

ABSTRACT
This report elucidates an E-cadherin-based force-transduction pathway that triggers changes in cell mechanics through a mechanism requiring epidermal growth factor receptor (EGFR), phosphoinositide 3-kinase (PI3K), and the downstream formation of new integrin adhesions. This mechanism operates in addition to local cytoskeletal remodeling triggered by conformational changes in the E-cadherin-associated protein α-catenin, at sites of mechanical perturbation. Studies using magnetic twisting cytometry (MTC), together with traction force microscopy (TFM) and confocal imaging identified force-activated E-cadherin-specific signals that integrate cadherin force transduction, integrin activation and cell contractility. EGFR is required for the downstream activation of PI3K and myosin-II-dependent cell stiffening. Our findings also demonstrated that α-catenin-dependent cytoskeletal remodeling at perturbed E-cadherin adhesions does not require cell stiffening. These results broaden the repertoire of E-cadherin-based force transduction mechanisms, and define the force-sensitive signaling network underlying the mechano-chemical integration of spatially segregated adhesion receptors.
KEY WORDS: E-cadherin, Integrin, Mechanotransduction, Traction force microscopy, Magnetic twisting cytometry, Cell signaling
Summary: Force transduction at E-cadherin junctions operates in addition to unfurling of cadherin-associated α-catenin. This pathway requires EGFR and triggers the downstream activation of integrin-dependent cell contractility.
INTRODUCTION
The transduction of mechanical stimuli to alter intracellular biochemical signals enables cells to modulate cell and tissue physiology in response to force fluctuations propagated through the cell environment or to changes in endogenous contractile, protrusive or compressive forces. Mechanotransduction involves an increasing repertoire of identified force-sensitive proteins found in both cells and the extracellular matrix (ECM), and cell surface adhesion complexes are common force-transducing elements. Integrins and their ECM ligands are well-established force transducers (Geiger and Yamada, 2011). Intercellular adhesive complexes not only regulate cell-to-cell cohesion and tissue barrier integrity, but also transduce force to instruct cell functions, alter cell shape and regulate tissue organization (Desai et al., 2013; Ladoux et al., 2010; le Duc et al., 2010; Leckband and de Rooij, 2014; Lecuit et al., 2011; Liu et al., 2010; Tepass et al., 2002; Tzima et al., 2005; Yonemura et al., 2010). Fluctuations in tension also trigger junction remodeling in endothelia and in epithelia, and these changes contribute to the mechanical reinforcement of adhesions (Barry et al., 2014, 2015; Liu et al., 2010; Thomas et al., 2013; Yonemura et al., 2010). Studies increasingly reveal how mechanotransduction at cell-to-cell adhesions in vivo impacts upon physiology in different mechanical contexts, such as at interendothelial junctions near regions of disturbed flow and during morphogenesis (Hahn and Schwartz, 2009; Schluck et al., 2013; Weber et al., 2012).
The rudiments of intercellular mechanotransduction mechanisms have been identified in only a few cases (Barry et al., 2014; Collins et al., 2012; Kim et al., 2015; le Duc et al., 2010; Tzima et al., 2005; Yonemura et al., 2010). E-cadherin complexes at epithelial intercellular junctions are force sensitive (Barry et al., 2014; le Duc et al., 2010; Thomas et al., 2013; Yonemura et al., 2010), and α-catenin is an identified force-transducing protein in these complexes (Yonemura et al., 2010). α-Catenin is a crucial mechanical link between homophilic intercellular E-cadherin bonds and the actin cytoskeleton (Barry et al., 2014; Buckley et al., 2014; Cavey et al., 2008; Desai et al., 2013; Nagafuchi et al., 1991). Experimental evidence supports a mechanism in which the force-dependent exposure of a cryptic binding site in α-catenin recruits vinculin, and enables localized actin polymerization through the Mena–VASP complex associated with vinculin (Barry et al., 2014; Buckley et al., 2014; le Duc et al., 2010; Leerberg et al., 2014; Thomas et al., 2013; Yao et al., 2014; Yonemura et al., 2010). This mechanism is consistent with measured force-activated changes in the viscoelasticity of E-cadherin adhesions (le Duc et al., 2010), but the stiffening response could also reflect additional force-transduction mechanism(s).
Force-independent cadherin ligation is well known to activate a number of signaling molecules including Src, phosphoinositide 3-kinase (PI3K), and Rho GTPases (Kovacs et al., 2002; McLachlan et al., 2007; McLachlan and Yap, 2007; Noren et al., 2003; Perez et al., 2008; Ratheesh et al., 2013; Tabdili et al., 2012a; Watanabe et al., 2009). Prior studies have also shown that passive E-cadherin ligation to E-cadherin-coated beads, without mechanical perturbation, altered focal adhesions through a mechanism that involved Src and PI3K (Jasaitis et al., 2012). However, a completely open question is whether mechanical perturbations activate any of these same signals. Moreover, the details of possible force-activated signaling pathway(s), the impact of signals on other adhesion proteins in the cell, and their relationship to force- and E-cadherin-dependent changes in measured cell mechanics have yet to be determined.
This current study identified an additional E-cadherin-based mechanotransduction mechanism that activates signal cascades that increase cell stiffness through integrin activation. Use of magnetic twisting cytometry (MTC), traction force microscopy (TFM) and fluorescence imaging identified a force-actuated, E-cadherin-ligand-specific signaling cascade that activates distant integrins and global cell contraction. By identifying early signaling cascades in E-cadherin mechanotransduction, these findings provide new insight into correlations between epithelial junction maturation and focal adhesions (Mertz et al., 2013), and elaborate potential details of signaling underlying force independent integrin–cadherin crosstalk (Al-Kilani et al., 2011; Jasaitis et al., 2012). Importantly, this study establishes an additional E-cadherin-based mechanotransduction mechanism, beyond proximal α-catenin conformation switching and local actin remodeling, that coordinates with integrins to regulate cell stiffening.
RESULTS
Force loading E-cadherin receptors affects cell traction forces
E-cadherin-mediated mechanotransduction triggers local vinculin recruitment and actin polymerization at force-loaded E-cadherin receptors (Barry et al., 2014; Kim et al., 2015; le Duc et al., 2010; Yonemura et al., 2010). This local cytoskeletal remodeling coincides with increased viscoelasticity of mechanically perturbed E-cadherin adhesions (le Duc et al., 2010). Here, we tested whether force-activated E-cadherin signals could also alter cell mechanics and possibly other adhesion proteins. Combining MTC with TFM (Fig. 1A), we first quantified effects of E-cadherin loading on global cell contractility and focal adhesion remodeling.
Fig. 1.

E-cadherin-based mechanotransduction alters cell traction and focal adhesions. (A) Illustration of the experimental setup combining magnetic twisting cytometry (MTC) and traction force microscopy (TFM). An oscillating magnetic field H generates a torque T, which displaces the magnetic beads. The amplitude of the bead displacement reflects the viscoelastic modulus of the bead–cell junction. Determined changes in cell stiffness or traction changes used cells with single beads, and excluded the majority of cells with multiple beads or beads at cell–cell contacts. (B) Time sequence of steps in combined MTC and TFM measurements. (C) Bar graph indicating changes in traction force (with or without load) exerted by MCF7 cells on collagen-coated polyacrylamide gels with elastic moduli of 8.8 kPa (−Load, n=7 cells; +Load, n=19 cells) and 34 kPa (−Load, n=11 cells, +Load, n=11 cells). (D) Bar graph showing changes in cell traction after force-loading beads modified with E-cadherin (E-cad, n=11 cells), poly-L-lysine (PLL, n=18 cells), neutral anti-E-cadherin antibody (Ntrl Ab, n=8 cells), or blocking anti-E-cadherin antibody (DECMA-1, n=8 cells). (E) Bar graph indicating traction changes (ΔRMS traction, Pa) after force-loading E-cadherin beads on cells adhered to collagen (n=11 cells), PLL (n=9 cells), or E-cadherin-coated polyacrylamide gels (34 kPa, n=7 cells). Results obtained with PLL-coated beads on cells adhered to PLL substrata are also shown (n=6 cells). With cells on either PLL- or E-cadherin-coated substrata, the medium contained integrin-blocking antibodies GOH3 and AIIB2. In C–E, the black bar denotes the same data used for statistical comparisons. Data presented are the mean±s.e.m. *P<0.01 (Student's t-test). Two or more independent experiments were performed. (F) Representative confocal immunofluorescence images of vinculin (green) and actin (gold) at the basal plane of cells on collagen-functionalized hydrogels. Cells were probed with E-cadherin (top) and DECMA-1 (bottom) functionalized beads, with (+Load) and without (−Load) 2 min of force loading. Scale bar: 10 µm.
TFM quantifies changes in the root mean square traction forces (ΔRMS traction), from displacement maps of fiduciary beads embedded in hydrogel substrata relative to positions prior to cell perturbations (Butler et al., 2002). Passive ligation of beads modified with recombinant E-cadherin ectodomains (Prakasam et al., 2006) to E-cadherin receptors on MCF7 cells on collagen-coated gels (−Load, Fig. 1B,C) increased cell traction, as reported previously (Jasaitis et al., 2012). However, applying an oscillating shear stress (force/area) of 8.4 Pa on E-cadherin bead/cell junctions triggered substantial increases in traction forces, from 144±18 Pa (−Load) to 249±31 Pa (+Load, Fig. 1B,C, Table 1). On fibronectin substrates, we similarly observed a 66% increase in ΔRMS traction (P<0.001, Table 1). The force-activated increase exceeded force-independent changes. Representative traction heat maps are in Fig. S1A and values are summarized in Table 1. Distributions of the magnitudes of the local substrate strain vectors illustrate the changes in cell traction following E-cadherin receptor loading (Fig. S1B). Force-loading E-cadherin receptors shifted the mode of the traction stress distribution from 25–50 pN (−Load) to 50–75 pN (+Load).
