

Abstract
Smoking is associated with increased incidence of chronic pain. However, the evidence is cross‐sectional in nature, and underlying mechanisms remain unclear. In a longitudinal observational study, we examined the relationship between smoking, transition to chronic pain, and brain physiology. In 160 subjects with subacute back pain (SBP: back pain lasting 4–12 weeks, and no prior back pain [BP] for at least 1 year) pain characteristics, smoking status, and brain functional properties were measured repeatedly over 1 year. Sixty‐eight completed the study, subdivided into recovering (SBPr, n = 31) and persisting (SBPp, n = 37), based on >20% decrease in BP over the year. Thirty‐two chronic back pain (CBP: duration > 5 years) and 35 healthy controls were similarly monitored. Smoking prevalence was higher in SBP and CBP but not related to intensity of BP. In SBP, smoking status at baseline was predictive of persistence of BP 1 year from symptom onset (differentiating SBPp and SBPr with 0.62 accuracy). Smoking status combined with affective properties of pain and medication use improved prediction accuracy (0.82). Mediation analysis indicated the prediction of BP persistence by smoking was largely due to synchrony of fMRI activity between two brain areas (nucleus accumbens and medial prefrontal cortex, NAc‐mPFC). In SBP or CBP who ceased smoking strength of NAc‐mPFC decreased from precessation to postcessation of smoking. We conclude that smoking increases risk of transitioning to CBP, an effect mediated by corticostriatal circuitry involved in addictive behavior and motivated learning. Hum Brain Mapp 36:683–694, 2015. © 2014 Wiley Periodicals, Inc.
Keywords: smoking, chronic pain, back pain, fMRI, accumbens, prefrontal cortex
INTRODUCTION
Chronic pain affects one in five adults [Harstall and Ospina, 2003] and has a staggeringly high annual cost [Medicine, 2011]. Back pain (BP) is one of the most common, disabling, and expensive chronic conditions, and compared to all other injuries and disabilities, it is the leading cause of years lived with disability in the United States, and the seventh leading cause worldwide [Murray and Lopez, 2013]. Many risk factors are associated with chronic back pain (CBP), including cigarette smoking [Andersson, 1999; Andersson et al., 1998; Heliovaara et al., 1991; Leino‐Arjas et al., 1998; Nagasu et al., 2007]. Strong associations have been repeatedly identified between smoking and CBP across multiple cross‐sectional studies, independent of other associated risk factors like obesity and activity level [Alkherayf et al., 2010; Deyo and Bass, 1989; Wright et al., 1995], and two meta‐analyses confirm this association [Goldberg et al., 2000; Shiri et al., 2010]. However, whether the relationship between smoking and the development of CBP is causal and whether this relationship is relevant to pain management strategies remains largely unknown.
End organ abnormalities have been the main focus of the search for causes for development of chronic pain, yet no dominant factors have emerged. For example, the probability that the cause of BP can be identified by spine radiography is less than 1% [van den Bosch et al., 2004]. Similarly, mechanisms that may explain the role of smoking in pain chronification have also been studied mainly from this peripheral and end organ viewpoint. High levels of circulating nicotine have been shown to decrease peripheral perfusion through vasoconstriction, causing damage to intervertebral disks [Uematsu et al., 2001]. Passive smoking in rats affects the gene expression of collagen and metalloproteinase‐1 in the intervertebral disks, and induces histologic changes of the nucleus pulposus and the anulus fibrosus [Uei et al., 2006]. Furthermore, smoking is shown to increase the circulation of systemic proinflammatory cytokines that may promote hyperalgesia [O'Loughlin et al., 2008; Watkins and Maier, 2000; Yanbaeva et al., 2007]. Nevertheless, studies in both smoking and non‐smoking human volunteers indicate nicotine has a moderate analgesic effect in multiple experimental paradigms [Fertig et al., 1986; Girdler et al., 2005; Pomerleau et al., 1984], and nicotine deprivation is associated with acute withdrawal hyperalgesia [Nastase et al., 2007; Pauli et al., 1993; Pomerleau et al., 1984], suggesting smoking may be a habit acquired to ameliorate ongoing pain rather than a contributing factor. Thus, peripheral effects of smoking remain equivocal.
Central mechanisms that support a role for smoking in the development of chronic pain have not been investigated. Given that smoking is a highly addictive behavior, it certainly engages brain addiction related pathways [Jasinska et al., 2014; Tolu et al., 2013; Zhou et al., 2001], and given that a longitudinal brain imaging study in subacute back pain (SBP: back pain lasting 4–12 weeks, and no prior BP for at least 1 year) indicates that components of this same circuitry, specifically information shared between nucleus accumbens (NAc) and medial prefrontal cortex (mPFC), are causally related to pain chronification [Baliki et al., 2012], we hypothesize smoking is related to pain chronification and that this relationship is mediated by the functional properties of the corticostriatal circuitry. This study addresses these hypotheses.
METHOD
Study Design
These data are derived from a longitudinal, brain imaging‐based observational study. The study was comprised of five visits over the course of 1 year. The first visit (designated baseline) consisted of a screening encounter, including a physical exam and medical assessment performed by a qualified physician. At each visit pain intensity was determined using a 100 mm visual analog scale (VAS; minimum rating represents “no pain,” maximum rating represents “worst pain imaginable”) and behavioral questionnaires were completed. At all visits after baseline (visits 1–4) structural and functional magnetic resonance imaging (MRI) brain scans were additionally collected. CBP subjects of more than 5 years duration, as well as healthy controls, were recruited for the study as positive and negative controls, respectively. All participants underwent identical procedures. The study was approved by the Institutional Review Board of Northwestern University, and informed consent was obtained prior to the initiation of study procedures.
Subjects
Participants were recruited through advertisements from the Chicago city area. CBP and healthy control subjects were also recruited. Of the 32 CBP at entry, 24 completed the study; while of 35 controls, 19 finished. Of the 160 SBP subjects screened (Table 1), 123 were recruited, with a subsequent retention rate of 61% (n = 68) at 1 year. The 68 SBP subjects who completed the study were divided into persisting (SBPp n = 38) and recovering (SBPr n = 31) groups based on whether or not pain decreased by 20% or more over 1 year (Table 2). A validation cohort was obtained by contacting 19 SBP subjects who had been screened and were eligible for the above study but elected to not enroll. Based on change in BP from interview to follow‐up phone call approximately 3 years later, 11 had pain decrease by >20% (SBPr'), while eight had not (SBPp').
Table 1.
