

Abstract
Alpha-carboxynucleoside phosphonates (α-CNPs) are novel viral DNA polymerase inhibitors that do not need metabolic conversion for enzyme inhibition. The prototype contains a cyclopentyl linker between nucleobase and α-carboxyphosphonate and preferentially (50- to 100-fold) inhibits HIV-1 RT compared with herpetic DNA polymerases. A synthetic methodology involving three steps has been developed for the synthesis of a series of novel α-CNPs, including a Rh(II) catalyzed O-H insertion which connects the carboxyphosphonate group to a linker moiety and attachment of a nucleobase to the other end of the linker by a Mitsunobu reaction followed by final deprotection. Replacing the cyclopentyl moiety in the prototype α-CNPs by a more flexible entity results in a selectivity shift of ~ 100-fold in favor of the herpetic DNA polymerases when compared to HIV-1 RT. The nature of the kinetic interaction of the acyclic α-CNPs against the herpetic DNA polymerases differs from the nature of the nucleobase-specific kinetic interaction of the cyclopentyl α-CNPs against HIV RT.
Graphical Abstract
1. INTRODUCTION
Viral DNA polymerases represent an approved target for antiviral drug therapy. HIV reverse transcriptase is the best known example for which several inhibitors have been discovered and applied in clinical practice. At least seven different nucleoside RT inhibitors (NRTIs), one nucleotide RT inhibitor (NtRTI), and five non-nucleoside RT inhibitors (NNRTIs) of HIV-1 RT have been formally approved for clinical use.1 In addition, several non-nucleoside competing RT inhibitors (NcRTIs) and RNase H inhibitors have been discovered that act against HIV-1 RT in a manner that is structurally and functionally different from that of N(t)RTIs and NNRTIs.2–7 With regard to herpesvirus therapy, a variety of structurally distinct inhibitors of herpetic DNA polymerases have been reported and several of them are clinically used.8 The best known examples are the acyclic nucleoside analogues acyclovir and its oral prodrug valacyclovir; penciclovir and its prodrug famciclovir; and ganciclovir and its prodrug valganciclovir.9 (E)-5-(2-Bromovinyl)-2′-deoxyuridine (BVDU) is endowed with potent activity against herpes simplex virus type 1 (HSV-1) and varicella-zoster virus (VZV),10 and phosphonoformic acid (PFA, foscarnet) is a pyrophosphate analogue that is clinically used for the treatment of human cytomegalovirus (HCMV) and herpesvirus infections.11 Among the above-cited agents, PFA was also shown to be active against HIV-1 RT,11 and acyclovir (as its 5′-triphosphate metabolite) has been shown to inhibit HIV-1 RT as well.12,13 Retrospective studies have shown some clinical efficacy of acyclovir in HIV-infected individuals that were co-infected with herpesviruses (enabling activation of the drug by the herpesvirus-encoded thymidine kinase or protein kinase).14,15
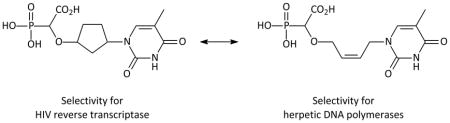
Recently, an entirely novel class of DNA polymerase inhibitors has been discovered and designated α-carboxynucleoside phosphonates (α-CNP).16,17 These compounds consist of a nucleobase, an α-carboxyphosphonate, and a linker moiety such as a cyclopentyl ring. These molecules were shown to be potent competitive inhibitors of HIV-1 RT without requiring phosphorylation to their triphosphate equivalent. This is in contrast to what is required for all NRTIs and the NtRTI (acyclic nucleoside phosphonate) tenofovir.18 Notably, α-CNPs are reversible competitive inhibitors of HIV-1 RT against the natural dNTP enzyme substrates with 50% inhibitory concentration (IC50) values in the high nanomolar range. Unlike the N(t)RTIs, α-CNPs are not incorporated into the nascent viral DNA chain.17 Interestingly, the α-CNPs were also shown to inhibit the DNA polymerases encoded by HSV-1, VZV and HCMV, albeit at higher concentrations (IC50 values in the low micromolar range).17 Based on this, the α-CNPs can be designated as a novel class of broad spectrum viral DNA polymerase inhibitors. Until now, our insight into the structure-activity relationships (SAR) of the α-CNPs has been quite limited, since the only analogues synthesized and explored thus far contain a cyclopentyl16 or (2′-deoxy)ribose moiety19 as the linker moiety between the nucleobase and α-carboxyphosphonate.16 Modifications in the carbohydrate (deoxyribose) part of antiviral nucleoside analogues have been shown to shift their selectivity towards particular viruses (and their encoded DNA polymerases). For instance, analogues with a 2′,3′-dideoxyribose have selectivity towards HIV-1 RT, whereas an acyclic aliphatic chain is typical in inhibitors of herpetic DNA polymerases. We therefore decided to modify the cyclopentyl part in the α-CNP scaffold and to evaluate their anti-DNA polymerase potency and selectivity. This approach turned out to be successful. Whereas the original cyclopentyl-containing α-CNPs showed ~50- to 100-fold selectivity for HIV-1 RT compared to herpetic and human DNA polymerases, the newly synthesized acyclic cis-butenyl-containing α-CNPs possessed 100-fold selectivity for herpetic DNA polymerases versus HIV-1 RT and human DNA polymerases.
2. RESULTS AND DISCUSSION
2.1. Design and chemical synthesis of novel α-CNPs
As recently reported16,17, the prototypic thymine-α-CNP markedly inhibits HIV-1 RT with activity in the higher nanomolar range (IC50: 0.40 μM) in an assay using poly rA.dT as the template/primer and [3H]dTTP as the competing natural dNTP substrate. Interestingly, this compound also inhibited the DNA polymerases encoded by HCMV, HSV-1, and VZV, albeit at ~ 50- to 100-fold higher concentrations (IC50: 26–38 μM). Importantly, the human DNA polymerases α and β were hardly inhibited by the thymine-α-CNP prototype compound (IC50: 229 μM and > 500 μM, respectively).17 All α-CNP analogues thus far studied contain a cyclopentyl linker moiety between the nucleobase and α-carboxyphosphonate. An earlier series of related α-CNPs containing a (2′-deoxy)ribose linker unit proved inactive.19 To further explore the structure-activity relationship for the α-CNPs, we now modified the linker moiety (Table 1). The connector was first altered by adding two methylene spacers in a syn-fashion on adjacent positions of the cyclopentyl ring which holds the nucleobase and the α-carboxyphosphonate (6a). Our synthetic strategy towards these modified α-CNPs involved three steps consisting of a rhodium-catalyzed O-H insertion, followed by a Mitsunobu reaction to attach the nucleobase, and a final step to remove the protecting groups (Scheme 1).
Table 1.
Generality of the O-H insertion reaction of rhodium carbenoid

|
Reaction conditions: (i) 1 (1.0 equiv.), 2 (1.05 equiv.), Rh2(OAc)4 (1 mol%), benzene, reflux, 16h.
Scheme 1.

Synthetic route towards modified α-CNPs (B = nucleobase) [(i) Rh(II)-catalyzed O-H insertion (ii) Mitsunobu reaction (iii) Deprotection].
We recently reported a facile method for attaching carboxyphosphonate groups to a suitably functionalized nucleobase via O-H insertion of rhodium carbenoid.16,19 Following these previously published reaction conditions, the reaction of cis-cyclopentyl-1,2-dimethanol 1a with trimethyl phosphonodiazoacetate 2 was carried out in the presence of 1 mol% of Rh2(OAc)4 in refluxing benzene. The reaction was complete in 16 hours and mono- (3a) and di-O-H inserted compounds were obtained in 60 and 21% yield, respectively. The reaction was also applied to other cyclic syn-dimethanols, i.e. cyclopropyl- (1b), cyclohexenyl- (1c), and benzene-1,2-dimethanol (1d). All reactions afforded the expected mono O-H inserted compounds as an equimolar mixture of (dia)stereoisomers in 60–65% yield (Table 1).
The next task was to attach the nucleobase to the carboxyphosphonylated linker moieties. A Mitsunobu reaction20 was performed to substitute the free hydroxyl group of 3 by thymine. The reaction was initiated by treating PPh3 and DIAD in dry THF at 0 °C to generate the betaine. The solution containing the PPh3:DIAD zwitterion was transferred to another reaction vessel containing mono O-H inserted compound 3 and 3-benzoyl-thymine 4a in THF at −40 °C which was allowed to warm to room temperature and further stirred for 24 hours. These reactions all afforded the expected nucleobase-substituted carboxyphosphonates 5a–5d in good yields (Table 2). Interestingly, we obtained the N-1 substituted compound 5 almost exclusively and the trace amount (<5%) of O2-alkylated product formed was not isolated.
Table 2.
Generality of the Mitsunobu reaction for attaching the nucleobase

|
Reaction conditions: (i) 3 (1.0 equiv.), 4a (1.2 equiv.), PPh3 (2.1 equiv.), DIAD (2.0 equiv.), THF, −40°C-RT, 24h.
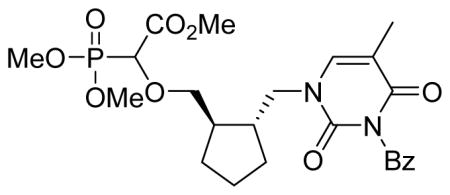
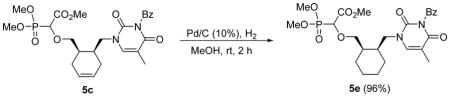
To check the influence of the ring conformation on anti-DNA polymerase activity we also prepared the reduced compound 5e by hydrogenation of 5c over Pd/C (Scheme 2).
Scheme 2.