Table 1.
Effect of bead coating, substrate modulus, and substrate coating on the RMS traction forces
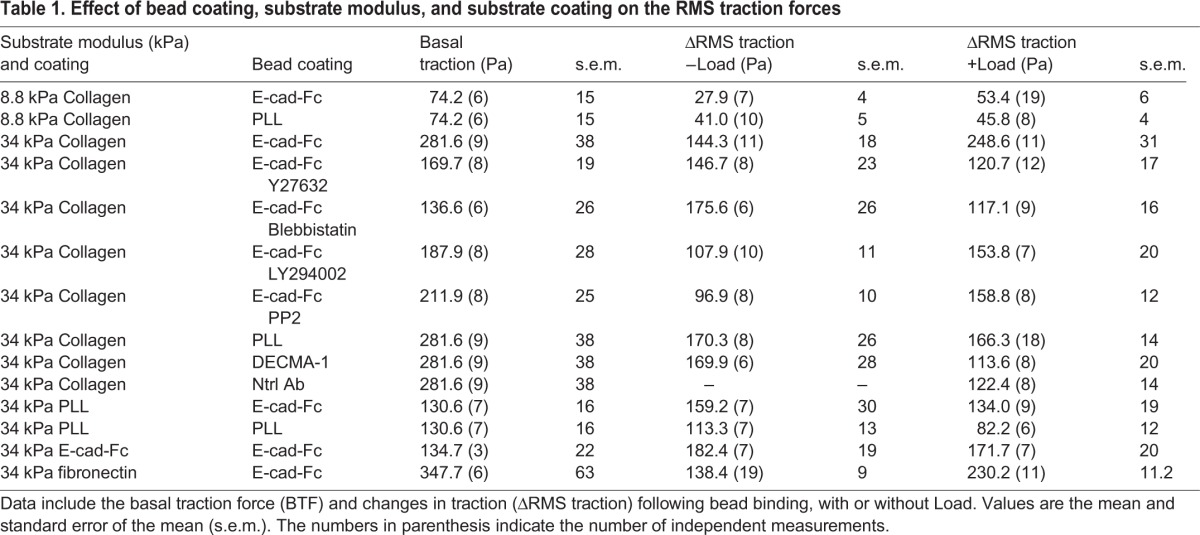
Use of different magnetic bead coatings demonstrated the E-cadherin-ligand specificity of the force-actuated changes in cell traction (Fig. 1D). Force-loading E-cadherin beads generated significantly greater traction increases than beads coated with (1) poly-L-lysine (PLL), (2) a neutral, non-blocking anti-E-cadherin antibody (Petrova et al., 2012) or (3) the blocking anti-E-cadherin antibody DECMA-1 (Ozawa et al., 1990) (Fig. 1D). The ΔRMS traction changes thus required specific E-cadherin ligation.
The traction changes also depended on the substrate coating (Fig. 1E). With cells cultured on PLL-coated gels, in the presence of integrin-blocking antibodies, E-cadherin force loading increased ΔRMS traction by only 134±19 Pa (mean±s.e.m.; Fig. 1E; Table 1). Force-loading PLL beads on cells on PLL-coated gels increased the ΔRMS traction by 82±12 Pa. Surprisingly, on E-cadherin-coated gels, the ΔRMS traction of 172±20 Pa was statistically similar to cells on PLL substrata (Fig. 1E, Table 1).
E-cadherin-specific mechanotransduction triggers focal adhesion remodeling
To determine whether force transduction signals altered focal adhesions, we quantified the number and area of focal adhesions (Gonon et al., 2005; Zamir et al., 1999) based on analyses of immunofluorescence images of vinculin at the basal plane. Focal adhesions were distributed mainly at the cell perimeter (Fig. 1F). Upon loading E-cadherin beads, the average focal adhesion area and number increased by 35% and 20%, respectively, relative to controls (−Load) (Fig. S1C). Controls with DECMA-1-coated beads, which do not trigger adaptive stiffening (Tabdili et al., 2012b) or changes in traction forces (Fig. 1D), did not alter focal adhesions significantly (Fig. S1C). Thus, focal adhesion remodeling was activated by ligand-dependent E-cadherin mechanotransduction and was not due to general tugging on cell membranes. These combined MTC and TFM measurements thus demonstrated that, in addition to proximal α-catenin-dependent cytoskeletal remodeling (Barry et al., 2014; le Duc et al., 2010; Yonemura et al., 2010), force-activated E-cadherin-mediated signals alter distal integrin-mediated traction forces.
E-cadherin-mediated adaptive stiffening requires the formation of new integrin adhesions
MTC measurements confirmed that cells exhibit E-cadherin-mediated adaptive stiffening when cultured on ECM proteins on either glass or on 34 kPa gels. Applying a modulated, orthogonal magnetic field induces a twisting torque, which displaces the beads. The bead displacements, D, are related to the storage G′ and loss G″ moduli of the bead–cell junctions by: T/D=G′+G″. In these studies, G″ did not change; decreasing displacement D reflected an increase in G′, or the stiffness (Wang et al., 1993). Changes in the percentage stiffness are reported relative to the initial modulus of the unperturbed junction. Perturbing E-cadherin beads with a shear stress of 7.2 Pa (force/area) increased MCF7 cell stiffness by ∼35% on glass and gels (Fig. 2A). Control, PLL-coated beads failed to induce cell stiffening (Fig. 2A).
Fig. 2.

E-cadherin-mediated adaptive stiffening requires integrin adhesion. (A) The mean±s.e.m. percentage stiffness change versus time during E-cadherin force loading, with cells on collagen-coated glass (black triangles, n=116 beads) or 34 kPa gels (black squares, n=67 beads). Control with poly-L-lysine (PLL) beads (black circles, n=27 beads) is also shown. Two independent experiments were performed for all conditions. (B) The mean±s.e.m. percentage stiffness change after 2 min of force loading with E-cadherin beads attached to cells adhered to 34-kPa gels coated with fibronectin (FN, n=9 cells), E-cadherin (E-cad, n=16 cells), poly-L-lysine (PLL, n=22 cells), or collagen (Col, n=13 cells). The negative control was with PLL beads bound to cells on PLL-coated gels (34 kPa) (n=16 cells). The x-axis labels indicate ‘bead coating/substrate coating’. *P<0.05 (Student's t-test). Two independent experiments were performed. (C) Representative confocal immunofluorescence images of vinculin (green, top) and actin (gold, bottom) in untreated MCF7 cells, or cells treated with function-blocking antibodies 16G3 or AIIB2 for 25 min. Scale bar: 10 µm. (D) The mean±s.e.m. cell area after brief antibody exposure is plotted for comparison with C (untreated, n=82 cells; AIIB2, n=79 cells; 16G3 treated, n=80 cells). (E) The mean±s.e.m. percentage stiffness change after 2 min of E-cadherin force loading on MCF7 cells subject to different treatments. Data shown are for untreated MCF7 cells on collagen-coated glass (black bar, n=116 beads), cells treated with antibodies [AIIB2 (n=83 beads), 16G3 (n=80 beads), or 13G12 (n=56 beads)] for 25 min prior to E-cadherin force loading, and cells treated for 25 min with 0.5 mM MnCl2 chloride (n=49 beads). *P<0.05 (Student's t-test). Two independent experiments were performed.
In endothelial cells, adaptive cell stiffening triggered by PECAM-1 requires the formation of new integrin adhesions (Collins et al., 2012). To test whether E-cadherin-mediated epithelial cell stiffening similarly requires integrins, MTC measurements were conducted with cells on different matrix proteins, in the presence of 10% serum (Fig. 2B). On fibronectin and on collagen I (Col I) substrata, E-cadherin bond loading increased cell stiffness by 23±2% and 32±5% (mean±s.e.m.), respectively. On PLL-coated gels in the presence of integrin-blocking antibodies AIIB2 and GOH3, the stiffness change was −3±5%. Somewhat surprisingly, with cells seeded on E-cadherin-coated gels, the stiffness change was −12±8%. Integrin-blocking antibodies were also included in the latter measurements.
To further test whether stiffening required the formation of new integrin adhesions, cells cultured on matrix-coated glass were treated with integrin- or fibronectin-blocking antibodies for 25 min, immediately prior to E-cadherin force loading. This brief antibody treatment (25-min exposure) did not affect the average spread area of cells or existing focal adhesions (Fig. 2C,D) (Collins et al., 2012; Hall et al., 1990). In cells treated with anti-β1 integrin AIIB2, or with anti-fibronectin antibody 16G3, the stiffening responses were −4±5% and 3±4%, respectively (Fig. 2E). The non-blocking, control (for 16G3) antibody 13G12 (Nagai et al., 1991) did not significantly alter cell stiffening (Fig. 2E).
The results with integrin function-blocking antibodies suggest that integrin conversion into an active conformation triggers increased cell contractility, such that the amplitude of the stiffening response would depend on the population of integrins undergoing this transition. We tested whether reducing the pool of integrins activated by E-cadherin-mediated signals would reduce the stiffening amplitude by pre-activating integrins with MnCl2 (0.5 mM) (Cluzel et al., 2005; Mould, 2002). This treatment reduced the force-dependent stiffening from 32±5% to 12±5% (Fig. 2E). This treatment also generated a higher (than untreated cells) basal traction force of 315±22 Pa, consistent with integrin activation.