Demographics, pain, and mood parameter differences between healthy controls (CON), SBP, and CBP, at baseline
| SBP N = 160 | CBP N = 32 | CON N = 35 | F‐value (P) | |
|---|---|---|---|---|
| Education (years) | 14.2 ± 0.197 | 13.8 ± 0.428 | 15.1 ± 0.533 | 1.92 (0.149) |
| Income (bracket) | 2.55 ± 0.1 | 2.34 ± 0.2 | 2.92 ± 0.2 | 1.98 (0.141) |
| PANAS (+) | 33.2 ± 0.688 | 31.1 ± 1.52 | 35.3 ± 1.5 | 1.35 (0.261) |
| PANAS (−) | 17.9 ± 0.556 | 20.5 ± 1.24 | 13.2 ± 0.73 | 11.4 (2 e − 5)* |
| BDI | 6.83 ± 0.434 | 8.65 ± 0.932 | 2.2 ± 0.539 | 13.3 (4 e − 6)* |
| MPQa | 2.92 ± 0.240 | 4.28 ± 0.530 | — | 4.48 (0.036) |
| MPQs | 13.4 ± 0.49 | 14.2 ± 0.893 | — | 0.451 (0.503) |
| Radiculopathy | 5.38 ± 0.178 | 5.78 ± 0.536 | — | 0.752 (0.387) |
| pDetect | 12.5 ± 0.575 | 13.8 ± 0.972 | — | 0.790 (0.375) |
| NPS | 46.1 ± 1.24 | 47.1 ± 2.76 | — | 0.0160 (0.900) |
| PDI | 27.7 ± 1.12 | 31.6 ± 1.90 | — | 2.52 (0.114) |
Clinical characteristics are shown as mean ± s.e.m, determined at baseline. *Corrected P < 0.05. Uncorrected P‐values are shown. Of the 32 CBP at entry 14 were female and group mean age was 45.1 ± 1.3 (s.e.m.). Of the 35 CON at entry 20 were female and group mean age was 36.2 ± 1.3 (s.e.m). Of the 160 SBP subjects, 80 were female, and group mean age was 42.06 ± 0.86 (s.e.m.); pain duration in weeks at baseline: 8.87 ± 4.5 (s.d.).
Table 2.
Demographics, pain, and mood parameters in SBPp and SBPr, at baseline
| SBPp N = 38 | SBPr N = 31 | t‐score (P) | |
|---|---|---|---|
| Education (yrs) | 14.0 ± 0.351 | 15.1 ± 0.448 | −1.92 (0.059) |
| Income (bracket) | 2.30 ± 0.159 | 2.90 ± 0.222 | −2.26 (0.027) |
| BDI | 6.79 ± 0.732 | 6.87 ± 0.931 | −0.0642 (0.949) |
| PANAS (+) | 33.6 ± 1.08 | 34.1 ± 1.39 | −0.274 (0.785) |
| PANAS (−) | 18.3 ± 1.08 | 16.0 ± 0.981 | 1.59 (0.116) |
| MPQs | 14.4 ± 1.08 | 10.6 ± 0.675 | 2.92 (4.8 e − 3) |
| MPQa | 3.78 ± 0.537 | 1.65 ± 0.340 | 3.22 (2.0 e − 3)* |
| Radiculopathy | 5.92 ± 0.360 | 5.03 ± 0.309 | 1.83 (0.072) |
| NPS | 48.9 ± 2.29 | 42.6 ± 2.34 | 1.90 (0.062) |
| PDI | 27.2 ± 2.37 | 25.2 ± 2.32 | 0.590 (0.557) |
| painDETECTa | 14.1 ± 1.04 | 9.00 ± 0.985 | 3.42 (1.2 e − 3)* |
| MQS | 3.01 ± 0.352 | 5.35 ± 0.753 | −2.97 (4.1 e − 3)* |
Values are shown as mean ± SEM.
N = 53 (22 SBPr, 31 SBPp).
*Corrected P < 0.05. Uncorrected P‐values are shown. Of the 68 SBP patients who completed the study (pain duration in weeks at baseline: 9.38 ± 4.4, visit 1: 12.0 ± 5.16, visit 2: 19.05 ± 6.0, visit 3: 39.79 ± 7.5, visit 4: 55.63 ± 6.1, mean ± s.d.), 36 were male (mean age 43.56 ± 11.71, s.d.) and 32 were female (mean age 43.16 ± 9.58, s.d.). Within the SBP subgroups, SBPp had mean pain duration of 9.14 ± 4.21 weeks at interview, mean age 42.8 ± 12.0 years, and consisted of 19 females while SBPr had mean pain duration of 9.68 ± 4.71 weeks, age of 43.9 ± 9.6 and 13 females (mean ± s.d.). The 19 SBP validation group (not shown) was comprised of 8 females, had mean age 40.7 ± 11.6, mean pain duration at baseline of 8.6 ± 3.5 weeks and duration of 171.20 ± 30.9 weeks from symptom onset at time of follow up (mean ± s.d.).
Questionnaires
All questionnaires were self‐reported. For all visits, either at screening or within 1 h prior to scanning, SBP and CBP subjects completed the short form of the McGill pain questionnaire (MPQ; separated into sensory, MPQs, and affective, MPQa, components) [Melzack, 1987], the Neuropathic Pain Scale (NPS) [Galer and Jensen, 1997], painDETECT [Freynhagen et al., 2006], and Pain Disability Index (PDI) [Tait et al., 1987]. Radiculopathy scores were quantified from locations on a body map that patients had shaded in on the MPQ form to indicate the location of their pain [Chanda et al., 2011]. Additionally, depression score Beck's Depression Inventory (BDI), Positive Affective Negative Affect Score (PANAS) [Crawford and Henry, 2004] and demographic information including education, income, and smoking status were collected from all participants. The validation cohort completed all questionnaires at the screening visit. In a follow‐up phone call they were asked to rate their current BP verbally from 0 to 100, and were instructed to consider 0 as “no pain” and 100 as “the worst pain imaginable.” This pain rating scale was modeled after the scale included in the MPQ.
The MPQ was separated into eight sensory descriptors such as throbbing, shooting, stabbing, and splitting (MPQs), and the four affective descriptors tiring‐exhausting, sickening, fearful, and punishing‐cruel (MPQa). Subjects indicate whether their experience of these descriptors is none, mild, moderate, or severe, and these responses are coded as 0–3. The sum of MPQs or MPQa questions is taken as the total MPQs/a score (resp). The NPS quantifies extent of intensity, sharpness, dullness, hotness/coldness, skin sensitivity to touch, itchiness, unpleasantness, and depth of pain for a total of eight questions each rated on a scale from 0 to 10. painDETECT asks subjects to rate seven types of sensations like “burning sensations,” “tingling or prickling sensations,” and pain form “light touch” according to a six ordinal‐categories of intensity, which like MPQ is coded as 0–5 from least intense to most. PDI asks subjects to rate their disability from 0 to 10 in six aspects of their life: family/home responsibilities, recreation, social activity, occupation, sexual behavior, and life‐support activity like feeding and bathing. PANAS asks subjects to indicate on a five ordinal‐categorical scale the extent to which 20 descriptors apply to their current affect, descriptors such as interested, distressed, excited, upset, strong, and guilty without making reference to their pain specifically. Like MPQ scores, all these ordinal‐categorical responses are summed for each questionnaire to produce a total score for NPS, painDETECT, PDI, and PANAS for further statistical analysis.