Hydrogenation of 5c to 5e [(i) Pd/C (10%), H2 (1 atm), MeOH, rt, 2 h]
The final deprotection of compounds 5a–5e was done in two steps but in a one-pot process, following the same procedure reported for analogous nucleoside carboxyphosphonates.16,19 Deprotection of the dimethoxyphosphonate group was done by treating with excess of TMSBr followed by water. The crude reaction mixture after the first step was treated with an excess of aqueous 1M NaOH solution for the saponification of the carboxyester and debenzoylation of the N-3-benzoylthymine. Purification of the phosphononucleosides 6a–6e was done with a charcoal column. Before adsorbing the crude reaction mixture on activated charcoal, acidification to pH ≈ 1.0–1.5 was done with a solution of 2M HCl, and benzoic acid was removed by extracting five times with dichloromethane. Finally, the acidic aqueous residue was adsorbed on an activated charcoal column and the desired phosphononucleosides were eluted as ammonium salts in good yields using a 1:1 mixture of EtOH and 20% aqueous ammonia (Table 3).
Table 3.
Deprotection of α-carboxy nucleoside phosphonates 5a–5e to 6a–6e.

|
Reaction conditions: (i) TMSBr (7.0 equiv) CH3CN, 0 °C-rt, 16 h.(ii) H2O, 30 min, rotary evaporation (iii) aq. NaOH, 50 °C, 12 h and then aq. NH3, charcoal column.
Next, we concentrated on changing the linker ring to a heterocycle, such as a 1,2,3-triazole moiety. Phosphononucleosides linked with a 1,2,3-triazole group were reported to exhibit interesting antiviral activity.21 The aim was to synthesize both the carboxyphosphonate moiety linked with an azide functionality 3f and N-1-propargyl thymine 4b, and subject these to Cu-catalyzed azide-alkyne cycloaddition. The Rh(II)-catalyzed reaction of 2-bromoethanol 1f with trimethyl phosphonodiazoacetate 2 followed by azidation of the O-H inserted compound furnished the azidocarboxyphosphonate 3f. N-3-benzoyl-N-1-propargyl-thymine 4b was treated with the azide 3f in the presence of 1 mol% of CuSO4.5H2O and sodium ascorbate which afforded the expected 1,4-substituted-1,2,3-triazole 5f in 89% yield. The deprotection procedure was repeated for 5f after which the final triazole-linked phosphononucleoside 6f was isolated in 50 % yield as an ammonium salt (Scheme 3).
Scheme 3.

Synthesis of α-carboxy nucleoside phosphonate 6f with a 1,2,3-triazole heterocycle as linker moiety [(i) a. Rh2(OAc)4 Benzene, reflux; b. NaN3 acetone/H2O 60 °C, 12 h (ii) CuSO4.5H2O, Sodium ascorbate, Dioxane/H2O, rt, 16 h, 89% (iii) a. TMSBr, CH3CN, 0 °C-rt, 16 h, H2O, 1h; b. aq. NaOH, 50 °C, 12 h, charcoal column]
In an attempt to synthesize potent and selective α-CNP inhibitors of herpetic DNA polymerases, we synthesized also a series of acyclic α-CNPs, since the acyclic nucleoside analogues acyclovir, ganciclovir and penciclovir are known selective inhibitors of herpesviruses.8,9 The synthesis of the acyclic derivative 6g was performed in the same way as described above. The O-H insertion products of cis-2-butenyl-1,4-diol was obtained in 58% (3g) yield. Mitsunobu reaction of carboxyphosphonylated compound 3g proceeded well with N-3-benzoylthymine 4a, affording the nucleobase-substituted compound 5g in good yield. Full deprotection of the Mitsunobu product 5g was carried out under similar conditions mentioned above, i.e. by initial treatment with excess TMSBr and water, followed by base hydrolysis to afford butenyl-linked phosphononucleosides in 61% (6g) yield (Scheme 4).
Scheme 4.

Synthesis of 2-butenyl-linked α-carboxy nucleoside phosphonate 6g [(i) Rh(II)-catalyzed O-H insertion (ii) 3g (1.0 equiv.), 4a (1.2 equiv.), PPh3 (2.1 equiv.), DIAD (2.0 equiv.), THF, −40 °C-RT, 24 h (iii) a. TMSBr, CH3CN, 0 °C-rt, 16 h, H2O, 1h; b. aq. NaOH, 50 °C, 12 h, charcoal column].
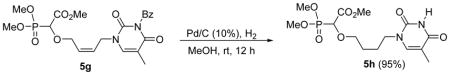
The n-butyl chain-linked phosphononucleoside 6h was synthesized starting from Mitsunobu product 5g which was subjected to hydrogenation over Pd/C to afford the reduced compound 5h in excellent yield. Interestingly, in the case of 5h, we observed that the N3-benzoyl group of the thymine part was also removed after hydrogenation. Complete deprotection of the reduced compound 5h afforded the n-butyl-linked thymine derivative 6h in good yield (Scheme 5).
Scheme 5.

Synthesis of n-butyl-linked α-carboxy nucleoside phosphonate 6h [(i) Pd/C (10%), H2 (1 atm), MeOH, rt, 12 h (ii) a. TMSBr, CH3CN, 0 °C-rt, 16 h, H2O, 1h; b. aq. NaOH, 50 °C, 12 h, charcoal column].
Given the fact that the thymine-butenyl-α-CNP 6g showed the strongest and most selective activity against herpetic DNA polymerases among all α-CNP derivatives synthesized thus far (see supra), we decided to prepare the butenyl analogues with different pyrimidine and purine bases (Scheme 6).
Scheme 6.

Synthesis of 2-butenyl linked α-carboxy nucleoside phosphonates 6i–6m with different nucleobases [(i) 3g (1.0 equiv.), 4 (1.2 equiv.), PPh3 (2.1 equiv.), DIAD (2.0 equiv.), THF, −40 °C-RT, 24 h (ii) Pd/C (10%), H2 (1 atm), MeOH, rt, 12 h (iii) a. TMSBr, CH3CN, 0 °C-rt, 16 h, H2O, 1h; b. aq. NaOH, 50 °C, 12 h, charcoal column].
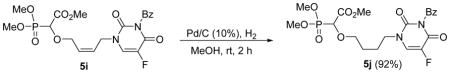
In the case of 5-fluorouracil, we synthesized the acyclic α-CNPs carrying either a butenyl (6i) or butyl moiety (6j) (Scheme 6).
To check whether the cis/syn geometry was important for the new class of acyclic α-CNPs, the carboxy phosphononucleoside 6o starting from cyclopentyl-1,2-anti-dimethanol 1o was synthesized. Following the three step synthetic strategy involving O-H insertion, Mitsunobu reaction and deprotection, the α-CNP 6q was prepared in good yield (Scheme 7).
Scheme 7.

Synthesis of α-carboxy nucleoside phosphonate 6o [(i) Rh(II)-catalyzed O-H insertion (ii) 3o (1.0 equiv.), 4a (1.2 equiv.), PPh3 (2.1 equiv.), DIAD (2.0 equiv.), THF, −40 °C-RT, 24 h (iii) a. TMSBr, CH3CN, 0 °C-rt, 16 h, H2O, 1h; b. aq. NaOH, 50 °C, 12 h, charcoal column].
Finally, an acyclic butenyl α-CNP derivative was synthesized lacking the thymine base linked to the butenyl, but instead containing a simple OH function. This was meant to reveal the importance of the presence of a nucleobase entity. The α-carboxyphosphonate 6n was obtained by the deprotection of O-H inserted compound 3g (Scheme 8).
Scheme 8.

Synthesis of carboxyphosphonate 6n [(i) Rh(II)-catalyzed O-H insertion (ii) a. TMSBr, CH3CN, 0 °C-rt, 16 h, H2O, 1h; b. aq. NaOH, 50 °C, 12 h].
2.2. Inhibitory activity of test compounds against DNA polymerases of different origin
2.2.1. Sensitivity of HIV-1 RT, herpetic and cellular DNA polymerases against (a)cyclic α-CNPs
To determine the impact of modifying the cyclic linker in the α-CNPs, the novel derivatives 6a–6f were first evaluated for their inhibitory activity against HIV-1 RT. It was found that these phosphononucleosides dramatically lost (≥300- to 1,000-fold) anti-HIV RT activity (IC50: 131 to ≥500 μM) when compared to the prototype thymine-α-CNP (IC50: 0.40 μM) (Table 4). Instead, when evaluated against three different herpetic DNA polymerases, we observed that, depending on the nature of the cyclic linker moiety, the inhibitory activity against herpetic DNA polymerases was preserved or even markedly enhanced. For example, compared to thymine-α-CNP, compounds 6b, 6c and 6e showed 8- to 13-fold superior activity against HCMV DNA polymerase (IC50: 3 to 4.6 μM). Compound 6d was 13-fold more active against VZV DNA polymerase and ~3-fold more active against HCMV and HSV-1 DNA polymerase, than our initial prototype inhibitor, i.e. thymine-α-CNP. The triazole linker-containing derivative 6f kept similar anti-herpetic DNA polymerase activity as the prototype thymine-α-CNP but markedly lost anti-HIV-1 RT activity (Table 4).
Table 4.