Integrin activation downstream of E-cadherin force transduction signals was further verified using the GST–fibronectin-III9-11 peptide, which specifically binds activated α5β1 integrin (Orr et al., 2006; Tzima et al., 2001). Quantitative comparisons of immunofluorescence images of whole cells revealed a force-dependent 34±14% increase in peptide labeling, relative to unperturbed (−Load) controls (Fig. 3A,B). Western blots indicated a 200±10% increase in peptide binding (Fig. 3C,D). The different amplitudes of these changes reflect differences in the sensitivities of the two assays, but both results confirmed integrin activation, relative to unperturbed controls.
Fig. 3.

E-cadherin-force transduction activates α5β1 integrin. (A) Representative fluorescence images of cells treated with the integrin activation marker GST–fibronectin (FN)-III9-11, after force loading, washing and then staining with anti-GST antibody. (B) The mean±s.e.m. fluorescence intensity (MFI) of whole cells stained with anti-GST-antibody, with or without load (−Load, n=112 beads; +Load, n=104 beads). Two independent experiments were performed. Data were normalized to the −Load condition. (C) Representative western blots for GST–fibronectin-III9-11 and actin. Mechanically stimulated cells were treated with GST-fibronectin-III9-11, lysed, and bound GST-fibronectin-III9-11 was assessed by blotting for GST. Two independent experiments were performed. (D) Comparison (mean±s.e.m.) of peptide binding in C with or without load. Blots were normalized to actin, and intensities were relative to the −Load condition. *P<0.01 (Student's t-test).
Force-activated E-cadherin-mediated signals required for traction changes
Diverse, independent studies have implicated non-muscle myosin II (MyII, also known as myosin-2), PI3K, Src and/or Rho-associated protein kinases (ROCK1 and ROCK2) in force-independent (−Load) crosstalk between cadherins and integrins, as well as in PECAM-1-mediated mechanotransduction (Collins et al., 2012; Jasaitis et al., 2012; Schwartz and DeSimone, 2008; Weber et al., 2011). Cell treatment with blebbistatin or with the ROCK inhibitor Y27632 reduced the load-dependent ΔRMS traction by 53% and 51%, respectively, relative to untreated cells (Fig. 4A,B). These traction changes were comparable to negative controls with antibody-coated beads (Table 1). Src or PI3K inhibitors reduced the load-dependent ΔRMS traction by 36% and by 38%, respectively, relative to untreated cells (Fig. 4B). Table 1 also summarizes the inhibitor effects on the basal traction (without beads).
Fig. 4.

Force-activated, E-cadherin-dependent cell stiffening requires Src, PI3K, myosin and ROCK. (A) Time sequence for measurements of the impact of inhibitors on ΔRMS Traction and adaptive stiffening. (B) The mean±s.e.m. change in the root mean square traction forces (ΔRMS Traction, Pa), after force-loading E-cadherin beads bound to cells treated with inhibitors of ROCK1 and ROCK2 (Y27632, n=12 cells), myosin II (Blebbistatin, n=9 cells), Src kinase (PP2, n=8 cells), or PI3K (LY294002, n=7 cells). Two independent experiments were performed. (C) The mean±s.e.m. percentage stiffness change, after 2 min of force-loading E-cadherin beads bound to cells treated with inhibitors of myosin II (blebbistatin, n=43 beads), ROCK1 (Y27632, n=33 beads), Src kinase (PP2, n=39 beads), PI3K (LY294002, n=22 beads; Wortmanin, n=47 beads). For the DMSO controls, n=50 beads, and more than 50 beads were analyzed in the PP3 controls. Two independent experiments were performed. *P<0.05 (Student's t-test).
The effects of inhibitors on adaptive stiffening were more pronounced (Fig. 4C). Blebbistatin (targeting MyII) and Y27632 (targeting ROCK1 and ROCK2) reduced adaptive stiffening from 32±5% to 5±6% and 0±3% (mean±s.e.m.), respectively. The Src inhibitor PP2 reduced the stiffening response to 6±4%. The PI3K inhibitors LY294002 and Wortmannin reduced the stiffening response to −4±6 and 7±5%, respectively. The Wortmannin concentration used reportedly does not affect other kinases or phospholipase A2 (Fruman et al., 1998; Orr et al., 2006).
Mechanical perturbation of E-cadherin does not increase Src activity
E-cadherin ligation (−Load) activates Src kinase (McLachlan et al., 2007), and the Src inhibitor PP2 inhibited E-cadherin actuated cell stiffening (Fig. 4C). However, visualizing changes with a FRET-based, membrane-associated KRas-Src reporter (Wang et al., 2005) indicated that E-cadherin force-loading did not enhance Src activity. The measured CFP-to-YFP emission ratio reports Src activity (Wang et al., 2005). In regions of interest (ROIs) around beads, the load-independent (−Load) Src activity peaked ∼30 min after bead ligation, and then dropped to initial levels within ∼45 min (data not shown). To minimize contributions from force-independent signaling, we measured the CFP-to-YFP ratio during force loading, at 40 min after bead ligation. However, ∼30 s of E-cadherin receptor loading did not significantly increase the CFP-to-YFP ratio, relative to the no-load control (Fig. S2). Thus, mechanically perturbing E-cadherin receptors does not increase Src activity, but a pool of active Src appears to be essential for E-cadherin-mediated cell stiffening.
Force-loading E-cadherin activates PI3K upstream of integrins
To test whether mechanically stimulated E-cadherin receptors activated PI3K, we quantified the accumulation of the PI3K reporter PH-Akt–GFP in ROIs around E-cadherin beads, relative to controls with PLL beads or cells expressing GFP (Fig. 5A–E). Force-loading E-cadherin resulted in a 41±0.1% (mean±s.e.m.) increase in the normalized, mean fluorescence intensity (Fig. 5A–F). Treatment with integrin or fibronectin-blocking antibodies abrogated adaptive stiffening (Fig. 2E), but not PH-Akt–GFP accumulation at force-loaded E-cadherin beads: mean fluorescence intensities (MFIs) increased by 54±0.1% and by 49±0.1% in cells treated with AIIB2 (Fig. 5F) or 16G3, respectively (Fig. 5D–F). GFP did not accumulate at E-cadherin beads, and PH-Akt–GFP did not accumulate at PLL beads (Fig. 5B,F). In addition, phosphorylation of endogenous Akt (at S473) increased by 21% in western blots of cell monolayers stimulated with E-cadherin beads, compared with a 2.6% change measured with PLL bead controls (data not shown). These results suggest that mechanically perturbed E-cadherin receptors activate PI3K upstream from integrins.
Fig. 5.

E-cadherin-dependent force transduction activates PI3K. (A–E) Representative confocal fluorescence images of PH-Akt–GFP-transfected cells with (+Load) and without (−Load) force-loading beads (white arrows indicate bead locations) coated with (A) E-cadherin (E-cad) or (B) PLL. (C) Controls used E-cadherin beads and cells expressing GFP. (D,E) PH-Akt-GFP recruitment to E-cadherin beads on MCF7 cells treated with (D) integrin-blocking antibody AIIB2, and (E) fibronectin-blocking antibody 16G3. Only cells laden with single beads were used for data analyses. (F) Bar graph of the normalized mean±s.e.m. fluorescence intensity of PH-Akt–GFP in ROIs surrounding E-cadherin beads, without force-loading (No Load, white) and after 4 min of force loading (+Load, black). Values were normalized to the No Load condition. Cells were grown on collagen-coated glass. Results show the normalized fluorescence in the presence of anti-integrin antibody AIIB2 (−Load, n=158 beads; +Load, n=186 beads, *P<0.001, Student's t-test), Gefitinib (−Load, n=205 beads; +Load, n=169 beads, *P=0.5, Student's t-test), or anti-EGF antibody (−Load, n=44 beads; +Load, n=50 beads, P=0.15, Student's t-test). Controls were with PLL-coated beads (−Load, n=133 beads; +Load, n=124 beads) or with cells expressing GFP (−Load, n=138 beads; +Load, n=106 beads). Two independent experiments were performed for all conditions. (G) The mean±s.e.m. percentage stiffness change after 2 min of E-cadherin bead loading for untreated cells (black) or cells treated with anti-EGF antibody (n=71) or Gefitinib (n=70). DMSO control is also shown (n=65). Two independent experiments were performed. *P<0.001 (Student's t-test).
To determine whether focal adhesion remodeling and cell stiffening corresponded with basal increases in PI3K activity, we imaged PH-Akt–GFP in regions of interest around focal adhesions at the basal plane. Despite changes in focal adhesion number and size, the sensor did not reveal any detectable force-dependent changes in PH-Akt–GFP distributions at the basal plane, above background.
MCF7 cells express a mutant form of PI3K (Vasudevan et al., 2009). Thus, similar studies were performed with MDCK cells – a non-tumorigenic epithelial cell line, which exhibits E-cadherin-mediated adaptive cell stiffening (Barry et al., 2014). With MDCK cells, PH-Akt–GFP similarly accumulated at force-loaded E-cadherin beads, relative to controls (Fig. S3).
EGFR is required for E-cadherin-mediated PI3K activation and adaptive stiffening
Receptor tyrosine kinases, which associate with different cadherins, activate PI3K (Cully et al., 2006), and VEGFR2 and EGFR co-immunoprecipitate with, respectively, VE-cadherin (Coon et al., 2015) and E-cadherin (Pece and Gutkind, 2000; Perrais et al., 2007). To test whether EGFR is required for E-cadherin-mediated PI3K activation and cell stiffening, cells were treated with the EGFR-specific inhibitor Gefitinib (Paez et al., 2004). At concentrations that block EGFR transphosphorylation (Yen et al., 2015), Gefitinib abrogated MCF7 cell stiffening, relative to vehicle controls (DMSO) or untreated cells (Fig. 5G). MDCK cell treatment reduced adaptive stiffening from 26±5% (mean±s.e.m.; n=47 cells, more than three experiments) to 9±3% (P<0.05; n=60 cells, three experiments). Gefitinib also abrogated PH-Akt–GFP recruitment to force-loaded E-cadherin beads on both MCF7 (Fig. 5F) and MDCK cells (data not shown).