Brain Imaging
Two types of scans, collected at visit 1, were analyzed for all 68 SBP who completed the study: MPRAGE type T1 anatomical images and fMRI data acquired at 3T. Brain data collection and analysis methods are described in detail in [Baliki et al., 2012]. The same preprocessing procedures were used here to extract strength of information sharing (functional connectivity) during a spontaneous pain rating task between NAc and mPFC, using coordinates derived from [Baliki et al., 2012] (functional connectivity designated as NAc‐mPFC; 6 × 6 × 6 mm areas were selected in the mPFC and NAc regions of interest, MNI coordinates 2,52,−2 and 10,12,−8, resp.). The BOLD timecourse was extracted from these ROIs and z‐Fisher transformed Pearson correlation coefficients were used to quantify connectivity strength, only between the two seeds.
Medication
Subjects participating in the study received no additional treatment, but we documented any treatment or medication they used for their pain. Drug consumption at each visit was quantified using the Medication Quantification Scale (MQS) [Harden et al., 2005].
Of the primary 68 SBP who completed the study, 42 took peripherally acting medications primarily aspirin, acetaminophen, NSAIDs (ibuprofen, naproxen), and steroid injections. Three took only centrally acting medications such as opiates (hydrocodone, oxycodone), the barbiturate butalbital, or centrally acting muscle relaxants (cyclobenzaprine). Fourteen took both centrally and peripherally acting medications. Finally, nine did not take any medications. Of the 16 SBP in the validation cohort 10 took peripherally acting medications primarily asprin, acetaminophen, and NSAIDs (ibuprofen, naproxen). Three took centrally acting medications: one was taking opiates (hydrocodone and morphine), another was taking SSRIs (paroxetine), and finally one was taking an anticonvulsant (pregabalin). Three were not taking any medication.
Statistics
We examined smoking between SBP, CBP, and controls using Fisher's exact test, two‐sided. ANCOVA was used, with gender and age as covariates, to identify differences in education, income, BDI, positive and negative PANAS. ANCOVA was additionally used to identify differences between CBP and SBP in MPQa and MPQs, radiculopathy, painDETECT, NPS, and PDI, none of which were collected in CON. Because we perform 14 comparisons a 5% false positive rate was enforced using Holm–Šidák correction for multiple comparisons.
One‐tailed t‐tests were used to evaluate if sensory (VAS, painDETECT, MPQs, NPS) or affective (MPQa) ratings of pain were different between SBP smokers and non‐smokers at baseline and visit 4. If smoking were analgesic it would suggest smoking behavior might be an effective substitute for medication use, so we also examined the relationship between MQS score and smoking. Lastly, some subjects reported smoking status at baseline but reported not smoking at some intermediate visit. Because nicotine withdrawal has been associated with acute hyperalgesia, we also examined change in VAS in these subjects between the first instance in which a smoking subject reported not smoking and the visit immediately prior using a one sample two‐tailed t‐test.
To test our a priori hypothesis, we investigated differences between persisting (SBPp) and recovering (SBPr) subjects. The divergence in pain (VAS ratings) between the two groups was quantified using a factorial repeated measure ANCOVA with gender and grouping as categorical factors and age as a continuous factor. We compared prevalence of smoking between SBPp and SBPr using Fisher's exact test, and generated a logistic regression model of BP persistence based on smoking status. We then compared the fit of this model to the fit of a logistic model which controlled for NAc‐mPFC connectivity, followed by formal mediation analysis.
Mediation analysis involved testing the relationship between NAc‐mPFC (mediator) and an independent variable (e.g. smoking) using linear regression, while probit regression was used to model the combined ability of NAc‐mPFC to predict pain outcome while controlling for independent variables. The product of standardized regression coefficients along the indirect path was taken as an estimated measure of the mediated effect size (e.g., α*β in Fig. 1C). Although reported odds ratios were derived from logistic models, the probit function is more appropriate for modeling dichotomous indicators of continuous latent variables, but more importantly allows for standard errors of the indirect effect to be estimated using a bias‐corrected bootstrap technique. This technique makes no assumptions about the otherwise highly skewed underlying statistical distributions of indirect effects, provides the most powerful method of testing for mediation and most importantly is the only robust method for estimating indirect effects using small samples [Mackinnon et al., 2004]. Bootstrapped 95% confidence intervals are reported. Mediation analysis was performed using Mplus 7.11.
Figure 1.
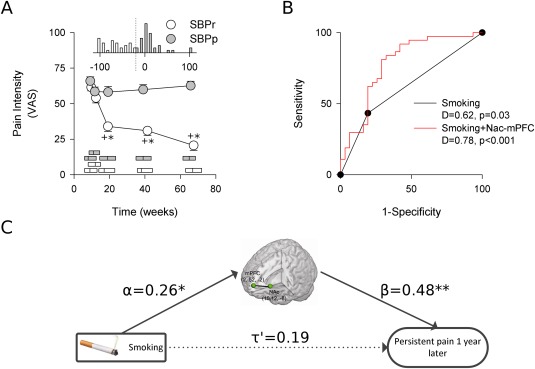
Smoking predicts pain persistence, mediated through brain functional connectivity of NAc‐mPFC. A) SBPr subjects (n = 31) report decreases in pain on a visual analog scale over the course of the study and ultimately differ significantly from SBPp subjects (n = 37). Horizontal bars show median and interquartile range of pain durations. Histogram insert shows distribution of percent pain change by visit 4 for each group. SBPp show a mean increase of 13% (median: 3% increase) while SBPr show variable pain decrease with 65% decrease on average (median: 68% decrease). Post hoc Tukey tests: +P < 0.001 group contrast at fixed time, *P < 0.001 within group comparison to visit 1. B) Smoking status at time of entry into the study was a significant predictor of pain persistence 1 year later. Modeling persistence in terms of smoking and NAc‐mPFC shows a significant improvement in predictive ability. Model output is a continuous variable, and a cut point needs to be selected to differentiate persisting from recovering subjects. Sensitivity and specificity is a function of such cut points. The ROC curve illustrates the entire spectrum of possible sensitivities and specificities for all unique cut points. D is discrimination accuracy, illustrated by AUC. Gray line indicates chance performance. C) Mediation analysis reveals smoking has a significant correlation with NAc‐mPFC. Although the direct effect of smoking on chronification is not significant, the indirect effect on pain chronification attributable to mediation by NAc‐mPFC is statistically significant. Standardized regression coefficients shown. Solid lines are significant effects. *P < 0.05, **P < 0.01.