Inhibitory activity against viral and human DNA polymerases of thymine-α-CNP derivatives with a modified cyclic linker moiety.
| Compound | IC50a (μM)
|
|||||
|---|---|---|---|---|---|---|
| HIV-1 RT | HCMV DNA Pol | HSV-1 DNA Pol | VZV DNA Pol | Human DNA Pol α | Human DNA Pol β | |
| 6a | ≥500 | 40±25 | 14±14 | 73±1 | 165±100 | >500 |
| 6b | 440±85 | 4.6±0.2 | 36±12 | 12±3 | 183±8 | 390±127 |
| 6c | 425±106 | 3.0±0.5 | 14±3 | 8.5±2.1 | 48±36 | >500 |
| 6d | 131±86 | 13±4 | 7.5±5.0 | 2.9±0.7 | 75±18 | 76±30 |
| 6e | ≥500 | 4.0±0.0 | 8.6±6.6 | 16±1 | 69±21 | >500 |
| 6f | >500 | 38±6 | 25±21 | 24±4 | 332±28 | 361±160 |
|
| ||||||
| Thymine α-CNP | 0.40±0.05 | 38±11 | 26±20 | 38±16 | 229±27 | >500 |
| PFA | 0.34±0.01 | 9.8±5.8 | 0.44±0.25 | 0.18±0.01 | 7.7±6.5 | >100 |
| BVDU-TP | 0.89±0.4 | 3.6±0.7 | 1.9±0.9 | 0.49±0.09 | 21±10 | 26±4 |
IC50 or 50% inhibitory concentration.
These findings demonstrated that not only (cyclo)aliphatic (i.e. 6a,b,e) but also a linker moiety containing unsaturated or aromatic functions (i.e. 6c,d) can preserve or even further increase the inhibitory potency of the compounds against herpetic DNA polymerases. Moreover, subtle differences between the different herpetic DNA polymerases must exist for optimal interaction of the compounds in the viral DNA polymerase binding sites. This results in varying degrees of inhibitory activities of the compounds against the particular herpetic DNA polymerases. For example, compounds 6a and 6b showed up to 5- to 8-fold differences in inhibitory potency among the individual herpetic DNA polymerases. In sharp contrast and as explained above, the cyclic linker-modified compounds 6a to 6e, had marginal, if any, activity against HIV-1 RT. On the other hand, it was interesting to see that human DNA polymerase α (IC50: 48 to 332 μM; Table 4) and in particular DNA polymerase β (IC50: 360 to > 500 μM [except for compound 6d (IC50: 76 μM)]) were hardly affected by compounds 6a–6d, pointing to a considerable degree of anti-(herpetic) DNA polymerase selectivity. For comparative reasons, the pyrophosphate analogue phosphonoformic acid (PFA, foscarnet) and the 5′-triphosphate of BVDU (brivudin) were included in the herpetic DNA polymerase assays. Depending the nature of the enzyme, some of the compounds were equally or even more inhibitory (i.e. HCMV DNA polymerase) or less inhibitory (i.e. HSV-1 and VZV DNA polymerase) than the reference compounds (Table 4).
Next, we assessed the biological activity of α-CNP derivatives with an acyclic aliphatic linker. Whereas the acyclic thymine butyl-α-CNP (6h) had 1,000-fold lower inhibitory activity against HIV-1 RT compared to the cyclopentyl-containing prototype thymine-α-CNP, this compound had comparable inhibitory activity against the herpetic DNA polymerases as thymine-α-CNP, irrespective of the nature of the herpetic DNA polymerase (IC50: 26 to 45 μM, respectively) (Table 5). Remarkably, the thymine-butenyl-α-CNP derivative 6g had markedly reduced (~600-fold lower than thymine α-CNP) anti-HIV-1 RT activity, yet showed 8- to 20-fold higher potency against the herpetic DNA polymerases (IC50: 2.1 to 3.7 μM) compared to its thymine butyl-α-CNP analogue 6h (Table 5). Thus, whereas the cyclic thymine-cyclopentyl-α-CNP inhibitor displays 50- to 100-fold preference for HIV-1 RT compared to herpetic DNA polymerases, the acyclic thymine-butenyl-α-CNP derivative 6g showed ~100-fold superior activity against herpetic DNA polymerases than against HIV-1 RT. Compound 6g also showed 4- to 7-fold selectivity for the herpetic DNA polymerases when compared to human DNA polymerase α.
Table 5.
Inhibitory activity of acyclic α-CNP derivatives carrying different pyrimidine or purine bases against viral and human DNA polymerases.
| Compound | IC50a (μM)
|
|||||
|---|---|---|---|---|---|---|
| HIV-1 RT | HCMV DNA Pol | HSV-1 DNA Pol | VZV DNA Pol | Human DNA Pol α | Human DNA Pol β | |
| 6g | 235±16 | 2.1±0.6 | 3.7±1.7 | 2.2±0.1 | 14±7 | >500 |
| 6h | 145±106 | 29±3 | 45±27 | 27±12 | 301±51 | 450±105 |
|
| ||||||
| 6i | >500 | 30±16 | 24±1 | 27±2 | 325±4 | >500 |
| 6j | 263±69 | 9.4±4.1 | 6.7±4.6 | 4.1±0.9 | 72±52 | 306±3 |
| 6k | 404±30 | 5.5±2.1 | 2.7±2.0 | 3.3±0.4 | ≥500 | 454±28 |
| 6l | ≥500 | 3.6±0.6 | 2.1±2.3 | 2.9±0.0 | 365±91 | >500 |
| 6m | 270±33 | 16±8 | 12±6 | 2.9±0.9 | 414±22 | >500 |
|
| ||||||
| Thymine α-CNP | 0.40±0.05 | 38±11 | 26±20 | 38±16 | 229±27 | >500 |
IC50 or 50% inhibitory concentration.
When the thymine nucleobase in 6h and 6g was replaced by 5-fluorouracil (i.e. 6i and 6j, respectively) the butyl derivative 6j showed consistently better inhibitory activity against the herpetic DNA polymerases than the butenyl derivative 6i, a phenomenon that was opposite to what was observed for the thymine derivatives 6h and 6g (Table 5). As for the anti-thymine-α-CNP 6o, this compound was found to have poor, if any, inhibitory activity against any of the herpetic or human DNA polymerases mentioned in Table 5.
On the other hand, when the thymine base in 6g was substituted by an adenine (6k) or guanine (6l), virtually equipotent inhibition of the herpetic DNA polymerases was observed, irrespective of the nature of the nucleobase or the herpetic (i.e. HSV-1, HCMV, or VZV) enzyme tested. Only the cytosine derivative 6m was somewhat (4- to 8-fold) less inhibitory against the HCMV and HSV-1 DNA polymerases, but not against VZV DNA polymerase. Interestingly, these compounds did not markedly affect the human DNA polymerases α and β (IC50: 365 to > 500 μM), nor HIV-1 RT (IC50: 235 to 500 μM), and thus, they are highly selective for targeting herpetic DNA polymerases.
2.2.2. Sensitivity of HIV-1 RT and different herpetic DNA polymerases against acyclic butenyl-α-CNPs in the presence of different radiolabeled dNTP substrates
It is important to note that in the inhibition studies shown in Tables 4 and 5, the inhibitory action of the compounds against the herpetic DNA polymerases was measured with activated calf thymus DNA as the template/primer and [3H]dTTP as the dNTP substrate, for HCMV and HSV-1 DNA polymerase, and a homopolymeric template/primer (poly dA.oligo dT) and [3H]dTTP as the dNTP substrate for VZV DNA polymerase. Given the different nature of the nucleobase in the butenyl carboxy nucleoside phosphonates 6g (thymine), 6k (adenine), 6l (guanine) and 6m (cytosine), we therefore performed similar experiments using radiolabeled dATP, dGTP or dCTP (instead of radiolabeled dTTP) to allow direct competition of the acyclic butenyl-α-CNPs with their corresponding natural dNTPs (Table 6).
Table 6.
Inhibitory activity of acyclic α-CNPs in herpetic DNA polymerase assays using different dNTP substrates
| Nucleobase | Compound | IC50a (μM)
|
||||
|---|---|---|---|---|---|---|
| HCMV DNA polymeraseb
|
VZV DNA polymerasec
|
|||||
| [3H]dATP | [3H]dCTP | [3H]dGTP | [3H]dTTP | [3H]dTTP | ||
| Thymine | 6g | 3.1 ± 1.1 | 3.0 ± 0.4 | 2.5 ± 0.3 | 2.1 ± 0.6 | 2.2 ± 0 |
| Adenine | 6k | 3.0 ± 2.0 | 2.9 ± 0.9 | 3.2 ± 0.4 | 5.5 ± 2.1 | 3.3 ± 0 |
| Guanine | 6l | 10 ± 12 | 11 ± 1 | 14 ± 11 | 3.6 ± 0.6 | 2.9 ± 0 |
| Cytosine | 6m | 33 ± 5 | 20 ± 14 | 30 ± 4 | 16 ± 8 | 2.9 ± 0 |
50% Inhibitory concentration.
Template/primer: activated calf thymus DNA; [3H]dNTP substrate + three non-radiolabeled dNTPs.
Template/primer: poly dA.oligo dT; [3H]dTTP substrate.
It was striking to observe that for HCMV DNA polymerase, each of the acyclic carboxy nucleoside phosphonates had virtually equal activity irrespective of the nature of the radiolabeled natural substrate used in the assay (i.e. IC50: 2.1 to 3.1 μM for the T-analogue 6g; 2.9 to 5.5 μM for the A-analogue 6k; 3.6 to 14 μM for the G-analogue 6l; and 16 to 33 μM for the C-analogue 6m; Table 6). Even more striking, the T-, A-, G- and C-analogues were equally inhibitory against VZV DNA polymerase tested with homopolymeric poly dA.oligo dT as the template/primer and [3H]dTTP as the radiolabeled substrate (IC50: 2.2 to 3.3 μM). Thus, surprisingly, the nature of the nucleobase did not play a discriminative inhibitory role even in the presence of a template/primer that can solely use dTTP as the incorporating substrate, as in the case of the VZV DNA polymerase assay. These findings were strongly suggestive that the nature of the particular nucleobase in the butenyl-α-CNP molecules is actually less important to achieve interaction with, and inhibition of, herpetic DNA polymerases. However, since both the viral and human enzymes were insensitive to the inhibitory potential of the a-basic α-CNP derivative 6n (data not shown), these observations suggest that a ring system at the opposite end of the α-carboxyphosphonate part of the acyclic α-CNP derivative is still necessary for inhibitory activity, but the precise nature of this cyclic entity seems to be less crucial to obtain anti-herpetic DNA polymerase activity. It would therefore be of interest to replace the purine or pyrimidine nucleobase by other aromatic or even non-aromatic entities to further explore the optimal nature of this part of the acyclic α-CNP molecule. Not surprisingly, none of the compounds proved significantly inhibitory to HIV-1 RT, irrespective of the nature of the nucleobase and the template/primer used (Table 7). For comparative reasons, the pyrophosphate analogue PFA and a number of ddNTPs have been included in the assays.