Gefitinib selectively inhibits EGFR, but not other EGFR-related receptor tyrosine kinases. However, it has been shown to inhibit other kinases with much higher IC50 values (Brehmer et al., 2005). As an alternative, we therefore treated cells with neutralizing anti-EGF antibody, which specifically inhibits EGF-dependent cell proliferation (Sakaguchi et al., 2008), and found that this treatment abolished PH-Akt–GFP recruitment to force-loaded E-cadherin receptors on MCF7 cells (Fig. 5F; n>40, two experiments). Anti-EGF antibody also blocked E-cadherin-mediated adaptive stiffening (Fig. 5G, P<0.05; n=71 cells, three experiments), indicating that cell stiffening is EGF (ligand) dependent.
Actin recruitment to perturbed E-cadherin junctions does not require integrin activation
We further tested whether actin accumulation at force-loaded E-cadherin beads – attributed to α-catenin unfurling and vinculin recruitment (Barry et al., 2014; le Duc et al., 2010; Thomas et al., 2013; Yonemura et al., 2010) – depends on cell stiffening. Although the anti-β1 antibody AIIB2 abrogated cell stiffening (Fig. 2E), it did not block actin accumulation at force-loaded E-cadherin beads (Fig. S4). The apparent difference in actin accumulation at E-cadherin beads on AIIB2-treated cells was not due to nonspecifically adsorbed matrix protein on the beads, because actin accumulation was unaffected by the presence or absence of serum (Fig. S4C). Conversely, there was no actin accumulation around PLL beads, in measurements with 10% serum. These results suggest that force-activated stiffening of bead–cell junctions mainly reflects changes in MCF7 cell contractility, rather than localized cytoskeletal remodeling.
DISCUSSION
Findings reported in this paper identified an additional E-cadherin mechanotransduction pathway, beyond force-dependent α-catenin switching and local cytoskeletal remodeling (Barry et al., 2014; Leckband and de Rooij, 2014; Leckband et al., 2011; Yonemura et al., 2010). This mechanism, summarized in Fig. 6, involves an EGFR-dependent signaling pathway that activates PI3K and downstream integrin ligation, followed by subsequent myosin-II-dependent cell stiffening.
Fig. 6.

Proposed model for E-cadherin-mediated global mechanotransduction. Mechanically stimulated E-cadherin receptors activate PI3K through an EGFR-dependent mechanism. PI3K activation is required for the downstream formation of new integrin adhesions and consequent ROCK-dependent activation of myosin II-dependent cell contractility.
The dependence of adaptive stiffening on PI3K, Src and integrins parallels mechanotransduction mediated by the immunoglobulin superfamily protein PECAM-1 in endothelia (Collins et al., 2012), but there are differences. One obvious difference is that E-cadherin activates force transduction in epithelia, which lack PECAM-1 and VE-cadherin. Importantly, these findings also revealed that EGFR is required for E-cadherin-mediated force-dependent PI3K activation and cell stiffening. Although we did not explicitly demonstrate that PI3K activates integrins, prior studies have reported that PI3K converts integrins into a high-affinity state independently of the ECM (Batra et al., 2012; Katsumi et al., 2005; Orr et al., 2006). Our results with the fibronectin-blocking antibody 16G3 showed that ECM ligation and the formation of new adhesions is required for the myosin II-dependent cell contractility.
The abrogation of stiffening in cells on E-cadherin substrata or following treatment with function blocking antibodies confirmed that cell stiffening is integrin dependent. The absence of stiffening on E-cadherin substrata was unexpected, because E-cadherin-based adhesion supports traction forces and activates GTPases, Src and PI3K signaling (Kovacs et al., 2002; le Duc et al., 2010; McLachlan and Yap, 2011; Tabdili et al., 2012a). In addition, E-cadherin receptors are mechanically coupled to actin, and could potentially transmit force through the cytoskeleton by stress focusing (Hu et al., 2003). Indeed, RGD-coated beads trigger changes in basal focal adhesions that are attributed to force transmission through microfilaments (Hu et al., 2003). The latter mechanical mechanism is, however, inconsistent with the inability of apical E-cadherin perturbations to alter basal E-cadherin adhesions. Similarly, perturbing E-cadherin receptors with DECMA-1-coated beads failed to either alter traction forces (Fig. 1D) or trigger stiffening by cells on ECM (le Duc et al., 2010). These results indicate that E-cadherin force transduction triggers integrin ligation primarily through biochemical signals. Results with fibronectin-blocking antibody in turn indicate that ECM ligation is required for subsequent cell stiffening.
E-cadherin force-loading does not appear to locally activate Rho-dependent contractility through RhoGEFs, as reported at focal adhesions (Guilluy et al., 2011), because the integrin blockade ablated the stiffening response. Similarly, the absence of stiffening by cells on PLL or on E-cadherin substrata also appears to rule this out.
We did not explicitly identify specific integrins or matrix proteins required for E-cadherin-mediated cell stiffening. In most studies, cells were initially seeded on collagen-coated substrata but were cultured with 10% serum for at least 6 h prior to measurements. Immunofluorescence images exhibited little fibronectin on the surrounding matrix, but the cells stained positive for fibronectin, confirming matrix secretion. The anti-β1-integrin antibody AIIB2, which blocks integrin receptors for laminin, collagens and fibronectin (Zaidel-Bar and Geiger, 2010), abrogated cell stiffening. However, the demonstrated activation of α5β1 integrin (fibronectin receptor) and the abrogation of stiffening by treatment with anti-fibronectin antibody 16G3 suggest that integrin activation and binding to fibronectin play a major roll in the stiffening response.
It is possible that α-catenin could also participate in this signaling pathway. In an obvious way, α-catenin is essential to sustain tension across cadherin adhesions required for force transduction (Barry et al., 2014; Buckley et al., 2014; Thomas et al., 2013). However, these results are intriguing in light of reports that E-cadherin-mediated adaptive stiffening, but not α-catenin conformation switching, requires the vinculin-binding site of α-catenin (Barry et al., 2014; Kim et al., 2015). Thus, α-catenin might play an additional role in this additional signaling pathway. Alternatively, its ligands such as vinculin might act as force-sensitive scaffolds for other signaling proteins.
The abrogation of cell stiffening correlated with some apparent reduction in actin accumulation at E-cadherin beads might suggest a positive feedback loop wherein endogenous contractile forces enhance actin polymerization at perturbed E-cadherin adhesions. Increased contractile forces might simply increase α-catenin unfurling at beads, with a concomitant increase in local actin polymerization (Leerberg et al., 2014). Alternatively, zyxin facilitates force-dependent actin polymerization at focal adhesions, and is enriched at stressed adherens junctions (Hirata et al., 2008; Oldenburg et al., 2015). Mechanically stimulating actin fibers also stimulated formin-dependent fiber elongation (Higashida et al., 2013; Riveline et al., 2001). Assessing whether such mechanisms underlie our observations is beyond the scope of this study.
The broader physiological implications of this EGFR-dependent mechanism have yet to be established, but our results suggest that force perturbs well-documented bidirectional signaling between E-cadherin and EGFR. Blocking EGFR function has been shown to promote desmosome formation and cell adhesion (Lorch et al., 2004; Wheelock and Johnson, 2003). Alternatively, in some contexts, EGFR activation leads to E-cadherin and β-catenin and p120 catenin phosphorylation and disruption of cytoskeletal interactions (Hazan and Norton, 1998; Mariner et al., 2004). Conversely, in contact-inhibited proliferation, E-cadherin adhesion suppresses EGFR signaling (Mariner et al., 2004; Perrais et al., 2007; Qian et al., 2004). E-cadherin appears to block EGFR trafficking to signaling compartments, by retaining it at the membrane (Curto et al., 2007; McClatchey and Yap, 2012), but cadherin might also suppress EGFR trans phosphorylation (Fedor-Chaikin et al., 2003). Our data suggest that mechanically perturbing E-cadherin might relieve contact-inhibited EGFR signaling. Intriguingly, our results might account for the increase in growth-factor-dependent cell proliferation with increasing substrate rigidity and junctional tension (Kim and Asthagiri, 2011; Kim et al., 2009).
In summary, these results support the model in Fig. 6, wherein E-cadherin-mediated force transduction is mechano-chemically coupled to integrin activation in epithelial cells through an EGFR-dependent pathway. This mechanism operates in addition to the proximal, α-catenin dependent cytoskeletal remodeling. Despite functional and biochemical differences between E-cadherin and PECAM-1, the similar mechanotransduction signaling suggests that these receptors exploit common pathways in different tissues to transduce force throughout mechano-chemically integrated networks.
MATERIALS AND METHODS
Cell lines, protein production and inhibitors
Michigan Cancer Foundation-7 (MCF7) human epithelial breast carcinoma and Madine Darby canine kidney (MDCK) cells were from the ATCC, and were used as received. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM 4.5 g/l glucose) supplemented with 10% (v/v) fetal bovine serum (FBS), 1 mM sodium pyruvate, and 1% (v/v) penicillin–streptomycin (Corning Cell Grow, Manassas, VA) unless otherwise stated. Recombinant E-cadherin extracellular fragment tagged with the C-terminal Fc domain of human IgG (Ecad-Fc) was produced as described previously (Prakasam et al., 2006).