We generated a more comprehensive model for predicting BP persistence in three steps [Hosmer and Lemeshow, 2000]. First, we identified parameters at baseline (gender, education, income, MQS, BDI, positive and negative PANAS, MPQs and MPQa, radiculopathy, NPS, PDI, and total painDETECT) that differentiated between SBP groups using a two‐sided t‐test for continuous parameters and Fisher's exact test for gender, correcting for multiple comparisons using the Holm–Šidák approach. Parameters that showed a significant difference between SBP groups at baseline were included in the preliminary model. Second, a preliminary model was generated using logistic multiple regression. Third, Wald's test was used to eliminate redundant parameters. We compared the final model fit to a model that also controlled for NAc‐mPFC, and followed up with formal mediation analysis. Comparisons between logistic models were performed using a χ2 test of likelihood ratios.
To inform the clinical relevance of the model, we investigated how well it performs when given input data from visits 1 to 4. We examined posterior probabilities and the receiver operator characteristic (ROC) curve for the model given data from each of these visits, and quantified performance in time using the area under the curve (AUC) as a measure of likelihood of correct discrimination. We further examined the effectiveness of the model in predicting outcome in the validation group according to the same measures.
To examine the effects of smoking cessation on functional connectivity, we identified SBP and CBP subjects who quit smoking after visit 1. Baseline connectivity data could not be obtained in subjects who quit smoking between baseline and visit 1, which excluded five SBP subjects from this analysis who had previously been used to examine effect of cessation on pain ratings. In addition to the remaining five SBP who quit between visits 1 and 4, one CBP subject quit between visits 3 and 4 and another three quit between visits 2 and 3. A paired t‐test was used to examine functional connectivity in these subjects to establish if NAc‐mPFC functional connectivity prior to cessation of smoking was higher than connectivity after cessation.
RESULTS
We have previously shown corticostriatal functional connectivity predicts who will persist with pain among the first 39 SBP subjects to complete the study [Baliki et al., 2012], and that this relationship is also captured by associated white matter tracks [Mansour et al., 2013]. Of 160 SBP enrolled, 68 SBP completed the study and are examined here together with concomitantly enrolled CBP and healthy controls. Consistent with previously published studies, we found a significant difference in prevalence of smoking at baseline between 160 SBP, 32 CBP, and 33 controls (Fisher's Exact test P = 0.003, df = 2). Controls and CBP in particular showed different prevalence of smoking (post hoc Fisher's exact test, P = 0.001; Table 3).
Table 3.
Contingency table for smoking in SBP, CBP, and controls (CON) at baseline
| CON | SBP | CBP | |
|---|---|---|---|
| Smoker | 4 | 49 | 16 |
| Non‐smoker | 29 | 111 | 16 |
To test the hypothesis that smoking might be analgesic to BP, we examined the relationship between pain and smoking in 160 SBP at baseline and in 68 SBP 1 year later. Smokers did not show reduced BP intensity as measured by VAS, and by various questionnaires scores (MPQa, MPQs, painDETECT, or NPS), at either time point. An alternative approach to test the analgesic efficacy of smoking is to examine its relationship with medication use. These two parameters were also not related, at baseline or 1 year later. Moreover, we found no evidence that cessation of smoking produced hyperalgesia in the 10 SBP who quit over the course of the study. Thus, there was no directly or indirectly measurable influence of active smoking on BP intensity.
In addition to smoking, we compared thirteen other clinical characteristics between the 160 SBP, CBP, and controls at baseline. The prevalence of a particular gender was not significantly different between groups. Neither were education, income or positive PANAS different between the three groups, however, negative PANAS and BDI were significantly different (F (2,219) = 11.4, P < 10−4; F (2,215) = 13.3, P < 10−5; resp.). Controls, who had the lowest mean BDI and negative PANAS scores, differed from SBP and CBP in both measures (Tukey test P < 10−3). CBP and SBP did not show any significant differences in MPQa, MPQs, painDETECT, NPS, or PDI (Table 1). These results illustrate how pain has a negative impact on a patient's affective state relative to controls, but there was no measurable difference in pain characteristics between SBP and CBP.
We next turned our attention to the SBP group specifically to test our a priori hypothesis that smoking should be a predictor of pain chronification and should be related to NAc‐mPFC functional connectivity. As expected, SBPp and SBPr subgroups showed significantly different progression of pain (VAS scores) in time, (rm‐ANCOVA group*time effect F (3,177) = 20.75, P < 10−6; group effect F (3,177) = 29.42, P = 10−6), but notably did not have different BP intensity or duration at baseline or visit 1 (Fig. 1A). Nevertheless, at baseline there were significant differences between the incidence of smoking in the two groups (Fisher's exact test, P = 0.042; Table 4), and a logistic model for persistence in terms of smoking further quantified its predictive value, with a discrimination accuracy (AUC) of 0.62 and odds ratio, OR, of 3.12 (Fig. 1B). Moreover, smokers showed greater NAc‐mPFC functional connectivity (one‐tailed t‐test, t = 2.17, P = 0.017). Including brain functional connectivity in the model significantly improved its fit (likelihood ratio test, χ2(1) = 11.0, P = 10−3), and reduced smoking effect to nonsignificance (P = 0.463; Table 5). Mediation analysis was used to formally infer if the effect of NAc‐mPFC on the predictive value of smoking could be attributed to an underlying interrelationship between smoking, corticostriatal connectivity, and pain persistence. Indeed, mediation analysis revealed that NAc‐mPFC was a significant mediator between smoking and pain persistence (indirect effect CI: [0.05, 0.66], mediation of 39% of total effect, nonsignificant direct effect, Fig. 1C). These analyses suggest NAc‐mPFC functional connectivity is part of the mechanism underlying the association between smoking and pain chronification, and thus, confirms our hypothesis.
Table 4.
Contingency table for smoking in SBPp and SBPr at baseline
| SBPr | SBPp | |
|---|---|---|
| Smoker | 6 | 16 |
| Non‐smoker | 25 | 21 |
Table 5.
Logistic models for pain persistence based on smoking, additional behavioral parameters and for mediation effect by brain NAc‐mPFC functional connectivity
| Parameter | OR (s.e.m.) | P | 95% CI |
|---|---|---|---|
| Model 1, smoking alone | |||
| Smoking | 3.17 (1.79) | 0.04 | [1.05, 9.57] |
| N = 68; AUC = 0.62; LR χ2 = 4.53, P = 0.03 | |||
| Model 2, smoking + NAc‐mPFC | |||
| Smoking | 1.56 (0.95) | 0.463 | [0.48, 5.14] |
| NAc‐mPFCa | 1.85 (0.37) | 0.002 | [1.25, 2.74] |
| Preliminary multiparameter model | |||
| Smoking | 5.86 (4.78) | 0.030 | [1.18, 29.01] |
| MPQa | 1.54 (0.34) | 0.052 | [1.00, 2.37] |
| painDETECT | 1.15 (0.11) | 0.137 | [0.96, 1.39] |
| MQS | 0.72 (0.11) | 0.028 | [0.54, 0.97] |
| N = 53; AUC = 0.89; LR χ2 = 27.8, P < 10−4 | |||
| Final multiparameter model | |||
| Smoking | 4.79 (3.21) | 0.019 | [1.29, 17.8] |
| MPQa | 1.48 (0.19) | 0.002 | [1.15, 1.92] |
| MQS | 0.771 (0.09) | 0.018 | [0.62, 0.96] |
| N = 68; AUC = 0.82; LR χ2 = 25.3, P < 10−4 | |||
| Final model + NAc‐mPFC | |||
| NAc‐mPFCa | 1.63 (0.35) | 0.021 | [1.08, 2.48] |
| Smoking | 2.96 (2.20) | 0.144 | [0.69, 12.70] |
| MPQa | 1.39 (0.19) | 0.013 | [1.07, 1.81] |
| MQS | 0.737 (0.10) | 0.019 | [0.57, 0.95] |
| N = 68; AUC = 0.86; LR χ2 = 31.8, P < 10−4 | |||
Odds Ratio represent a 0.1 increment in correlation between NAc‐mPFC, range: [−0.23,0.66].