Table 7.
Inhibitory activity of acyclic α-CNPs in HIV-1 RT assays using different homopolymeric template/primer and dNTP testing conditions.
| Nucleobase | Compound | IC50a(μM)
|
|||
|---|---|---|---|---|---|
| Poly rA.dT ([3H]dTTP) | Poly rU.dA ([3H]dATP | Poly rI.dC ([3H]dCTP) | Poly rC.dG ([3H]dGTP) | ||
| Thymine | 6g | 235 ± 16 | ≥500 | >500 | >500 |
| Adenine | 6k | 404 ± 30 | 40 ± 8 | >500 | >500 |
| Guanine | 6l | ≥500 | 369 ± 48 | >500 | >500 |
| Cytosine | 6m | 270 ± 33 | >500 | 273 ± 54 | >500 |
|
| |||||
| ddTTP | - | 0.05 ± 0.01 | >20 | >20 | >20 |
| ddATP | - | >20 | 0.46 ± 0.12 | >20 | >20 |
| ddCTP | - | >20 | >20 | 3.1 ±2.2 | >20 |
| ddGTP | - | >20 | >20 | >20 | 0.26 ±0.17 |
| PFA | - | 0.34 ± 0.01 | 0.75 ± 0.13 | 6.8 ± 2.0 | 17 ± 2 |
50% Inhibitory concentration, obtained in the presence of homopolymeric template/primers and their corresponding [3H]dNTPs.
2.2.3. Kinetic properties of the (a)cyclic α-CNPs against VZV DNA polymerase
In our previous studies, the HIV-1 RT inhibition by the prototype cyclopentyl thymine-α-CNP analogue was found to be competitive with respect to the natural substrate (dTTP).17 Clearly, in all HIV-1 RT assays, the α-CNPs behaved highly nucleobase-specific in terms of interaction with the template/primer. This behavior was ascertained and confirmed in a variety of kinetic, biochemical and structural studies.17 Strikingly, an entirely different behavior is seen with the acyclic butenyl-α-CNP inhibitors of herpetic DNA polymerases since these enzymatic assays (Table 6) rather suggest a lack of nucleobase-specific interaction with these enzymes. Therefore, the kinetics of the interaction of the most potent acyclic butenyl-α-CNP 6g against VZV DNA polymerase were investigated in the presence of varying dTTP concentrations and a fixed (saturating) concentration of poly dA.oligo dT as the template/primer. The Lineweaver-Burk graph (reciprocal plots of substrate concentration versus velocity values) revealed that the nature of interaction of compound 6g with VZV DNA polymerase was non-competitive with respect to the natural substrate (Fig. 1, panel A). (E)-5-(2-Bromovinyl)-2′-deoxyuridine-5′-triphosphate (BVDU-TP), a well-known inhibitor of VZV DNA polymerase, showed purely competitive kinetics with respect to the natural dTTP substrate (Fig. 1, panel C). It should be noticed that for the thymine-cyclopentyl α-CNP prototype compound a competitive interaction was earlier found against HIV-1 RT.17
Fig. 1.

Lineweaver-Burk plots for inhibition of VZV DNA polymerase by compound 6g, thymine-α-CNP and BVDU-TP using poly dA.oligo dT as the template/primer and dTTP as the natural substrate.
Thus, the non-competitive kinetics are suggestive for an independent (mutually non-exclusive) binding of the acyclic butenyl α-CNP and the natural substrate (dNTP) to the herpetic DNA polymerase, and are strikingly different from our earlier competitive findings for the cyclopentyl α-CNPs against HIV-1 RT. In order to reveal whether the difference in kinetic interaction is due to a structural difference between the cyclic versus acyclic α-CNPs, or a functional difference between HIV-1 RT versus herpetic DNA polymerases, the kinetic behavior of the prototype cyclopentyl thymine-α-CNP was examined against VZV DNA polymerase. It was found that both the cyclic and acyclic α-CNPs behaved identically (non-competitively versus the natural substrate) against this herpetic DNA polymerase (Fig. 1, panel B). Thus, the type of kinetic interaction of the α-CNPs, regardless of being cyclopentyl or acyclic butenyl derivatives, depends solely on the nature of the enzyme, being either reverse transcriptase (competitive mechanism) or a herpetic DNA polymerase (non-competitive mechanism).
2.2.4. Biochemical properties of the interaction of α-CNPs with HCMV DNA polymerase-related enzymes
To further gain additional information on the kinetic/biochemical interaction of the acyclic α-CNPs with the herpetic DNA polymerases, template/primer-extension reactions were performed with the adenine α-CNP (6k) using three different HCMV DNA polymerase-related enzymes, namely wild-type HCMV-UL54 (DNA polymerase encoded by HCMV), the wild-type (WT) bacteriophage RB69 DNA polymerase gp43 and RB69/ABC5, a hybrid DNA polymerase enzyme containing an engineered nucleotide-binding site derived from HCMV UL54.22,23 In all enzymes, the 3′,5′-exonuclease activity had been deleted to eliminate confounding effects of editing activities in the enzyme assays. When evaluated for their anti-DNA polymerase activity, the adenine (and also thymine)-butenyl-α-CNPs were highly inhibitory against HCMV-UL54 (IC50: ≤0.5 μM) but completely inactive against bacteriophage WT RB69 and the hybrid RB69/ABC5 (IC50: > 100 μM). These findings are suggestive for binding of the α-CNP inhibitors to a HCMV DNA polymerase site different from the dNTP substrate-binding active site.
Thus, primer extension reactions on a heteropolymeric template revealed dose-dependent inhibition of purified HCMV-UL54, while no significant changes in DNA synthesis are seen with the WT RB69 WT and the RB69/ABC5 chimera (Fig. 2). Inhibition of HCMV UL54 is seen during the first four to five nucleotide incorporation events, which points to a mechanism that is independent of Watson-Crick base-pairing. Unlike WT RB69, the chimera RB69/ABC5 is sensitive to active-site inhibitors phosphonoformic acid and acyclovir triphosphate, and thus, represents a fully active enzyme with a functional substrate-active site.23,24 Hence, our combined observations suggest that this compound binds to an enzyme site that is different from the polymerase active-site. Although we utilized enzymes with (knock-out) mutations in the 3′-5′ exonuclease active-site, we cannot exclude that 6k binds selectively to the 3′-5′ exonuclease active center of wild-type HCMV DNA polymerase UL54 or to a yet unknown binding site.
Fig 2.

Primer extension in the presence of increasing concentrations of adenine-butenyl-α-CNP (6k). (A) Dose response analysis of primer extensions with DNA polymerase RB69 gp43 (left), RB69 ABC5 (middle) and HCMV UL54 (right). (B) Sequences of primer and template used in primer extension assay.
As shown above, the anti-herpetic DNA polymerase activity of the acyclic α-CNPs proved independent from the nature of the nucleobase. Since traditional Watson-Crick base-pairing seems not to be strictly required to afford inhibition of herpetic DNA polymerase, it seems relevant to optimize the aromatic ring system in the acyclic α-CNP inhibitors, with the potential to move away from a nucleobase entity.
2.2.5. Sensitivity of mutant HIV-1 M184I/V RT against the acyclic α-CNPs
The prototype cyclopentyl α-CNPs are sensitive to HIV-1 RT amino acid mutations adjacent to the cyclopentyl ring. In particular, the mutation M184V confers 1.7- to 16-fold resistance to these α-CNPs depending on the nature of the nucleobase in the α-CNP.17 According to the crystal structure of the thymine α-CNP-HIV-1 RT complex, this resistance was likely due to steric hindrance of the L-sugar ring by the 184-valine side-chain,17 a phenomenon that has also been earlier reported for the L-enantiomers of 3TC (lamivudine) and FTC (emtricitabine).25 Although the acyclic α-CNPs only show a moderate inhibitory activity against wild-type HIV-1 RT (IC50: 320–1,250 μM), their activity was virtually annihilated when exposed to mutant M184I and M184V HIV-1 RT irrespective the nature of the nucleobase (IC50 ≥ 2,500 μM) (Table 8). Instead, introduction of the Y115F RT mutation that also is part of the binding pocket of the α-CNPs,17 did not markedly influence the binding affinity of the acyclic α-CNPs as also observed earlier for the cyclopentyl α-CNPs.17 Thus, although the acyclic α-CNPs are markedly less inhibitory to HIV-1 RT than the prototype cyclopentyl α-CNPs, they still seem to bind to the same pocket in HIV-1 RT as the cyclopentyl α-CNPs.
Table 8.
Inhibitory activity of acyclic α-CNPs against wild-type and mutant HIV-1 RTs
| Nucleobase | Compound | IC50 (μM)
|
|||
|---|---|---|---|---|---|
| Wild-type | M184I | M184V | Y115F | ||
| Thymine | 6g | 320 ± 89 | >2,500 | >2,500 | 655 ± 409 |
| Adenine | 6k | 531 ± 154 | ≥2,500 | >2,500 | 630 ± 37 |
| Guanine | 6l | ≥1,250 | >1,250 | >1,250 | ≥1,250 |
| Cytosine | 6m | 318 ± 73 | >1,250 | >1,250 | 408 ± 16 |
2.3. Molecular modeling
In order to understand the molecular and structural basis for the different potencies of the cyclic and acyclic α-CNPs against HIV-1 RT we took a molecular modeling approach.