Src kinase was blocked by incubating cells with 10 μM PP2 (Calbiochem, UK) for 30 min, prior to mechanical measurements. PI3K was inhibited by treating cells with 50 μM LY294002 (Cell Signaling Technologies, Boston, MA) or with 5 nM Wortmannin (Santa Cruz Biotechnology, CA) for 1 h and 0.5 h, respectively (Orr et al., 2006). At 5 nM, Wortmanin reportedly does not inhibit other kinases or phospholipase A2 (Orr et al., 2006; Vlahos et al., 1994). Treatment with 10 μM Y27632 (Tocris Biosciences, Bristol, UK) for 30 min inhibits ROCK (Shewan et al., 2005). Treatment with 0.05 mM blebbistatin for 1 h inhibits myosin II (Barry et al., 2014). EGFR was blocked with 15 μM of the non-competitive EGFR-specific inhibitor Gefitinib (IRESSA) (Selleckchem, Houston, TX) for 2 h (Yen et al., 2015). Gefitinib is an EGFR-specific, non-competitive inhibitor that blocks EGFR kinase activity (Brehmer et al., 2005; Paez et al., 2004). EGF was neutralized with 5 µg/ml human monoclonal anti-EGF antibody (R&D Systems, Minneapolis, MN: Mouse IgG1 Clone #10825) for 30 min (Sakaguchi et al., 2008). When blocking EGF during MTC measurements, cells were bathed in medium containing anti-EGF antibody.
Integrin and ECM antibodies
Integrins were blocked with anti-α6 antibody (GOH3) (20 μg/ml; Santa Cruz Biotechnology, SC-19622) and with anti-β1-integrin antibody AIIB2 (1:20 dilution) (Barry et al., 2014). The hybridoma producing AIIB2 was from Johan de Rooij (Hubrecht Institute, The Netherlands). The anti-fibronectin antibody 16G3 and the control antibody 13G12, which does not inhibit integrin binding to fibronectin, were from Kenneth Yamada (NIH, Bethesda, MD) (Nagai et al., 1991). As a positive integrin-activation control, surface-expressed unbound integrin dimers were activated with 0.5 mM MnCl2 in DMEM (with 10% FBS) for 25 min (Cluzel et al., 2005; Orr et al., 2006).
Magnetic twisting cytometry
MTC was used to mechanically perturb E-cadherin receptors, as described previously (Barry et al., 2014; le Duc et al., 2010; Tabdili et al., 2012b). Beads were twisted with an oscillating field of 60 Gauss at 0.33 Hz, which generates a shear stress (force/area) of 7.4 Pa. Prior reports showed that the stiffening increases with the applied twisting torque (Barry et al., 2015; le Duc et al., 2010). In controls, beads were coated with either poly-L-Lysine (PLL), blocking anti-E-cadherin antibody DECMA-1 (Sigma-Aldrich, St Louis, MO, U3254) (Ozawa et al., 1990), or with neutral (non-blocking) anti-E-cadherin antibody (Clone 76D5, from Barry Gumbiner, University of Virginia, Charlottesville, VA). The neutral antibody binds the ectodomain, but does not alter the E-cadherin-binding affinity (Petrova et al., 2012; Shashikanth et al., 2015). In all cases, the bead coatings were optimized to bind cells and undergo displacements sufficient to measure the viscoelastic moduli of bead–cell junctions (Barry et al., 2014, 2015; le Duc et al., 2010; Tabdili et al., 2012b; Twiss et al., 2012).
We quantified stiffening or traction changes in cells loaded with single beads, although cells with multiple beads were included infrequently. Some images in this paper show cells with multiple bound beads, but only cells with single attached beads were primarily included in the analyses (see Fig. S4, for example). In MTC, the specific modulus follows a log-normal distribution, from which we obtained the mean and s.e.m. To determine the stiffness of individual cells in MTC and TFM measurements, for each condition, at least 40 cells were analyzed to obtain sufficient statistics.
Traction force microscopy
Traction force microscopy (TFM) measurements used polyacrylamide hydrogels with Young's moduli of 34 or 8.8 kPa (Tse and Engler, 2010). Proteins were immobilized on Sulfo-SANPAH-activated gels, by overnight incubations at 4°C with (1) 0.2 mg/ml collagen type 1 (Sigma, St Louis, MO), (2) fibronectin (0.2 mg/ml) or (3) PLL (0.2 mg/ml) in immobilization buffer (100 mM HEPES, 100 mM NaCl, and 5 mM CaCl2 at pH 8) (Tabdili et al., 2012b). Fc-tagged E-cadherin extracellular domains were immobilized as described previously (Tabdili et al., 2012b). After protein immobilization, the substrates were rinsed twice with 1× phosphate buffered saline (PBS), and sterilized by irradiation (365 nm), for at least 15 min, before seeding cells. E-cad-Fc substrates were blocked with 1% (w/v) BSA (in 1× PBS) for 20 min prior to rinsing and irradiation. MCF7 cells were harvested using 3.5 mM EDTA in PBS containing 1% (w/v) BSA (Takeichi and Nakagawa, 2001), seeded at 5000–8000 cells/ml onto hydrogels, and allowed to adhere and spread for 6 h at 37°C under 5% CO2, on polyacrylamide gels with embedded fluorescent microspheres. The absolute basal RMS traction force (BTF) was determined from fiduciary bead displacements, relative to the traction-free bead positions after cell removal (lysis) (Butler et al., 2002). Constrained traction maps and the RMS traction stress (Pa; N/m2) were determined from the bead displacement maps (Butler et al., 2002).
Combined MTC and traction force microscopy
MTC measurements were performed as described previously (Barry et al., 2014; le Duc et al., 2010; Tabdili et al., 2012b), except harvested cells (Takeichi and Nakagawa, 2001) were cultured on 34-kPa modulus polyacrylamide gels with embedded fiduciary fluorescent beads (Tse and Engler, 2010). In combined MTC and TFM measurements, fiduciary bead positions were imaged after incubating cells with ferromagnetic beads for 40 min (∼one bead per cell). The twisting torque was generated with an oscillating (0.3 Hz) field of 70 Gauss, which generates a shear stress (force/area) of 8.4 Pa. The force-induced traction changes (ΔRMS, +Load) were determined from bead displacement maps (Butler et al., 2002) measured immediately before and after 2 min of force-loading (ΔRMS, +Load). Controls imaged beads after 2 min, but without load (ΔRMS, −Load). Figs 1B and 4A indicate the measurement time sequences. The reported values for the BTF and ΔRMS traction are averages of results obtained with different cells and replicate experiments. The ΔRMS tractions were determined from the change in traction forces upon stimulating cells with E-cadherin beads. By contrast, the absolute BTF was measured with cells free of ferromagnetic beads, and required cell lysis.
Imaging
Confocal immunofluorescence
Immediately after bead loading, cells were fixed with 4% (w/v) paraformaldehyde for 15 min at room temperature. Control cells were not subjected to bead twisting, but all conditions were otherwise identical. Cells were permeabilized with 0.1% Triton X-100 for 5 min, blocked in 1% (w/v) BSA for 20 min, and stained with primary antibodies, followed by rinsing, and treatment with secondary antibodies and phalloidin in 1% (w/v) BSA for 1 h.
Focal adhesions were visualized by immunofluorescence imaging of vinculin at the basal plane. Vinculin was stained with monoclonal mouse anti-vinculin antibody (1:100, Sigma-Aldrich, V9131), and Alexa-Fluor-488-conjugated goat anti-mouse-IgG (1:200, Sigma-Aldrich, SAB4600388). F-actin was stained with Rhodamine–phalloidin (Invitrogen). Coverslips were mounted using ProLong Gold (Invitrogen), and images were acquired with a laser scanning confocal microscope (LSM 700, Zeiss) with a 40× and 1.3 NA oil immersion objective and 488-nm and 555-nm lasers. Confocal images of immunostained vinculin were analyzed with a custom MATLAB code (from Brenton Hoffman, Duke University), based on the watershed algorithm (Zamir et al., 1999). Focal adhesion size and number were quantified using the ‘Analyze Particles’ function in ImageJ (NIH) with a minimum threshold area of 0.3 μm2 (Gonon et al., 2005).
Cell areas were analyzed in ImageJ from acquired DIC images of respective single MCF7 cells (n=80), with appropriate magnification-specific pixel-to-micron conversion factors. The cells were cultured in medium containing integrin-blocking antibody (AIIB2) or Mn2+ for 25 min to inhibit or activate integrins, respectively. Cell areas were similarly quantified after treatment with the fibronectin-blocking antibody 16G3.
Integrin activation assays
Cell incubation with 20 μg/ml of GST–fibronectin-III9-11 peptide (from Martin Schwartz, Yale School of Medicine, New Haven, CT) for 30 min was used to assess α5β1 integrin activation (Orr et al., 2006; Tzima et al., 2001). For immunofluorescence imaging, cells were fixed and stained with mouse anti-GST antibody (1:100, Thermo Scientific, MA4-004), and with Alexa-Fluor-488-conjugated goat anti-mouse-IgG antibody (1:200, Sigma-Aldrich, SAB4600388). Cover slips were mounted with Prolong Gold (Invitrogen), and imaged by laser scanning confocal microscopy (Zeiss LSM 700, 40×1.3 NA oil immersion objective). Total cell fluorescence was quantified with ImageJ. Western blots similarly assessed peptide binding (Tzima et al., 2001). Controls used unperturbed cells (−Load). To activate integrins, cells were bathed in DMEM (with 10% FBS) containing 0.5 mM MnCl2 for 25 min before E-cadherin-force loading (Cluzel et al., 2005; Orr et al., 2006). The BTF and stiffening response of MnCl2-activated cells was determined as described above.