To build a more comprehensive, multivariate model for predicting persistence, we first modeled persistence in terms of MQS, painDETECT, and MPQa scores together with smoking, because all three differentiated between SBPp and SBPr at baseline (Table 2). This resulting model showed high discriminative accuracy (0.89), with significant or borderline significant contributions from all factors except painDETECT (Table 5). Indeed, a regression analysis revealed painDETECT reflects much of the same information as MPQa (r 2 = 0.42, P > 10−4, n = 53). The data was refit without painDETECT, yielding our final predictive model which showed high discrimination accuracy (0.82), performance comparable to the preliminary model (likelihood ratio test, χ2(1) = 2.5, P = 0.11), a significant contribution from every constituent parameter, and identified smoking as a dominant risk factor (OR 4.79; Fig. 2, Table 5). This model could be applied to CBP subjects as well to determine if they show characteristics consistent with persistent pain, and a comparison of model posterior probabilities across SBPr, SBPp, and CBP showed a significant difference across groups and singled out SBPr as different from SBPp and CBP (ANOVA F = 17.1, P < 10−6; post hoc Tukey SBPr vs. CBP: P = 10−4, SBPr vs. SBPp: P = 10−4; Fig. 2B).
Figure 2.
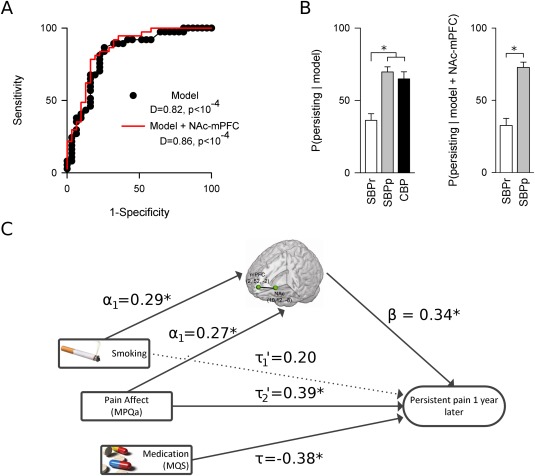
Three parameters smoking, pain affect, and medication use yield a highly predictive model for pain persistence, where smoking and pain affect are mediated through brain functional connectivity of NAc‐mPFC. A) ROC curve for the three‐parameter model shows baseline parameter values significantly and accurately (0.82) differentiate between SBPp and SBPr 1 year later. Inclusion of NAc‐mPFC in the model minimally improves its predictive abilities. D indicates AUC. Gray line represents chance classification. B) Although the behavioral model yields similar posterior probabilities for SBPp and CBP, both pain groups differ significantly from SBPr (left), suggesting the model should be stable as subjects become chronic. Posterior probabilities obtained by including NAc‐mPFC in the model are comparable to those obtained without it (right). Errors represent SEM. C) Mediation analysis reveals both smoking and pain affect predictions are significantly mediated by NAc‐mPFC connectivity, while medication use confers a protective effect. Standardized regression coefficients shown. Solid lines are significant effects. *P < 0.05.
To test the contribution of NAc‐mPFC in the multivariate model, we performed a mediation analysis. Because we have previously shown MPQa is related to NAc‐mPFC [Baliki et al., 2012], we expect NAc‐mPFC connectivity mediates the contributions of both smoking and MPQa. Controlling for brain connectivity significantly alters the fit of the model (likelihood ratio test, χ2(1) = 6.5, P = 0.01) but produces little improvement in predictive accuracy (AUC = 0.86; paired t‐test P(SBPp|model) vs. P(SBPp| model, NAc‐mPFC): t < 10−5, P > 0.99; Fig. 2), despite reducing the significance of the contribution from smoking (P = 0.114, Table 5). Mediation analysis identifies a significant indirect effect for both smoking (indirect effect CI: [0.03,0.72], mediation of 33% of total effect, nonsignificant direct effect) and MPQa (indirect effect CI: [0.01,0.11], mediation of 19% of total effect, direct effect CI: [0.04,0.39]).
The model showed consistency over time (Fig. 3A), which together with the comparison of posterior probabilities to CBP (Fig. 2B), suggests it captures stable traits. In addition, assessing persistence at 3 years from baseline in a small validation SBP group using this model yielded a discrimination accuracy of 0.68, and showed greater posterior probabilities in SBPp' compared to SBPr' (Fig. 3B). Overall, the model shows a consistent pattern in identifying subjects more vulnerable to pain chronification across groups and for different time intervals.
Figure 3.
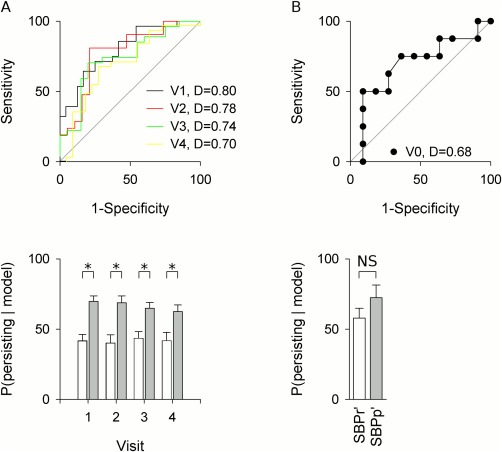
Longitudinal analysis and tests against a novel SBP group suggest model is stable and valid. A) The model was tested against data collected at visits 1–4. ROC analysis and posterior probabilities showed performance similar to that obtained at baseline. D indicates AUC. Gray line indicates chance performance. B) Testing the model on a validation group reveals consistent results despite poor model calibration. The three‐parameter model shows a predictive accuracy of 0.68 when tested in the validation group. The posterior probabilities within each group show reduced discriminative abilities, but continue to assess the likelihood of persistence of SBPr' lower than for SBPp'. Errors represent SEM. *P < 0.05.