To determine the basis for the markedly reduced HIV-1 RT inhibition by the acyclic α-CNPs (i.e. 6g) compared to the cyclopentyl α-CNPs, we modeled 6g into the crystal structure of the RT/dsDNA/T-α-CNP complex.17 Analogous to the binding of cyclopentyl T-α-CNP, the carboxyphosphonate moiety of 6g is required to chelate the active site metal ions, and the thymine base should base-pair with the template overhang. Modeled conformations of the acyclic linker did not simultaneously satisfy the base-pairing and metal chelation at the two ends of the molecule (Fig. 3).
Fig. 3.
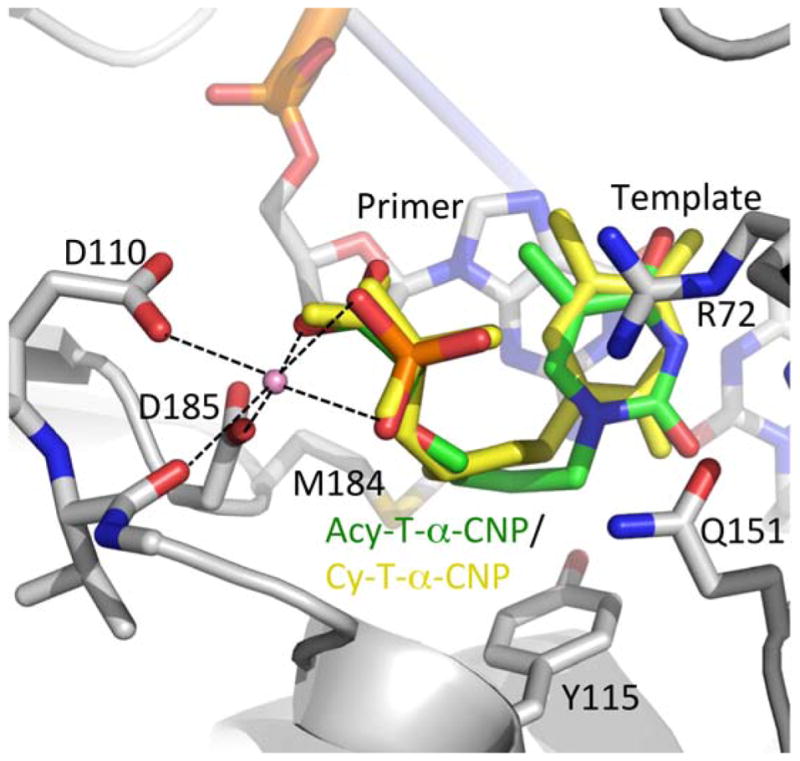
Modeling of acyclic T-α-CNP (6g) in the polymerase active site of HIV-1 RT/DNA/cyclic T-α-CNP complex (PDB ID. 4R5P17). The protein and DNA structures are shown in gray; active site metal ions as pink spheres with chelation indicated by dashed lines; and the acyclic T-α-CNP (6g) in green. The acyclic linker of 6h (green) does not permit favorable binding when compared to the binding of the cyclopentyl T-α-CNP (yellow) in the crystal structure: the acyclic linker develops a short contact with Y115 and forbids the thymine base to align well with the base of cyclopentyl T-α-CNP while maintaining the metal chelation.
We also compared the HIV-1 RT binding mode of the acyclic α-CNP with that of another acyclic nucleotide, i.e. tenofovir diphosphate (TFV-DP). This analysis revealed that the acyclic linker of 6g, which is different in chemical composition and length compared to that of TFV-DP, does not permit binding to HIV-1 RT in a favorable mode, unlike the favorable binding of TFV-DP observed by cocrystallization.26 Our modeling results therefore suggest that the acyclic linker of 6g and 6i–6m is not optimized for binding at the polymerase active site of HIV-1 RT, which apparently is the primary reason for the remarkably lower inhibitory activity (Table 5).
2.4. Antiviral activity
The compounds 6a–6m have been evaluated for antiviral activity against herpes simplex virus type 1 (KOS) and type 2 (G) in HEL cell cultures. None of them were endowed with significant antiviral activity presumably due to their poor, if any, cellular uptake due to the highly polar nature of the compounds.
3. CONCLUSION
In this study, a variety of new α-CNP derivatives are synthesized by modifying the linker moiety between the nucleobase and the α-carboxyphosphonate moiety. Whereas the potent inhibitory activity against HIV-1 RT by the prototype thymine-α-CNP was not maintained, a markedly increased anti-herpetic DNA polymerase activity was observed, in particular for the (acyclic) butenyl-α-CNP derivatives regardless of the nature of the nucleobase. These modifications in the linker moiety shifted the ~50- to 100-fold selectivity for HIV-1 RT towards herpetic DNA polymerase selectivity. The novel compounds inhibited the herpetic enzymes in a mechanistically different manner (non-competitive with respect to the natural substrate) than found for the prototype thymine-α-CNP against HIV-1 RT (competitive with respect to the natural substrate). Further optimization of the compounds will now focus on modifying the nucleobase part. Also, prodrugs will be designed to achieve efficient uptake of these novel type of DNA polymerase inhibitors by virus-infected cell cultures.
4. EXPERIMENTAL SECTION
NMR spectra were acquired on commercial instruments (Bruker Avance 300 MHz, Bruker AMX 400 MHz or Bruker Avance II+ 600 MHz) and chemical shifts (δ) are reported in parts per million (ppm) referenced to tetramethylsilane (1H), or the internal (NMR) solvent signal (13C). Mass spectra were run using a HP5989A apparatus (EI, 70 eV ionisation energy) with Apollo 300 data system, a Micromass Quattro II apparatus (ESI) with MASSLYNX data system or a Thermo Finnigan LCQ Advantage apparatus (ESI). Exact mass measurements were acquired on a Kratos MS50TC instrument (performed in the EI mode at a resolution of 10000. Melting points (not corrected) were determined using a Reichert Thermovar apparatus. For column chromatography 70–230 mesh silica 60 (E. M. Merck) was used as the stationary phase. Chemicals received from commercial sources were used without further purification. Reaction solvents (toluene) were used as received from commercial sources. The compounds that have been subject to biological evaluation are at least 95 % pure as estimated by HPLC.
4.1. General experimental procedure for O-H insertion reactions
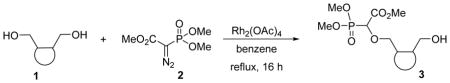
To a degassed solution of the diol 1 (1.0 g) and trimethylphosphonodiazoacetate 2 (1.2 equiv.) in benzene (100 mL) 1 mol% of rhodium(II)acetate was added and the mixture was refluxed under nitrogen for 16 hours. After completion of the reaction as seen from thin layer chromatography, the solvent was evaporated under reduced pressure. Purification was done by flash chromatography (Silica, 5% methanol in ethyl acetate) yielding the O-H insertion product 3 in 56–65% yield.
The following compounds were prepared following the protocol for O-H insertion reactions:
Methyl-2-(dimethoxyphosphoryl)-2-cis-2(hydroxymethyl)cyclopentyl)methoxy) acetate 3a
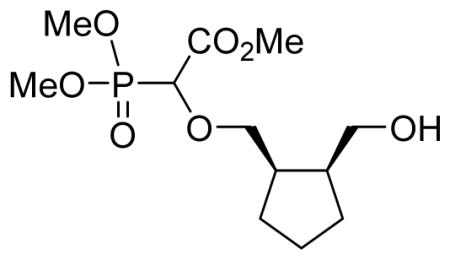
Light yellow oil (60%, 1.42 g, diastereomeric mixture); δH (300 MHz, CDCl3): 1.24–1.40 (m, 2H), 1.46–1.77 (m, 4H), 2.18–2.31 (m, 1H), 2.35–2.49 (m, 1H), 2.98–3.09 (m, 1H), 3.39–3.44 (m, 0.5H), 3.57–3.81 (m, 3.5H), 3.83–3.89 (m, 9H), 4.31–4.37 (m, 1H) ppm; δC (75 MHz, CDCl3): 23.2, 23.3, 27.7, 27.8, 28.4, 28.8, 40.2, 40.7, 44.5, 44.9, 52.9, 53.9–54.4 (m), 63.1, 63.3, 74.3–74.6 (m), 75.4, 75.6, 167.7, 167.8 ppm. δP (160 MHz, CDCl3): 17.0, 17.5 ppm; HRMS (EI) mass calculated for C12H23O7P (M)+ 310.1181; Found: 310.1189.
Methyl-2-(dimethoxyphosphoryl)-2-((2-(hydroxymethyl)cyclopropyl)methoxy)acetate 3b
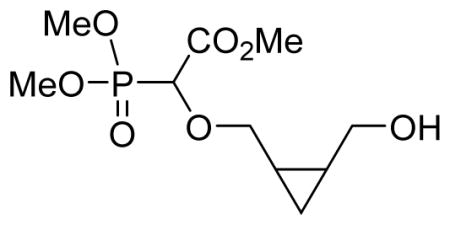
Light yellow oil (61%, 1.68 g, diastereomeric mixture); δH (400 MHz, CDCl3): 0.18–0.20 (m, 1H), 0.81–0.85 (m, 1H), 1.36–1.38 (m, 2H), 3.26–3.37 (m, 2H), 3.84–3.90 (m, 9H), 3.94–4.01 (m, 1.5H), 4.07–4.11 (m, 0.5H), 4.37 (d, 0.5H, JPH= 18.8 Hz), 4.43 (d, 0.5H, JPH= 18.8 Hz) ppm; δC (75 MHz, CDCl3): 7.6, 7.7, 14.7, 14.9, 18.7, 18.8, 53.0, 53.1, 54.0–54.5 (m), 62.3, 73.2–73.6 (m), 74.5, 74.8, 167.6, 167.8 ppm. δP (160 MHz, CDCl3): 15.9, 17.0 ppm. HRMS (EI) mass calculated for C10H19O7P (M)+ 282.0868; Found: 282.0853.