PI3K activation and actin accumulation at force-loaded E-cadherin beads
PI3K activation was visualized with the GFP-tagged pleckstrin homology domain of Akt (PH-Akt–GFP) (Collins et al., 2012). MCF7 or MDCK cells on collagen-coated glass were transiently transfected with PH-Akt–GFP (plasmid from Yingxiao Wang, UCSD, San Diego, CA). The accumulation of PH-Akt–GFP and of Phalloidin-stained actin with or without load (4 min) was quantified from background-subtracted fluorescence images (Barry et al., 2014, 2015). Background-subtracted mean fluorescence intensities (MFI), were normalized by measurements under no-load conditions (−Load). PH-Akt–GFP controls also used cells transiently transfected with GFP (EGFP-N1, Clontech, Mountain View, CA).
To confirm that PH-Akt–GFP accumulation reflected an increase in endogenous Akt phosphorylation during E-cadherin force-loading, we lysed the serum-starved MCF7 monolayer after 4 min of E-cadherin bead twisting. Lysates were blotted for Akt phosphorylation at S473 with a pan-specific antibody (1:1000, R&D Systems, Minneapolis, MN: catalog #AF887) overnight at 4°C. Densitometry scans, using ImageJ quantified changes in phospho-Akt levels, relative to PLL bead controls (Tzima et al., 2001). Actin was used as a loading control.
Dynamic fluorescence imaging of Src activity
Dynamic changes in Src kinase activity at cadherin adhesions were visualized with a membrane-targeted FRET-based KRas-Src reporter (Yingxiao Wang, UCSD, San Diego CA) (Seong et al., 2013; Wang et al., 2005). Transiently transfected MCF7 cells were cultured on collagen-I-coated glass-bottomed dishes (Glass #1.5, Cell E&G, Houston TX) in Phenol-Red-free DMEM supplemented with 0.5% FBS, 1 mM sodium pyruvate, and 1% (v/v) penicillin-streptomycin (Corning Cell Grow, Manassas, VA), for 6 h prior to the measurements, in order to reduce serum-induced Src kinase activity (McLachlan et al., 2007; Wang et al., 2005). Cells were then incubated with E-cadherin functionalized ferromagnetic beads for 40 min. An inverted Leica microscope (40× magnification, 0.55 NA air objective) coupled with a Dual View system (Optical Insights) acquired CFP and YFP images simultaneously with a CCD camera (Hamamatsu). The filter sets included CFP (excitation, S430/25, emission S470/30) and YFP (excitation, S500/20, emission S535/30). The emission filter set uses a 515-nm dichroic mirror to split the two emission images, and the exposure time was 300 ms.
The time-dependent CFP-to-YFP ratio, which is proportional to active Src, was monitored for 30 s. The 30 s time frame includes ∼10 s prior to force-loading, a time delay of ∼10 s between switching on the oscillating magnetic field and acquiring FRET images, and an additional 10 s during bead twisting. The acquired stacked images were then uniformly contrast-enhanced, using ImageJ. Next, the ratios of the CFP to YFP emission in a region of interest (ROI) around the beads were calculated, using a custom MATLAB code (available on request). Each image was background corrected, using the custom MATLAB program, which negates manual bias when choosing an arbitrary region for background correction. The program uniformly subtracted the minimal fluorescent pixel intensity from each of the acquired CFP and YFP images. Control data were acquired under identical conditions, but without bead loading. The CFP-to-YFP ratios in the ROIs were normalized to the average CFP-to-YFP ratio prior to bond shear. In additional controls, cells were treated with the Src inhibitor PP2.
Statistics
The mean±s.e.m. are reported in the text and figure legends. P-values were calculated from two-tailed Student's t-tests, using Microsoft Excel, with *P<0.05 defining statistically significant differences. The standard errors for a set of replicate experiments were calculated using the pooled standard deviations of each experiment, as indicated in the text and figure legends. The sample size was not computed when designing the study. The number of beads analyzed to assess protein recruitment (confocal immunofluorescence) or adaptive stiffening and the number of replicate experiments required for statistical significance were based on extensive prior studies. Similarly, the number of traction force measurements required for statistical significance (n≥6) was based on prior experience (Barry et al., 2014) and published literature (Butler et al., 2002).
Acknowledgements
We thank Saiko Rosenberg for technical assistance, Ryan Huang for code development, Prof. Hoffman (Duke University) for focal adhesion analysis software, and Kenneth Yamada for 16G3 and 13G12 antibodies.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
I.M. designed and conducted experiments, analyzed data, edited and wrote the manuscript. J.W. designed, conducted, analyzed experiments and edited the manuscript. P.S. designed, conducted and analyzed results. X.K. assisted with data analysis, A.T. assisted with experiments, N.W. provided technical advice, D.E.L. designed experiments, analyzed data, and edited, wrote and finalized the manuscript.
Funding
This work was supported by the National Science Foundation [grant numbers CMMI 1029871 and 1462739 to D.E.L.]; and National Institutes of Health [grant number R01 GM097443 to D.E.L. and N.W.]. J.W. was supported by the Shen Postdoctoral Fellowship. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.185447/-/DC1
References
- Al-Kilani A., de Freitas O., Dufour S. and Gallet F. (2011). Negative feedback from integrins to cadherins: a micromechanical study. Biophys. J. 101, 336-344. 10.1016/j.bpj.2011.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry A. K., Tabdili H., Muhamed I., Wu J., Shashikanth N., Gomez G. A., Yap A. S., Gottardi C. J., de Rooij J., Wang N. et al. (2014). alpha-Catenin cytomechanics: role in cadherin-dependent adhesion and mechanotransduction. J. Cell Sci. 127, 1779-1791. 10.1242/jcs.139014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry A. K., Wang N. and Leckband D. E. (2015). Local VE-cadherin mechanotransduction triggers long-ranged remodeling of endothelial monolayers. J. Cell Sci. 128, 1341-1351. 10.1242/jcs.159954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra N., Burra S., Siller-Jackson A. J., Gu S., Xia X., Weber G. F., DeSimone D., Bonewald L. F., Lafer E. M., Sprague E. et al. (2012). Mechanical stress-activated integrin alpha5beta1 induces opening of connexin 43 hemichannels. Proc. Natl. Acad. Sci. USA 109, 3359-3364. 10.1073/pnas.1115967109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehmer D., Greff Z., Godl K., Blencke S., Kurtenbach A., Weber M., Muller S., Klebl B., Cotten M., Keri G. et al. (2005). Cellular targets of gefitinib. Cancer Res. 65, 379-382. [PubMed] [Google Scholar]
- Buckley C. D., Tan J., Anderson K. L., Hanein D., Volkmann N., Weis W. I., Nelson W. J. and Dunn A. R. (2014). The minimal cadherin-catenin complex binds to actin filaments under force. Science 346, 1254211 10.1126/science.1254211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J. P., Tolic-Norrelykke I. M., Fabry B. and Fredberg J. J. (2002). Traction fields, moments, and strain energy that cells exert on their surroundings. Am. J. Physiol. Cell Physiol. 282, C595-C605. 10.1152/ajpcell.00270.2001 [DOI] [PubMed] [Google Scholar]
- Cavey M., Rauzi M., Lenne P.-F. and Lecuit T. (2008). A two-tiered mechanism for stabilization and immobilization of E-cadherin. Nature 453, 751-756. 10.1038/nature06953 [DOI] [PubMed] [Google Scholar]
- Cluzel C., Saltel F., Lussi J., Paulhe F., Imhof B. A. and Wehrle-Haller B. (2005). The mechanisms and dynamics of (alpha)v(beta)3 integrin clustering in living cells. J. Cell Biol. 171, 383-392. 10.1083/jcb.200503017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C., Guilluy C., Welch C., O'Brien E. T., Hahn K., Superfine R., Burridge K. and Tzima E. (2012). Localized tensional forces on PECAM-1 elicit a global mechanotransduction response via the integrin-RhoA pathway. Curr. Biol. 22, 2087-2094. 10.1016/j.cub.2012.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon B. G., Baeyens N., Han J., Budatha M., Ross T. D., Fang J. S., Yun S., Thomas J.-L. and Schwartz M. A. (2015). Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J. Cell Biol. 208, 975-986. 10.1083/jcb.201408103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully M., You H., Levine A. J. and Mak T. W. (2006). Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 6, 184-192. 10.1038/nrc1819 [DOI] [PubMed] [Google Scholar]
- Curto M., Cole B. K., Lallemand D., Liu C.-H. and McClatchey A. I. (2007). Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol. 177, 893-903. 10.1083/jcb.200703010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai R., Sarpal R., Ishiyama N., Pellikka M., Ikura M. and Tepass U. (2013). Monomeric alpha-catenin links cadherin to the actin cytoskeleton. Nat. Cell Biol. 15, 261-273. 10.1038/ncb2685 [DOI] [PubMed] [Google Scholar]
- Fedor-Chaikin M., Hein P. W., Stewart J. C., Brackenbury R. and Kinch M. S. (2003). E-cadherin binding modulates EGF receptor activation. Cell Commun. Adhes. 10, 105-118. 10.1080/cac.10.2.105.118 [DOI] [PubMed] [Google Scholar]