The statistical interrelationship between NAc‐mPFC functional connectivity and smoking in predicting transition to chronic pain implies NAc‐mPFC functional connectivity strength should decrease in participants who ceased smoking. A small group of SBP and CBP (n = 9) participants ceased smoking at different times from start of the study. In this pooled group of SBP and CBP who quit smoking there was a significant reduction in NAc‐mPFC functional connectivity strength between before and after cessation of smoking (paired t‐test, t = 3.1 P = 0.007, mean difference: 0.17) (Fig. 4), consistent with our statistical models.
Figure 4.
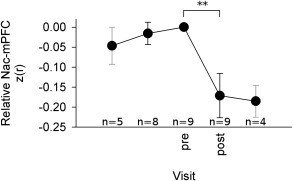
Smoking cessation coincides with a prominent decrease in NAc‐mPFC functional connectivity in a mixed group of SBP and CBP (n = 9). Functional connectivity was normalized with respect to the visit immediately preceding cessation, and measurements were aligned at the same visit (“pre”). Of these nine subjects, there were four CBP, three SBPp, one SBPr, and one SBP subject who was lost to follow‐up and could not be classified as persisting or recovering. The latter subject quit smoking between visits 1 and 2, three CBP quit smoking between visits 2 and 3, and the remaining subjects all quit between visits 3 and 4. This variability in cessation visit reduces the number of observations at visits distant from the date of cessation. Nevertheless, the only major source of variability throughout this period coincides with smoking cessation, which was statistically significant (P = 0.007, n = 9, paired t‐test). Mean ± SEM. Gray error bars indicate sparse data and limited inferential abilities. Number of observations indicated for each visit.
DISCUSSION
We demonstrate that smoking is an important risk factor for transitioning from SBP to CBP. Smoking status, when combined with affective properties of BP and with medication use, accurately and consistently predicts future rate of transition to chronic pain in discovery and validation SBP groups, as well as in CBP. Moreover, our results indicate smoking during the transition to chronicity is related to, and may be considered a surrogate marker for, brain functional connectivity between NAc and mPFC, a circuit critical for addictive behavior [Everitt and Robbins, 2005; Kalivas and McFarland, 2003; Shaham et al., 2003], motivated learning and pain chronification [Apkarian, 2008; Baliki et al., 2012]. Recent results in a whole brain analysis of resting state data in otherwise healthy subjects suggests such abnormally high connectivity between mPFC and ventral striatum may characterize smokers in general [Janes et al., 2012]. These results have broad clinical implications for the identification of individuals vulnerable to transitioning to CBP, including interventions to prevent this transition, and reveal the relative importance of central mechanisms in pain chronification.
It is remarkable that age, gender, education, and income do not seem to be critical factors in determining transition to chronic pain. Moreover, anxiety and depression were significantly higher in SBP and CBP as compared to healthy controls but did not increase with persistence of pain either in SBPp at 1 year compared to baseline nor in SBP compared to CBP. Such parameters have been repeatedly associated, mostly in cross‐sectional studies and based on recruitment from secondary or tertiary health clinics, with chronic pain, for example [Andersson, 1999; Andersson et al., 1998; Nagasu et al., 2007]. Here, we observe demographics, mood, and sensory properties of BP are not important influences on chronification of BP, when SBP and CBP are recruited from the population at large, and when SBPp and SBPr are matched for BP intensity, duration, and prior history at entry into the study. It is also possible that distinct vulnerabilities may underlie the transition to versus maintenance of chronic pain, or that additional other factors that were not collected may also influence transition to chronic pain.
Cigarette smoking is the major preventable cause of death and disability worldwide. In the United States it is responsible for 20% of deaths, and the economic burden of cigarette use is more than US$150 billion annually [Services, 2010]. Smoking is highly addictive, as best exemplified by human behavior. Although 70% of smokers in the United States report they want to quit, and approximately 44% report that they try to quit each year [Fiore and Jaen, 2008], of those who try, only 3–5% remain abstinent without the use of nicotine replacement therapies, and less than 30% are successful [Dome et al., 2010; Stead et al., 2008]. Smoking promotes addiction by activating nicotinic acetylcholine receptors in reward‐related dopamine systems [De Biasi and Dani, 2011; Jasinska et al., 2014; Koob and Volkow, 2010; Tolu et al., 2013; Zhou et al., 2001]. Interaction between NAc, the amygdala, and the prefrontal cortex is thought to mediate reinforcement for addiction [Everitt and Robbins, 2005] and drug relapse [Kalivas and McFarland, 2003; Shaham et al., 2003].
In contrast to the end organ viewpoint, present results show that smoking in SBP is related to the strength of NAc‐mPFC functional connectivity, and thus, it reflects the state of the brain corticostriatal circuitry in these individuals. We found no evidence that tobacco smoking had analgesic or hyperalgesic effects. However, we cannot rule out that smoking may also have an impact on end organ integrity. Conversely, our results provide strong evidence in support of our a priori hypothesis that smoking behavior is related to properties of the corticostriatal circuitry and that this relationship may be the primary reason for the association between smoking and CBP. Still, the specific mechanisms underlying the interaction between NAc‐mPFC connectivity and smoking behavior during pain chronification needs to be established. One possibility is that smoking is indicative of a pre‐existing enhanced corticostriatal functional connectivity. Alternatively, alterations to this circuitry following nicotine addiction could increase risk of pain chronification in smokers. Lastly, active smoking during the development of BP may potentiate NAc‐mPFC connectivity by cholinergic enhancement of excitability of corticostriatal circuitry. All three alternatives are consistent with studies highlighting the presence of increased levels of dopamine in NAc following acute exposure to nicotine [Brody, 2006; Zhou et al., 2001], and evidence of abnormal experiential reward prediction error signals within the mesocorticolimbic circuitry of smokers, including NAc and mPFC [Chiu et al., 2008]. Nevertheless, decreased functional connectivity in CBP and SBP who quit smoking establishes that this physiological property is at least partially reversible. The latter further suggests that cessation of smoking may reduce propensity to develop chronic pain, and recent evidence shows that smoking cessation diminishes BP intensity [Behrend et al., 2012]. Consistent with all three alternatives is the meta‐analysis result showing increased risk of chronic pain in current smokers than non‐smokers, and in current smokers compared to never smokers [Shiri et al., 2010]. However, we observe that smoking cessation decreases functional connectivity but not intensity of BP. This result suggests that functional connectivity is more directly linked to smoking, and that perhaps decrease in BP is a more delayed response or that BP intensity is a more crude measure and thus small changes remain undetected when the number of observations is small.