Methyl-2-(dimethoxyphosphoryl)-2-cis-6-(hydroxymethyl)cyclohex-3-enyl)methoxy)acetate 3c
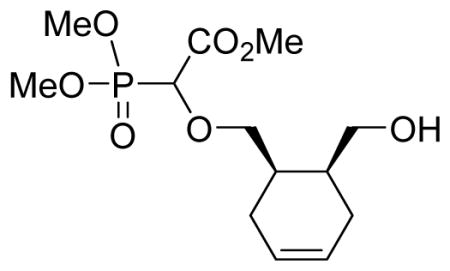
Light yellow oil (62%, 1.41 g, diastereomeric mixture); δH (400 MHz, CDCl3): 1.90–2.22 (m, 5H), 2.34–2.40 (m, 1H), 3.39–3.43 (m, 0.5H), 3.56–3.62 (m, 1H), 3.64–3.79 (m, 3H), 3.85–3.90 (m, 9.5H), 4.32–4.40 (m, 1H), 5.59–5.66 (m, 2H) ppm; δC (75 MHz, CDCl3): 25.8, 26.7, 26.9, 27.8, 33.7, 34.4, 36.8, 37.4, 52.9, 53.9–54.3 (m), 64.1, 64.2, 74.6, 74.7, 75.2, 75.4, 75.7, 125,1, 125.3, 125.6, 125.8, 167.7, 167.8 ppm; δP (160 MHz, CDCl3): 16.5, 16.8 ppm. HRMS (EI) mass calculated for C13H23O7P (M)+ 322.1181; Found: 322.1164.
Methyl-2-(dimethoxyphosphoryl)-2-(2-(hydroxymethyl)benzyloxy)acetate 3d
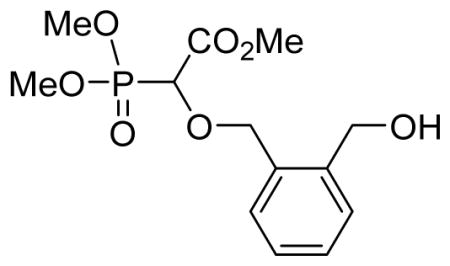
Light yellow oil (60%, 1.38 g); δH (400 MHz, CDCl3): 3.80–3.85 (m, 9H), 4.43 (d, 1H, JPH= 18.8 Hz), 4.67–4.72 (m, 2H), 4.82 (d, 1H, J= 12.4 Hz), 4.97 (d, 1H, J= 11.6 Hz), 7.28–7.32 (m, 2H), 7.37–7.41 (m, 1H), 7.47–7.49 (m, 1H) ppm; δC (75 MHz, CDCl3): 53.0, 54.0–54.5 (m), 62.9, 73.0–73.3 (m), 75.4, 127.9, 129.6, 130.5, 130.6, 133.5, 141.1, 167.6 ppm; δP (160 MHz, CDCl3): 17.3 ppm. HRMS (EI) mass calculated for C13H19O7P (M)+ 318.0868; Found: 318.0856.
Methyl 2-(2-azidoethoxy)-2-(dimethoxyphosphoryl)acetate 3f
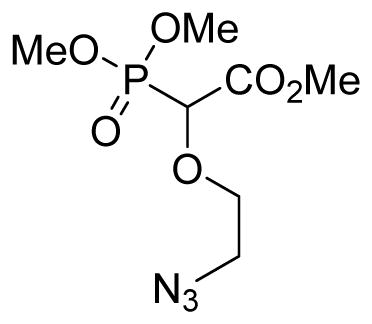
The O-H inserted product from the reaction of 2-bromoethanol and trimethylphosphonodiazoacetate was then treated with 3 equivalents of sodium azide in a 4:1 mixture of acetone/H2O and stirred at 60 °C for 6 h. After the completion of the reaction, acetone was evaporated and 25 mL of water were added. The aqueous layer was extracted with diethyl ether (3 × 20 mL). The organic layer was dried over anhydrous MgSO4 and the solvent was evaporated to obtain the azido compound 3f as a light yellow oil (65% over two steps, 1.38 g); δH (400 MHz, CDCl3): 3.47–3.49 (m, 2H), 3.70–3.75 (m, 1H), 3.85–3.89 (m, 10H), 4.44 (d, 1H, J= 17.6 Hz) ppm; δC (100 MHz, CDCl3): 50.7, 52.9, 54.1–54.3 (m), 71.3–71.4 (m), 75.9, 167.3 ppm; δP (160 MHz, CDCl3): 16.6 ppm. HRMS (ESI) mass calculated for C7H14O6P (M+H) 268.0693; Found: 268.0689.
cis-Methyl 2-(dimethoxyphosphoryl)-2-(4-hydroxybut-2-enyloxy)acetate 3g
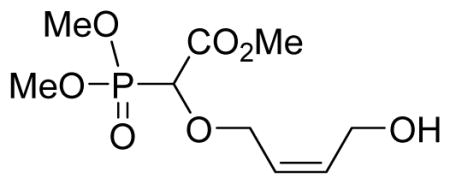
Light yellow oil (58%, 1.76 g); δH (300 MHz, CDCl3): 3.83–3.89 (m, 9H), 4.10–4.28 (m, 3H), 4.35–4.39 (m, 1H), 4.45 (d, 1H, JPH= 18.9 Hz), 5.66–5.74 (m, 1H), 5.91–5.99 (m, 1H) ppm; δC (75 MHz, CDCl3): 52.9, 54.1–54.4 (m), 58.4, 67.4, 67.6, 73.8, 75.9, 125.9, 135.1, 167.8 ppm. δP (160 MHz, CDCl3): 16.9 ppm. HRMS (ESI) mass calculated for C9H17O7P (M+H) 269.079; Found: 269.0788.
Methyl-2-(dimethoxyphosphoryl)-2-trans-2(hydroxymethyl)cyclopentyl)methoxy) acetate 3o
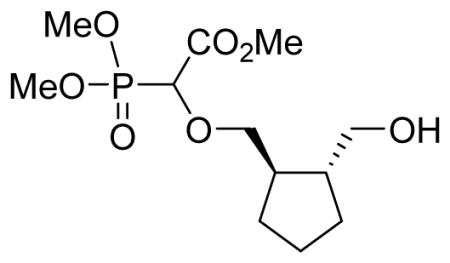
Light yellow oil (61%, 1.45 g, diastereomeric mixture); δH (300 MHz, CDCl3): 1.25–1.36 (m, 2H), 1.51–1.60 (m, 2H), 1.73–1.83 (m, 2H), 1.86–2.03 (m, 2H), 3.38–3.52 (m, 2H), 3.58–3.70 (m, 2H), 3.81–3.87 (m, 9H), 4.33–4.42 (m, 1H) ppm; δC (75 MHz, CDCl3): 24.2, 24.4, 29.7, 29.8, 29.9, 43.4, 43.6, 46.6, 46.8, 52.9, 54.1–54.3 (m), 66.7, 66.8, 75.2, 75.6, 167.6, 167.7 ppm. δP (160 MHz, CDCl3): 17.1, 17.2 ppm; HRMS (ESI) mass calculated for C12H23O7P (M+Na) 333.1078; Found: 333.1073.
4.2. General experimental procedure for Mitsunobu reactions
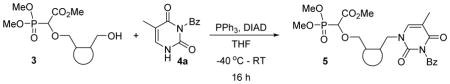
To a flame dried flask, PPh3 (2.1 equiv.) was added. After degassing and flushing nitrogen for 10 minutes, dry THF was added and the solution was cooled in an ice bath. Then, diisopropylazodicarboxylate (DIAD) (2.0 equiv.) was added in a drop wise manner to the reaction mixture and a colour change was observed. This was allowed to stir for 30 minutes. In another flask the alcohol 3 (1.0 equiv.) and N-3-benzoyl thymine 4a (1.3 equiv.) were taken; degassed, flushed with nitrogen and then dissolved in THF and allowed to cool to −40 °C in an acetonitrile-dry ice bath. To this flask the PPh3-DIAD solution was added and allowed to attain room temperature gradually, furtherstirred for approximately 16 hours or until complete consumption of the alcohol 3. After completion of the reaction, the solvent was evaporated under reduced pressure and purification of the residue was done by flash chromatography (Silica, 3% methanol in ethyl acetate) affording phosphonucleoside products 5 in 45–66% yield.