- Fruman D. A., Meyers R. E. and Cantley L. C. (1998). Phosphoinositide kinases. Annu. Rev. Biochem. 67, 481-507. 10.1146/annurev.biochem.67.1.481 [DOI] [PubMed] [Google Scholar]
- Geiger B. and Yamada K. M. (2011). Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol. 3, a005033 10.1101/cshperspect.a005033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonon E. M., Skalski M., Kean M. and Coppolino M. G. (2005). SNARE-mediated membrane traffic modulates RhoA-regulated focal adhesion formation. FEBS Lett. 579, 6169-6178. 10.1016/j.febslet.2005.09.090 [DOI] [PubMed] [Google Scholar]
- Guilluy C., Swaminathan V., Garcia-Mata R., O'Brien E. T., Superfine R. and Burridge K. (2011). The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 13, 722-727. 10.1038/ncb2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn C. and Schwartz M. A. (2009). Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 10, 53-62. 10.1038/nrm2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D. E., Reichardt L. F., Crowley E., Holley B., Moezzi H., Sonnenberg A. and Damsky C. H. (1990). The alpha 1/beta 1 and alpha 6/beta 1 integrin heterodimers mediate cell attachment to distinct sites on laminin. J. Cell Biol. 110, 2175-2184. 10.1083/jcb.110.6.2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan R. B. and Norton L. (1998). The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J. Biol. Chem. 273, 9078-9084. 10.1074/jbc.273.15.9078 [DOI] [PubMed] [Google Scholar]
- Higashida C., Kiuchi T., Akiba Y., Mizuno H., Maruoka M., Narumiya S., Mizuno K. and Watanabe N. (2013). F- and G-actin homeostasis regulates mechanosensitive actin nucleation by formins. Nat. Cell Biol. 15, 395-405. 10.1038/ncb2693 [DOI] [PubMed] [Google Scholar]
- Hirata H., Tatsumi H. and Sokabe M. (2008). Mechanical forces facilitate actin polymerization at focal adhesions in a zyxin-dependent manner. J. Cell Sci. 121, 2795-2804. 10.1242/jcs.030320 [DOI] [PubMed] [Google Scholar]
- Hu S., Chen J., Fabry B., Numaguchi Y., Gouldstone A., Ingber D. E., Fredberg J. J., Butler J. P. and Wang N. (2003). Intracellular stress tomography reveals stress focusing and structural anisotropy in cytoskeleton of living cells. Am. J. Physiol. Cell Physiol. 285, C1082-C1090. 10.1152/ajpcell.00159.2003 [DOI] [PubMed] [Google Scholar]
- Jasaitis A., Estevez M., Heysch J., Ladoux B. and Dufour S. (2012). E-cadherin-dependent stimulation of traction force at focal adhesions via the Src and PI3K signaling pathways. Biophys. J. 103, 175-184. 10.1016/j.bpj.2012.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumi A., Naoe T., Matsushita T., Kaibuchi K. and Schwartz M. A. (2005). Integrin activation and matrix binding mediate cellular responses to mechanical stretch. J. Biol. Chem. 280, 16546-16549. 10.1074/jbc.C400455200 [DOI] [PubMed] [Google Scholar]
- Kim J.-H. and Asthagiri A. R. (2011). Matrix stiffening sensitizes epithelial cells to EGF and enables the loss of contact inhibition of proliferation. J. Cell Sci. 124, 1280-1287. 10.1242/jcs.078394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-H., Kushiro K., Graham N. A. and Asthagiri A. R. (2009). Tunable interplay between epidermal growth factor and cell-cell contact governs the spatial dynamics of epithelial growth. Proc. Natl. Acad. Sci. USA 106, 11149-11153. 10.1073/pnas.0812651106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.-J., Zheng S., Sun J., Muhamed I., Wu J., Lei L., Kong X., Leckband D. E. and Wang Y. (2015). Dynamic visualization of alpha-catenin reveals rapid, reversible conformation switching between tension states. Curr. Biol. 25, 218-224. 10.1016/j.cub.2014.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs E. M., Ali R. G., McCormack A. J. and Yap A. S. (2002). E-cadherin homophilic ligation directly signals through Rac and Phosphatidylinositol 3-kinase to regulate adhesive contacts. J. Biol. Chem. 277, 6708-6718. 10.1074/jbc.M109640200 [DOI] [PubMed] [Google Scholar]
- Ladoux B., Anon E., Lambert M., Rabodzey A., Hersen P., Buguin A., Silberzan P. and Mege R.-M. (2010). Strength dependence of cadherin-mediated adhesions. Biophys. J. 98, 534-542. 10.1016/j.bpj.2009.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Duc Q., Shi Q., Blonk I., Sonnenberg A., Wang N., Leckband D. and de Rooij J. (2010). Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a MyosinII dependent manner. J. Cell Biol. 189, 1107-1115. 10.1083/jcb.201001149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leckband D. E. and de Rooij J. (2014). Cadherin adhesion and mechanotransduction. Annu. Rev. Cell Dev. Biol. 30, 291-315. 10.1146/annurev-cellbio-100913-013212 [DOI] [PubMed] [Google Scholar]
- Leckband D. E., le Duc Q., Wang N. and de Rooij J. (2011). Mechanotransduction at cadherin-mediated adhesions. Curr. Opin. Cell Biol. 23, 523-530. 10.1016/j.ceb.2011.08.003 [DOI] [PubMed] [Google Scholar]
- Lecuit T., Lenne P.-F. and Munro E. (2011). Force generation, transmission, and integration during cell and tissue morphogenesis. Annu. Rev. Cell Dev. Biol. 27, 157-184. 10.1146/annurev-cellbio-100109-104027 [DOI] [PubMed] [Google Scholar]
- Leerberg J. M., Gomez G. A., Verma S., Moussa E. J., Wu S. K., Priya R., Hoffman B. D., Grashoff C., Schwartz M. A. and Yap A. S. (2014). Tension-sensitive actin assembly supports contractility at the epithelial zonula adherens. Curr. Biol. 24, 1689-1699. 10.1016/j.cub.2014.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Tan J. L., Cohen D. M., Yang M. T., Sniadecki N. J., Ruiz S. A., Nelson C. M. and Chen C. S. (2010). Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl. Acad. Sci. USA 107, 9944-9949. 10.1073/pnas.0914547107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorch J. H., Klessner J., Park J. K., Getsios S., Wu Y. L., Stack M. S. and Green K. J. (2004). Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J. Biol. Chem. 279, 37191-37200. 10.1074/jbc.M405123200 [DOI] [PubMed] [Google Scholar]
- Mariner D. J., Davis M. A. and Reynolds A. B. (2004). EGFR signaling to p120-catenin through phosphorylation at Y228. J. Cell Sci. 117, 1339-1350. 10.1242/jcs.01001 [DOI] [PubMed] [Google Scholar]
- McClatchey A. I. and Yap A. S. (2012). Contact inhibition (of proliferation) redux. Curr. Opin. Cell Biol. 24, 685-694. 10.1016/j.ceb.2012.06.009 [DOI] [PubMed] [Google Scholar]
- McLachlan R. W. and Yap A. S. (2007). Not so simple: the complexity of phosphotyrosine signaling at cadherin adhesive contacts. J. Mol. Med. 85, 545-554. 10.1007/s00109-007-0198-x [DOI] [PubMed] [Google Scholar]
- McLachlan R. W. and Yap A. S. (2011). Protein tyrosine phosphatase activity is necessary for E-cadherin-activated Src signaling. Cytoskeleton 68, 32-43. 10.1002/cm.20492 [DOI] [PubMed] [Google Scholar]
- McLachlan R. W., Kraemer A., Helwani F. M., Kovacs E. M. and Yap A. S. (2007). E-cadherin adhesion activates c-Src signaling at cell-cell contacts. Mol. Biol. Cell 18, 3214-3223. 10.1091/mbc.E06-12-1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz A. F., Che Y., Banerjee S., Goldstein J. M., Rosowski K. A., Revilla S. F., Niessen C. M., Marchetti M. C., Dufresne E. R. and Horsley V. (2013). Cadherin-based intercellular adhesions organize epithelial cell-matrix traction forces. Proc. Natl. Acad. Sci. USA 110, 842-847. 10.1073/pnas.1217279110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould A. P. (2002). Analyzing integrin-dependent adhesion. Curr. Protoc. Cell Biol. Chapter 9, Unit 9 4 10.1002/0471143030.cb0904s15 [DOI] [PubMed] [Google Scholar]
- Nagafuchi A., Takeichi M. and Tsukita S. (1991). The 102 kd cadherin-associated protein: similarity to vinculin and posttranscriptional regulation of expression. Cell 65, 849-857. 10.1016/0092-8674(91)90392-C [DOI] [PubMed] [Google Scholar]
- Nagai T., Yamakawa N., Aota S., Yamada S. S., Akiyama S. K., Olden K. and Yamada K. M. (1991). Monoclonal antibody characterization of two distant sites required for function of the central cell-binding domain of fibronectin in cell adhesion, cell migration, and matrix assembly. J. Cell Biol. 114, 1295-1305. 10.1083/jcb.114.6.1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren N. K., Arthur W. T. and Burridge K. (2003). Cadherin engagement inhibits RhoA via p190RhoGAP. J. Biol. Chem. 278, 13615-13618. 10.1074/jbc.C200657200 [DOI] [PubMed] [Google Scholar]
- Oldenburg J., van der Krogt G., Twiss F., Bongaarts A., Habani Y., Slotman J. A., Houtsmuller A., Huveneers S. and de Rooij J. (2015). VASP, zyxin and TES are tension-dependent members of Focal Adherens Junctions independent of the alpha-catenin-vinculin module. Sci. Rep. 5, 17225 10.1038/srep17225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr A. W., Ginsberg M. H., Shattil S. J., Deckmyn H. and Schwartz M. A. (2006). Matrix-specific suppression of integrin activation in shear stress signaling. Mol. Biol. Cell 17, 4686-4697. 10.1091/mbc.E06-04-0289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa M., Hoschutzky H., Herrenknecht K. and Kemler R. (1990). A possible new adhesive site in the cell-adhesion molecule uvomorulin. Mech. Dev. 33, 49-56. 10.1016/0925-4773(90)90134-8 [DOI] [PubMed] [Google Scholar]