By defining recovering subjects as >20% decrease in BP over 1 year, we observe that SBPp remain at the same level of BP from entry to 1 year later, and SBPr show a large decrease in group mean BP. The divergence in BP intensity occurs early on (within 3 months), corresponds to a dichotomization of objectively measureable brain anatomy between groups [Mansour et al., 2013], and yields an SBPr group that continuously decreases in BP intensity to levels that clinically suggest relief from BP [van Tulder et al., 2000], and matches the trajectory for acute BP intensity identified in a systematic review of the literature [Pengel et al., 2003]. The resultant model indicates that the dominant factor for pain persistence is smoking, whereas medication use is protective. In addition, more affective properties of BP also indicate likelihood for persistence. The mediation analyses show how smoking and affective properties of BP reflect brain corticostriatal circuitry. The model should be validated in a larger population to establish clinical utility. Given that the participants were recruited from the population at large, this model also needs to be tested in the setting of a standard pain clinic, where depression might have a more important influence.
Multiple groups have developed tools for predicting chronification of BP, see review [Chou and Shekelle, 2010]. However, differences in definitions of risk factors, variability in thresholds used, populations studied, durations of assessment, and outcomes assessed, all of which are inconsistencies recognized in a prior systematic review [Hayden et al., 2009], obviate direct comparisons with the current model. Yet our model exhibits performance comparable to those reported by these other instruments (accuracy of our model ranges between 0.68 and 0.89, while for the studies reviewed by [Chou and Shekelle, 2010] the range is 0.72–0.92 across six different tools). The review [Chou and Shekelle, 2010] concluded that the influence of smoking was not significant for BP chronification; however, this conclusion was based on only three studies, with assessments lasting 3 to 6 months, and none of which used change in BP intensity as a primary outcome.
CONCLUSION
In a highly vulnerable subject population, SBP (55% exhibit persistent BP over 1 year), we observe that smoking is a major risk factor for transition to chronic pain. We also show this factor is mediated by corticostriatal brain properties. This is the first evidence explicitly linking smoking, pain chronification, and brain addiction/motivation circuitry. When smoking was incorporated in a logistic model, together with emotional properties of BP, and with medication use we obtain a simple and accurate predictive tool for pain chronification. The results suggest that smoking cessation may be a viable option to diminish propensity to transition to chronic pain.
Author Contributions
AVA, TJS, and MNB conceived of the study and its initial design. ST and TJS recruited participants, evaluated participant eligibility, and ensured participant retention. ST, HK, AM, BA, MNB collected questionnaire and brain imaging data. BP and MNB analyzed brain activity. BP, ST, AM analyzed questionnaire data. BP, JWG, GDO performed statistical analyses. BP, JWG performed mediation analyses. All authors contributed to and edited the report. All authors approved the report before submission.
ACKNOWLEDGMENTS
Todd Parrish, PhD, and Brian Williams for technical support. We thank all participants in the study, Apkarian lab members for reading and commenting on the manuscript. All authors declare no competing interests.
REFERENCES
- Alkherayf F, Wai EK, Tsai EC, Agbi C (2010): Daily smoking and lower back pain in adult Canadians: The Canadian Community Health Survey. J Pain Res 3:155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson GB (1999): Epidemiological features of chronic low‐back pain. Lancet 354:581–585. [DOI] [PubMed] [Google Scholar]
- Andersson H, Ejlertsson G, Leden I (1998): Widespread musculoskeletal chronic pain associated with smoking. An epidemiological study in a general rural population. Scand J Rehabil Med 30:185–191. [PubMed] [Google Scholar]
- Apkarian AV (2008): Pain perception in relation to emotional learning. Curr Opin Neurobiol 18:464–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, Fields HL, Apkarian AV (2012): Corticostriatal functional connectivity predicts transition to chronic back pain. Nat Neurosci 15:1117–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrend C, Prasarn M, Coyne E, Horodyski M, Wright J, Rechtine GR (2012): Smoking cessation related to improved patient‐reported pain scores following spinal care. J Bone Joint Surg Am 94:2161–2166. [DOI] [PubMed] [Google Scholar]
- Brody AL (2006): Functional brain imaging of tobacco use and dependence. J Psychiatr Res 40:404–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda ML, Alvin MD, Schnitzer TJ, Apkarian AV (2011): Pain characteristic differences between subacute and chronic back pain. J Pain 12:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu PH, Lohrenz TM, Montague PR (2008): Smokers' brains compute, but ignore, a fictive error signal in a sequential investment task. Nat Neurosci 11:514–520. [DOI] [PubMed] [Google Scholar]
- Chou R, Shekelle P (2010): Will this patient develop persistent disabling low back pain? JAMA 303:1295–1302. [DOI] [PubMed] [Google Scholar]
- Crawford JR, Henry JD (2004): The positive and negative affect schedule (PANAS): Construct validity, measurement properties and normative data in a large non‐clinical sample. Br J Clin Psychol, 43:245–265. [DOI] [PubMed] [Google Scholar]
- De Biasi M, Dani JA (2011): Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci 34:105–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyo RA, Bass JE (1989): Lifestyle and low‐back pain. The influence of smoking and obesity. Spine 14:501–506. [DOI] [PubMed] [Google Scholar]
- Dome P, Lazary J, Kalapos MP, Rihmer Z (2010): Smoking, nicotine and neuropsychiatric disorders. Neurosci Biobehav Rev 34:295–342. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW (2005): Neural systems of reinforcement for drug addiction: From actions to habits to compulsion. Nat Neurosci 8:1481–1489. [DOI] [PubMed] [Google Scholar]
- Fertig JB, Pomerleau OF, Sanders B (1986): Nicotine‐produced antinociception in minimally deprived smokers and ex‐smokers. Addict Behav 11:239–248. [DOI] [PubMed] [Google Scholar]
- Fiore MC, Jaen CR (2008): A clinical blueprint to accelerate the elimination of tobacco use. JAMA 299:2083–2085. [DOI] [PubMed] [Google Scholar]
- Freynhagen R, Baron R, Gockel U, Tolle TR (2006): Paindetect: A new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin 22:1911–1920. [DOI] [PubMed] [Google Scholar]
- Galer BS, Jensen MP (1997): Development and preliminary validation of a pain measure specific to neuropathic pain: The Neuropathic Pain Scale. Neurology 48:332–338. [DOI] [PubMed] [Google Scholar]