The following compounds where prepared following the protocol for Mitsunobu reactions:
Methyl-2-(dimethoxyphosphoryl)-2-cis-2(3-benzoyl-2,4-dioxo-5-methyl-pyrimidin-1-yl)cyclopentyl)methoxy) acetate 5a
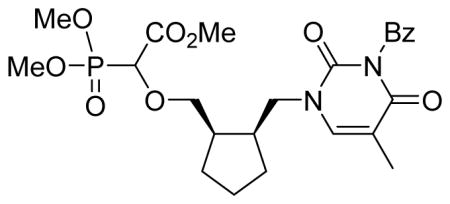
Light yellow oil (60%, 1.01 g, diastereomeric mixture); δH (400 MHz, CDCl3): 1.37–1.59 (m, 4H), 1.71–1.81 (m, 2H), 2.00 (s, 3H), 2.42–2.43 (m, 2H), 3.47–3.55 (m, 1H), 3.64–3.72 (m, 1H), 3.85–3.89 (m, 9H), 3.94–4.05 (m, 2H), 4.29–4.37 (m, 1H), 7.46–7.52 (m, 3H), 7.63–7.67 (m, 1H), 7.92–7.94 (m, 2H) ppm; δC (75 MHz, CDCl3): 12.3, 22.3, 22.6, 27.7, 27.9, 28.4, 28.8, 40.9, 41.0, 41.8, 41.9, 48.5, 53.0, 53.9–54.3 (m), 73.9, 75.3, 110.4, 110.5, 129.1, 130.4, 131.8, 134.9, 141.0, 141.3, 150.3, 163.3, 163.4, 167.8, 169.4 ppm; δP (160 MHz, CDCl3): 17.1, 17.5 ppm. HRMS (EI) mass calculated for C24H31N2O9P (M)+ 522.1767; Found: 522.1755.
Methyl-2-(dimethoxyphosphoryl)-2-((2-(3-benzoyl-2,4-dioxo-5-methyl-pyrimidin-1-yl)cyclopropyl)methoxy)acetate 5b
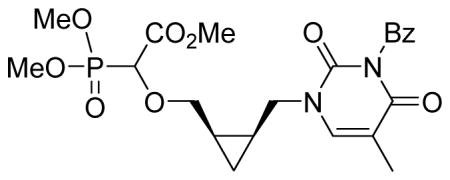
Light yellow oil (50%, 875 mg, diastereomeric mixture); δH (400 MHz, CDCl3): 0.29–0.33 (m, 0.6H), 0.46–0.49 (m, 0.4H), 0.89–0.95 (m, 1H), 1.27–1.49 (m, 2H), 2.01 (s, 3H), 3.44–3.68 (m, 2H), 3.83–3.89 (m, 9.5H), 3.94–4.00 (m, 1H), 4.16–4.21 (m, 0.5H), 4.34–4.43 (m, 1H), 7.48–7.53 (m, 3H), 7.64–7.68 (m, 1H), 7.94–7.97 (m, 2H) ppm; δC (150 MHz, CDCl3): 7.2, 7.7, 11.9, 12.0, 15.1, 15.4, 15.5, 15.6, 46.5, 47.1, 52.7, 53.6–53.9 (m), 72.2, 72.3, 72.5, 72.6, 75.5, 110.1, 110.3, 128.8, 130.2, 131.5, 134.6, 140.2, 140.7, 149.8, 149.8, 163.0, 163.2, 167.3, 167.4, 169.0, 169.1 ppm. δP (160 MHz, CDCl3): 16.9, 17.6 ppm. HRMS (EI) mass calculated for C22H27N2O9P (M)+ 494.1454; Found: 494.1443.
Methyl 2-(dimethoxyphosphoryl)-2-cis-6-(3-benzoyl-2,4-dioxo-5-methyl-pyrimidin-1-yl)cyclohex-3-enyl)methoxy)acetate 5c
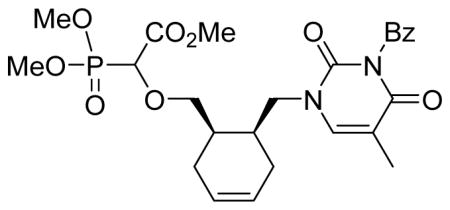
Light yellow oil (63%, 1.04 g, diastereomeric mixture); δH (300 MHz, CDCl3): 1.79–1.92 (m, 2H), 1.98–1.99 (m, 3H), 2.08–2.42 (m, 4H), 3.46–3.51 (m, 0.5H), 3.60–3.65 (m, 0.5H), 3.71–3.79 (m, 1.5H), 3.82–3.87 (m, 9H), 3.90–4.13 (m, 1.5H), 4.33 (d, 1H, JPH= 18.9 Hz), 5.61–5.64 (m, 2H), 7.45–7.51 (m, 2H), 7.61–7.66 (m, 2H), 7.89–7.93 (m, 2H) ppm; δC (75 MHz, CDCl3): 12.1, 12.2, 26.0, 26.2, 27.4, 27.8, 33.9, 34.4, 34.8, 49.9, 53.0, 53.8–54.4 (m), 74.0, 74.2, 75.2, 110.1, 125.1, 125.3, 129.1, 130.4, 131.8, 134.8, 141.9, 142.1, 150.2, 150.3, 163.4, 167.5, 167.8, 169.3 ppm; δP (160 MHz, CDCl3): 17.1, 17.5 ppm; HRMS (EI) mass calculated for C25H31N2O9P (M)+ 534.1767; Found: 534.1786.
Methyl 2-(dimethoxyphosphoryl)-2-(2-(3-benzoyl-2,4-dioxo-5-methyl-pyrimidin-1-yl)benzyloxy)acetate 5d
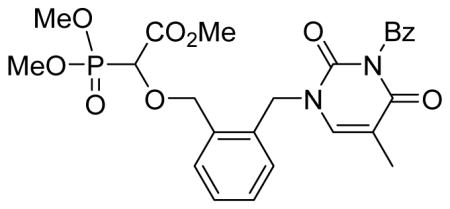
Light yellow oil (60%, 1.00 g); δH (400 MHz, CDCl3): 1.96 (s, 3H), 3.81–3.85 (m, 9H), 4.45 (d, 1H, JPH= 19.2 Hz), 4.65 (d, 1H, J= 10.8 Hz), 4.90 (d, 1H, J= 11.2 Hz), 5.17–5.27 (m, 2H), 7.32–7.34 (m, 3H), 7.40–7.43 (m, 2H), 7.50–7.54 (m, 2H), 7.64–7.68 (m, 1H), 7.97 (d, 2H, J= 8.0 Hz) ppm; δC (75 MHz, CDCl3): 12.3, 47.5, 53.0, 54.1–54.3 (m), 72.9, 73.1, 74.1, 111.1, 128.3, 128.6, 129.1, 130.0, 130.5, 131.2, 131.7, 133.6, 134.9, 135.8, 140.6, 150.5, 163.2, 167.4, 167.5, 169.1 ppm; δP (160 MHz, CDCl3): 17.1 ppm; HRMS (EI) mass calculated for C25H27N2O9P (M)+ 530.1454; Found: 530.1440.
cis-Methyl 2-(dimethoxyphosphoryl)-2-(4-(3-benzoyl-2,4-dioxo-5-methyl-pyrimidin-1-yl)but-2-enyloxy)acetate 5g
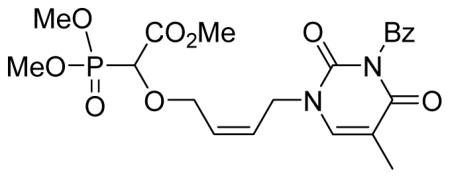
Light yellow oil (51%, 910 mg); δH (300 MHz, CDCl3): 1.96 (s, 3H), 3.81–3.86 (m, 9H), 4.19–4.25 (m, 1H), 4.59–4.35 (m, 4H), 5.71–5.80 (m, 1H), 5.85–5.93 (m, 1H), 7.43–7.52 (m, 3H), 7.62–7.67 (m, 1H), 7.92 (d, 2H, J= 7.5 Hz) ppm; δC (75 MHz, CDCl3): 12.4, 42.9, 52.9, 54.1–54.4 (m), 58.4, 67.4, 67.6, 75.9, 110.9, 125.9, 129.1, 131.8, 141.0, 150.3, 163.3, 167.8, 169.6 ppm. δP (160 MHz, CDCl3): 17.1 ppm. LRMS (EI) mass calculated for C21H25N2O9P (M)+ 480.1298; Found: 480.1301.
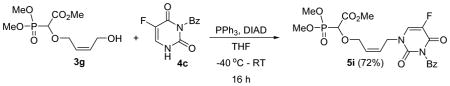
Methyl-2-(4-(3-benzoyl-5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)but-2-enyloxy)-2-(dimethoxyphosphoryl)acetate 5i
Colourless oil (72%, 1.4 g); δH (300 MHz, CDCl3): 3.85–3.87 (m, 9H), 4.19–4.22 (m, 1H), 4.34–4.37 (m, 1H), 4.43 (d, 1H, JPH= 9.4 Hz), 4.65–4.56 (m, 2H), 5.81–5.83 (m, 1H), 5.95–5.96 (m, 1H), 7.51–7.54 (m, 2H), 7.67–7.69 (m, 1H), 7.90–7.95 (m, 3H); δC (75 MHz, CDCl3): 45.3, 53.1, 53.0, 54.1–54.3 (m), 67.4, 67.6, 74.5, 128.4, 128.8, 129.2, 129.3, 129.6, 130.6, 131.1, 135.4, 138.5, 148.5, 167.4, 167.5 ppm; δP (240 MHz, CDCl3): 16.2 ppm; HRMS (ESI) mass calculated for C20H22FN2O9P (M+H) 485.1125; Found: 485.1119.
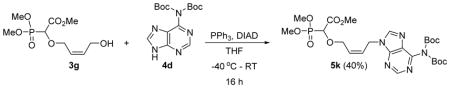
Methyl-2-(4-(6-(bis(tert-butoxycarbonyl)amino)-9H-purin-9-yl)but-2-enyloxy)-2-(dimethoxyphosphoryl)acetate 5k
Light yellow oil (40%, 920mg); δH (300 MHz, CDCl3): 1.46 (s, 18H), 3.84–3.89 (m, 9H), 4.34–4.40 (m, 1H), 4.50–4.56 (m, 1H), 4.57 (d, 1H, JPH= 18.9 Hz ), 5.07 (d, 2H, J= 6 Hz), 5.86–5.98 (m, 2H), 8.26 (s, 1H), 8.87 (s, 1H); δP (240 MHz, CDCl3): 16.3 ppm; HRMS (ESI) mass calculated for C24H36N5O10P (M+H) 586.2277; Found: 586.2272.