- Paez J. G., Janne P. A., Lee J. C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F. J., Lindeman N., Boggon T. J. et al. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497-1500. 10.1126/science.1099314 [DOI] [PubMed] [Google Scholar]
- Pece S. and Gutkind J. S. (2000). Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J. Biol. Chem. 275, 41227-41233. 10.1074/jbc.M006578200 [DOI] [PubMed] [Google Scholar]
- Perez T. D., Tamada M., Sheetz M. P. and Nelson W. J. (2008). Immediate-early signaling induced by E-cadherin engagement and adhesion. J. Biol. Chem. 283, 5014-5022. 10.1074/jbc.M705209200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais M., Chen X., Perez-Moreno M. and Gumbiner B. M. (2007). E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol. Biol. Cell 18, 2013-2025. 10.1091/mbc.E06-04-0348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova Y. I., Spano M. M. and Gumbiner B. M. (2012). Conformational epitopes at cadherin calcium-binding sites and p120-catenin phosphorylation regulate cell adhesion. Mol. Biol. Cell 23, 2092-2108. 10.1091/mbc.E11-12-1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakasam A. K., Maruthamuthu V. and Leckband D. E. (2006). Similarities between heterophilic and homophilic cadherin adhesion. Proc. Natl. Acad. Sci. USA 103, 15434-15439. 10.1073/pnas.0606701103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X., Karpova T., Sheppard A. M., McNally J. and Lowy D. R. (2004). E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 23, 1739-1784. 10.1038/sj.emboj.7600136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratheesh A., Priya R. and Yap A. S. (2013). Coordinating Rho and Rac: the regulation of Rho GTPase signaling and cadherin junctions. Prog. Mol. Biol. Transl. Sci. 116, 49-68. 10.1016/B978-0-12-394311-8.00003-0 [DOI] [PubMed] [Google Scholar]
- Riveline D., Zamir E., Balaban N. Q., Schwarz U. S., Ishizaki T., Narumiya S., Kam Z., Geiger B. and Bershadsky A. D. (2001). Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J. Cell Biol. 153, 1175-1186. 10.1083/jcb.153.6.1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M., Sonegawa H., Murata H., Kitazoe M., Futami J.-I., Kataoka K., Yamada H. and Huh N.-H. (2008). S100A11, an dual mediator for growth regulation of human keratinocytes. Mol. Biol. Cell 19, 78-85. 10.1091/mbc.E07-07-0682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluck T., Nienhaus U., Aegerter-Wilmsen T. and Aegerter C. M. (2013). Mechanical control of organ size in the development of the Drosophila wing disc. PLoS ONE 8, e76171 10.1371/journal.pone.0076171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M. A. and DeSimone D. W. (2008). Cell adhesion receptors in mechanotransduction. Curr. Opin. Cell Biol. 20, 551-556. 10.1016/j.ceb.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong J., Tajik A., Sun J., Guan J.-L., Humphries M. J., Craig S. E., Shekaran A., Garcia A. J., Lu S., Lin M. Z. et al. (2013). Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc. Natl. Acad. Sci. USA 110, 19372-19377. 10.1073/pnas.1307405110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashikanth N., Petrova Y. I., Park S., Chekan J., Maiden S., Spano M., Ha T., Gumbiner B. M. and Leckband D. E. (2015). Allosteric regulation of E-cadherin adhesion. J. Biol. Chem. 290, 21749-21761. 10.1074/jbc.M115.657098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewan A. M., Maddugoda M., Kraemer A., Stehbens S. J., Verma S., Kovacs E. M. and Yap A. S. (2005). Myosin 2 is a key Rho kinase target necessary for the local concentration of E-cadherin at cell-cell contacts. Mol. Biol. Cell 16, 4531-4542. 10.1091/mbc.E05-04-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabdili H., Barry A. K., Langer M. D., Chien Y.-H., Shi Q., Lee K. J. and Leckband D. E. (2012a). Cadherin point mutations alter cell sorting and modulate GTPase signaling. J. Cell Sci. 125, 3299-3309. 10.1242/jcs.087395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabdili H., Langer M., Shi Q., Poh Y.-C., Wang N. and Leckband D. (2012b). Cadherin-dependent mechanotransduction depends on ligand identity but not affinity. J. Cell Sci. 125, 4362-4371. 10.1242/jcs.105775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. and Nakagawa S. (2001). Cadherin-dependent cell-cell adhesion. Curr. Protoc. Cell Biol. Chapter 9, Unit 9 3 10.1002/0471143030.cb0903s00 [DOI] [PubMed] [Google Scholar]
- Tepass U., Godt D. and Winklbauer R. (2002). Cell sorting in animal development: signalling and adhesive mechanisms in the formation of tissue boundaries. Curr. Opin. Genet. Dev. 12, 572-582. 10.1016/S0959-437X(02)00342-8 [DOI] [PubMed] [Google Scholar]
- Thomas W. A., Boscher C., Chu Y.-S., Cuvelier D., Martinez-Rico C., Seddiki R., Heysch J., Ladoux B., Thiery J. P., Mege R.-M. et al. (2013). alpha-catenin and vinculin cooperate to promote high E-cadherin-based adhesion strength. J. Biol. Chem. 288, 4957-4969. 10.1074/jbc.M112.403774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse J. R. and Engler A. J. (2010). Preparation of hydrogel substrates with tunable mechanical properties. Curr. Protoc. Cell Biol. Chapter 10, Unit 10 16 10.1002/0471143030.cb1016s47 [DOI] [PubMed] [Google Scholar]
- Twiss F., Le Duc Q., Van Der Horst S., Tabdili H., Van Der Krogt G., Wang N., Rehmann H., Huveneers S., Leckband D. E. and De Rooij J. (2012). Vinculin-dependent Cadherin mechanosensing regulates efficient epithelial barrier formation. Biol. Open 1, 1128-1140. 10.1242/bio.20122428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima E., del Pozo M. A., Shattil S. J., Chien S. and Schwartz M. A. (2001). Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 20, 4639-4647. 10.1093/emboj/20.17.4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima E., Irani-Tehrani M., Kiosses W. B., Dejana E., Schultz D. A., Engelhardt B., Cao G., DeLisser H. and Schwartz M. A. (2005). A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426-431. 10.1038/nature03952 [DOI] [PubMed] [Google Scholar]
- Vasudevan K. M., Barbie D. A., Davies M. A., Rabinovsky R., McNear C. J., Kim J. J., Hennessy B. T., Tseng H., Pochanard P., Kim S. Y. et al. (2009). AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 16, 21-32. 10.1016/j.ccr.2009.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos C. J., Matter W. F., Hui K. Y. and Brown R. F. (1994). A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269, 5241-5248. [PubMed] [Google Scholar]
- Wang N., Butler J. P. and Ingber D. E. (1993). Mechanotransduction across the cell surface and through the cytoskeleton. Science 260, 1124-1127. 10.1126/science.7684161 [DOI] [PubMed] [Google Scholar]
- Wang Y., Boltvinick E. L., Zhao Y., Berns M. W., Usami S., Tsien R. Y. and Chien S. (2005). Visualizing the mechanical activation of Src. Nature 434, 1040-1045. 10.1038/nature03469 [DOI] [PubMed] [Google Scholar]
- Watanabe T., Sato K. and Kaibuchi K. (2009). Cadherin-mediated intercellular adhesion and signaling cascades involving small GTPases. Cold Spring Harb. Perspect. Biol. 1, a003020 10.1101/cshperspect.a003020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G. F., Bjerke M. A. and DeSimone D. W. (2011). Integrins and cadherins join forces to form adhesive networks. J. Cell Sci. 124, 1183-1193. 10.1242/jcs.064618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G. F., Bjerke M. A. and DeSimone D. W. (2012). A mechanoresponsive cadherin-keratin complex directs polarized protrusive behavior and collective cell migration. Dev. Cell 22, 104-115. 10.1016/j.devcel.2011.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock M. J. and Johnson K. R. (2003). Cadherins as modulators of cellular phenotype. Annu. Rev. Cell Dev. Biol. 19, 207-235. 10.1146/annurev.cellbio.19.011102.111135 [DOI] [PubMed] [Google Scholar]
- Yao M., Qiu W., Liu R., Efremov A. K., Cong P., Seddiki R., Payre M., Lim C. T., Ladoux B., Mege R.-M. et al. (2014). Force-dependent conformational switch of alpha-catenin controls vinculin binding. Nat. Commun. 5, 4525 10.1038/ncomms5525 [DOI] [PubMed] [Google Scholar]
- Yen H.-Y., Liu Y.-C., Chen N.-Y., Tsai C.-F., Wang Y.-T., Chen Y.-J., Hsu T.-L., Yang P.-C. and Wong C.-H. (2015). Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc. Natl. Acad. Sci. USA 112, 6955-6960. 10.1073/pnas.1507329112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S., Wada Y., Watanabe T., Nagafuchi A. and Shibata M. (2010). alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 12, 533-542. 10.1038/ncb2055 [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R. and Geiger B. (2010). The switchable integrin adhesome. J. Cell Sci. 123, 1385-1388. 10.1242/jcs.066183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir E., Katz B. Z., Aota S., Yamada K. M., Geiger B. and Kam Z. (1999). Molecular diversity of cell-matrix adhesions. J. Cell Sci. 112, 1655-1669. [DOI] [PubMed] [Google Scholar]