregulates the expression of the nuclear cofactor p300 and its interaction with C/EBPbeta in cultured myotubes. Journal of Cellular Biochemistry. 2005;94:1058–1067. doi: 10.1002/jcb.20371. [DOI] [PubMed] [Google Scholar]
- Yang H, Wei W, Menconi M, Hasselgren PO. Dexamethasone-induced protein degradation in cultured myotubes is p300/HAT dependent. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2007;292:R337–R344. doi: 10.1152/ajpregu.00230.2006. [DOI] [PubMed] [Google Scholar]
- Yin HN, Chai JK, Yu YM, Shen CA, Wu YQ, Yao YM, Liu H, Liang LM, Tompkins RG, Sheng ZY. Regulation of signaling pathways downstream of IGF-I/insulin by androgen in skeletal muscle of glucocorticoid-treated rats. Journal of Trauma. 2009;66:1083–1090. doi: 10.1097/TA.0b013e31817e7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CY, Mayba O, Lee JV, Tran J, Harris C, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS ONE. 2010;5:e15188. doi: 10.1371/journal.pone.0015188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, McFarlane C, Lokireddy S, Bonala S, Ge X, Masuda S, Gluckman PD, Sharma M, Kambadur R. Myostatin-deficient mice exhibit reduced insulin resistance through activating the AMP-activated protein kinase signalling pathway. Diabetologia. 2011;54:1491–1501. doi: 10.1007/s00125-011-2079-7. [DOI] [PubMed] [Google Scholar]
- Zhao W, Qin W, Pan J, Wu Y, Bauman WA, Cardozo C. Dependence of dexamethasone-induced Akt/FOXO1 signaling, upregulation of MAFbx, and protein catabolism upon the glucocorticoid receptor. Biochemical and Biophysical Research Communications. 2009;378:668–672. doi: 10.1016/j.bbrc.2008.11.123. [DOI] [PubMed] [Google Scholar]
- Zheng B, Ohkawa S, Li H, Roberts-Wilson TK, Price SR. FOXO3a mediates signaling crosstalk that coordinates ubiquitin and atrogin-1/MAFbx expression during glucocorticoid-induced skeletal muscle atrophy. The FASEB Journal. 2010;24:2660–2669. doi: 10.1096/fj.09-151480. [DOI] [PMC free article] [PubMed] [Google Scholar]