- Girdler SS, Maixner W, Naftel HA, Stewart PW, Moretz RL, Light KC (2005): Cigarette smoking, stress‐induced analgesia and pain perception in men and women. Pain 114:372–385. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Scott SC, Mayo NE (2000): A review of the association between cigarette smoking and the development of nonspecific back pain and related outcomes. Spine (Phila Pa 1976) 25:995–1014. [DOI] [PubMed] [Google Scholar]
- Harden RN, Weinland SR, Remble TA, Houle TT, Colio S, Steedman S, Kee WG (2005): Medication Quantification Scale Version III: Update in medication classes and revised detriment weights by survey of American Pain Society Physicians. J Pain 6:364–371. [DOI] [PubMed] [Google Scholar]
- Harstall C, Ospina M (2003): How prevalent is chronic pain? Pain Clin Updates 11:1–4. [Google Scholar]
- Hayden JA, Chou R, Hogg‐Johnson S, Bombardier C (2009): Systematic reviews of low back pain prognosis had variable methods and results: Guidance for future prognosis reviews. J Clin Epidemiol 62:781–796 e1. [DOI] [PubMed] [Google Scholar]
- Heliovaara M, Makela M, Knekt P, Impivaara O, Aromaa A (1991): Determinants of sciatica and low‐back pain. Spine (Phila Pa 1976) 16:608–614. [DOI] [PubMed] [Google Scholar]
- Hosmer DW, Lemeshow S (2000): Applied Logistic Regression, Vol. xii New York: Wiley; 373p. [Google Scholar]
- Janes AC, Nickerson LD, Bde BF, Kaufman MJ (2012): Prefrontal and limbic resting state brain network functional connectivity differs between nicotine‐dependent smokers and non‐smoking controls. Drug Alcohol Depend 125:252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasinska AJ, Zorick T, Brody AL, Stein EA (2014): Dual role of nicotine in addiction and cognition: A review of neuroimaging studies in humans. Neuropharmacology 84:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, McFarland K (2003): Brain circuitry and the reinstatement of cocaine‐seeking behavior. Psychopharmacology (Berl) 168:44–56. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND (2010): Neurocircuitry of addiction. Neuropsychopharmacology 35:217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leino‐Arjas P, Hanninen K, Puska P (1998): Socioeconomic variation in back and joint pain in Finland. Eur J Epidemiol 14:79–87. [DOI] [PubMed] [Google Scholar]
- Mackinnon DP, Lockwood CM, Williams J (2004): Confidence limits for the indirect effect: Distribution of the product and resampling methods. Multivariate Behav Res 39:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour AR, Baliki MN, Huang L, Torbey S, Herrmann KM, Schnitzer TJ, Apkarian AV (2013): Brain white matter structural properties predict transition to chronic pain. Pain 154:2160–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicine IO (2011): Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. In: Medicine IO, editor. Washington, DC: National Academies Press (US). [PubMed]
- Melzack R (1987): The short‐form McGill pain questionnaire. Pain 30:191–197. [DOI] [PubMed] [Google Scholar]
- Murray CJ, Lopez AD (2013): Measuring the global burden of disease. N Engl J Med 369:448–457. [DOI] [PubMed] [Google Scholar]
- Nagasu M, Sakai K, Ito A, Tomita S, Temmyo Y, Ueno M, Miyagi S (2007): Prevalence and risk factors for low back pain among professional cooks working in school lunch services. BMC Public Health 7:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nastase A, Ioan S, Braga RI, Zagrean L, Moldovan M (2007): Coffee drinking enhances the analgesic effect of cigarette smoking. Neuroreport 18:921–924. [DOI] [PubMed] [Google Scholar]
- O'Loughlin J, Lambert M, Karp I, McGrath J, Gray‐Donald K, Barnett TA, Delvin EE, Levy E, Paradis G (2008): Association between cigarette smoking and C‐reactive protein in a representative, population‐based sample of adolescents. Nicotine Tob Res 10:525–532. [DOI] [PubMed] [Google Scholar]
- Pauli P, Rau H, Zhuang P, Brody S, Birbaumer N (1993): Effects of smoking on thermal pain threshold in deprived and minimally‐deprived habitual smokers. Psychopharmacology (Berl) 111:472–476. [DOI] [PubMed] [Google Scholar]
- Pengel LH, Herbert RD, Maher CG, Refshauge KM (2003): Acute low back pain: Systematic review of its prognosis. BMJ 327:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerleau OF, Turk DC, Fertig JB (1984): The effects of cigarette smoking on pain and anxiety. Addict Behav 9:265–271. [DOI] [PubMed] [Google Scholar]
- Services U.S. Department of Health and Human Services (2010): A Report of the Surgeon General: How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking‐Attributable Disease. Washington, DC. [PubMed]
- Shaham Y, Shalev U, Lu L, De Wit H, Stewart J (2003): The reinstatement model of drug relapse: History, methodology and major findings. Psychopharmacology (Berl) 168:3–20. [DOI] [PubMed] [Google Scholar]
- Shiri R, Karppinen J, Leino‐Arjas P, Solovieva S, Viikari‐Juntura E (2010): The association between smoking and low back pain: A meta‐analysis. Am J Med 123:87 e7–e35. [DOI] [PubMed] [Google Scholar]
- Stead LF, Bergson G, Lancaster T (2008): Physician advice for smoking cessation. Cochrane Database Syst Rev:CD000165. [DOI] [PubMed] [Google Scholar]
- Tait RC, Pollard CA, Margolis RB, Duckro PN, Krause SJ (1987): The Pain Disability Index: Psychometric and validity data. Arch Phys Med Rehabil 68:438–441. [PubMed] [Google Scholar]
- Tolu S, Eddine R, Marti F, David V, Graupner M, Pons S, Baudonnat M, Husson M, Besson M, Reperant C, Zemdegs J, Pages C, Hay YA, Lambolez B, Caboche J, Gutkin B, Gardier AM, Changeux JP, Faure P, Maskos U (2013): Co‐activation of VTA DA and GABA neurons mediates nicotine reinforcement. Mol Psychiatry 18:382–393. [DOI] [PubMed] [Google Scholar]
- Uei H, Matsuzaki H, Oda H, Nakajima S, Tokuhashi Y, Esumi M (2006): Gene expression changes in an early stage of intervertebral disc degeneration induced by passive cigarette smoking. Spine (Phila Pa 1976) 31:510–514. [DOI] [PubMed] [Google Scholar]
- Uematsu Y, Matuzaki H, Iwahashi M (2001): Effects of nicotine on the intervertebral disc: An experimental study in rabbits. J Orthop Sci 6:177–182. [DOI] [PubMed] [Google Scholar]
- van den Bosch MA, Hollingworth W, Kinmonth AL, Dixon AK (2004): Evidence against the use of lumbar spine radiography for low back pain. Clin.Radiol 59:69–76. [DOI] [PubMed] [Google Scholar]
- van Tulder MW, Scholten RJ, Koes BW, Deyo RA (2000): Nonsteroidal anti‐inflammatory drugs for low back pain: A systematic review within the framework of the Cochrane Collaboration Back Review Group1. Spine 25:2501–2513. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF (2000): The pain of being sick: Implications of immune‐to‐brain communication for understanding pain. Annu Rev Psychol 51:29–57. [DOI] [PubMed] [Google Scholar]
- Wright D, Barrow S, Fisher AD, Horsley SD, Jayson MI (1995): Influence of physical, psychological and behavioural factors on consultations for back pain. Br J Rheumatol 34:156–161. [DOI] [PubMed] [Google Scholar]
- Yanbaeva DG, Dentener MA, Creutzberg EC, Wesseling G, Wouters EF (2007): Systemic effects of smoking. Chest 131:1557–1566. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA (2001): Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci 4:1224–1229. [DOI] [PubMed] [Google Scholar]