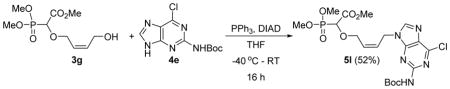
Methyl-2-(4-(2-(tert-butoxycarbonylamino)-6-chloro-9H-purin-9-yl)but-2-enyloxy)-2-(dimethoxyphosphoryl)acetate 5l
Colourless oil (52%, 804mg); δH (300 MHz, CDCl3): 1.55 (s, 9H), 3.83–3.89 (m, 9H), 4.39–4.45 (m, 1H), 4.53–4.59 (m, 1H), 4.58 (d, 1H, JPH= 18.6 Hz ), 4.96–4.98 (m, 2H), 5.83–5.96 (m, 2H), 7.79 (s, 1H), 8.09 (s, 1H)ppm; δC (75 MHz, CDCl3): 28.2, 40.9, 53.0, 54.1–54.4(m), 67.7, 74.4, 81.6, 127.7, 130.0, 144.2, 150.3, 151.2, 152.5, 152.7, 167.5, 167.6 ppm; δP (240 MHz, CDCl3): 16.4 ppm; HRMS (ESI) mass calculated for C19H27ClN5O8P (M+H) 520.1364; Found: 520.1365.
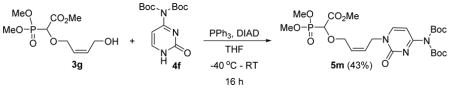
Methyl 2-(4-(4-(bis(tert-butoxycarbonyl)amino)-2-oxopyrimidin-1(2H)-yl)but-2-enyloxy)-2-(dimethoxyphosphoryl)acetate 5m
Colourless oil (43%, 805mg); δH (300 MHz, CDCl3): 1.55 (s, 18H), 3.83–3.88 (m, 9H), 4.21–4.26 (m, 1H), 4.39–4.44 (m, 1H), 4.44 (d, 1H, JPH= 18.6 Hz ), 4.54–4.70 (m, 2H), 5.77–5.92 (m, 2H), 7.02 (d, 1H, J=7.5 Hz), 7.80 (d, 1H, J= 7.5 Hz) ppm; δC (75 MHz, CDCl3): 27.7, 47.0, 53.0, 54.2–54.3 (m), 67.5, 67.7, 74.5, 84.8, 96.5, 128.9, 129.0, 147.9, 149.6, 155.0, 162.4, 167.6 ppm; δP (240 MHz, CDCl3): 16.3 ppm; HRMS (ESI) mass calculated for C23H36N3O11P (M+H) 562.2165; Found: 562.2161.
Methyl-2-(dimethoxyphosphoryl)-2-trans-2(3-benzoyl-2,4-dioxo-5-methyl-pyrimidin-1-yl)cyclopentyl)methoxy) acetate 5o
Light yellow oil (45%, 758 mg, diastereomeric mixture); δH (400 MHz, CDCl3): 1.54–1.68 (m, 4H), 1.75–1.88 (m, 2H), 1.94–2.22 (s, 3H & m, 2H), 3.37–3.46 (m, 1H), 3.65–3.93 (m, 12H), 4.31–4.38 (m, 1H), 7.46–7.51 (m, 3H), 7.61–7.66 (m, 1H), 7.91–7.93 (m, 2H) ppm; δC (75 MHz, CDCl3): 12.3, 24.3, 24.4, 28.7, 0.9, 31.0, 42.4, 44.1, 45.0, 51.9, 52.9, 54.0–54.1 (m), 75.2, 75.4, 110.5, 110.6, 129.1, 130.4, 131.8, 134.8, 141.0, 141.1, 150.4, 163.2, 167.7, 167.8, 169.3 ppm; δP (160 MHz, CDCl3): 17.3, 17.4 ppm. HRMS (EI) mass calculated for C24H31N2O9P (M)+ 522.1767; Found: 522.1755.
4. 3. Hydrogenation of compounds 5c, 5g and 5i
The hydrogenation of phosphonucleosides 5c, 5g and 5i was done in the presence of palladium (5% on carbon) catalyst. The catalyst was added to a hydrogenation vessel followed by a solution of phosphonucleosides in methanol. The reaction mixture was shaken under hydrogen under 30 psi at room temperature for 2–12 hours. The completion of the reaction was monitored by checking the 1H NMR of the crude reaction mixture. After completion the reaction mixture was filtered through a short column of celite and the filtrate was concentrated in vacuo. Purification of the residue was done by flash chromatography (Silica, 3% methanol in ethyl acetate) which yielded the reduced phosphonucleoside products 5e, 5h and 5j in excellent yields.
Methyl 2-(dimethoxyphosphoryl)-2-cis-6-(3-benzoyl-2,4-dioxo-5-methyl-pyrimidin-1-yl)cyclohexyl)methoxy)acetate 5e
Colourless oil (96%, 142 mg, diastereomeric mixture); δH (300 MHz, CDCl3): 1.39–1.48 (m, 6H), 1.67–1.71 (m, 2H), 1.98 (s, 3H), 2.12–2.20 (m, 2H), 3.43–3.47 (m, 0.5H), 3.63–3.80 (m, 2.5H), 3.84–3.88 (m, 9H), 4.04–4.17 (m, 1H), 4.29–4.39 (m, 1H), 7.46–7.51 (m, 2H), 7.56–7.66 (m, 2H), 7.90–7.94 (m, 2H) ppm; δC (75 MHz, CDCl3): 12.2, 22.9, 23.7, 25.9, 27.0, 36.8, 37.7, 53.0, 54.1–54.3 (m), 73.9, 74.1, 110.3, 110.5, 129.1, 130.4, 131,8, 134.8, 141.3, 141.6, 150.5, 163.4, 167.9, 169.4 ppm; δP (160 MHz, CDCl3): 17.1, 17.6 ppm; HRMS (EI) mass calculated for C25H33N2O9P (M)+ 536.1924; Found: 536.1895.
Methyl 2-(dimethoxyphosphoryl)-2-(4-(3-benzoyl-2,4-dioxo-5-methyl-pyrimidin-1-yl) butyloxy)acetate 5h
It was observed that the N3-benzoyl group of the thymine part was also removed after hydrogenation.
Colourless oil (95%, 105 mg); δH (300 MHz, CDCl3): 1.67–1.71 (m, 2H), 1.82–1.87 (m, 2H), 1.93 (s, 3H), 3.61–3.71 (m, 2H), 3.78–3.80 (m, 2H), 3.85–3.88 (m, 9H), 4.37 (d, 1H, JPH= 19.2 Hz), 7.18 (s, 1H), 9.67 (brs, 1H) ppm; δC (75 MHz, CDCl3): 12.3, 25.9, 26.0, 48.0, 52.9, 54.0–54.2 (m), 72.6, 72.7, 110.5, 140.9, 150.7, 163.9, 167.7 ppm; δP (160 MHz, CDCl3): 17.3 ppm; LRMS (EI) mass calculated for C14H23N2O8P (M)+ 378.1; Found: 378.6.
Methyl-2-(4-(3-benzoyl-5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)butoxy)-2-(dimethoxyphosphoryl)acetate 5j
Colourless oil (92%, 170 mg); δH (300 MHz, CDCl3): 1.69–1.73 (m, 2H), 1.87–1.94 (m, 2H), 3.65–3.70 (m, 2H), 3.76–3.93 (m, 11H), 4.35 (d, 1H, JPH= 19.2 Hz), 7.49–7.54 (m, 2H), 7.65–7.70 (m, 1H), 7.79 (d, 1H, J = 6 Hz), 7.90–7.94 (m, 2H) ppm; δC (75 MHz, CDCl3): 25.6, 26.0, 48.9, 53.0, 54.1–54.2 (m), 72.7, 72.9, 75.2, 129.3, 129.5, 129.9, 130.6, 131.1, 135.4, 138.2, 141.4, 148.5, 156.3, 156.7, 167.5, 167.7 ppm; δP (240 MHz, CDCl3): 16.6 ppm. HRMS (EI) mass calculated for C20H24FN2O9P (M)+ 486.1203; Found: 486.1190.
4.4. Synthesis of 1,2,3-triazole linked phosphonucleoside 5f by copper catalyzed click reaction
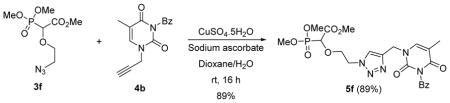
The dipolar cycloaddition of 3f (1.0 equiv.) and 4b (1.0 equiv.) was done in the presence of 5 mol% of CuSO4.5H2O and 10 mol% of sodium ascorbate in 2:1 mixture of dioxane and H2O at room temperature for 16 h. After the completion of the reaction, the solvent was evaporated off under reduced pressure and purification was done by flash chromatography (Silica, 3% methanol in ethyl acetate) affording 1,2,3-triazole linked phosphonucleoside product 5f in 89% yield. Light yellow oil (89%, 420 mg); δH (600 MHz, CDCl3): 1.95 (s, 3H), 3.76–3.80 (m, 9H), 3.98–4.08 (m, 2H), 4.34 (d, 1H, JPH= 18.0 Hz), 4.62–4.63 (m, 2H), 4.99–5.05 (m, 2H), 7.48–7.50 (m, 2H), 7.63–7.66 (m, 1H), 7.91 (d, 1H, J = 6 Hz), 7.97 (s, 1H) ppm; δC (75 MHz, CDCl3): 12.4, 42.9, 50.2, 53.1, 54.1–54.3 (m), 70.5, 70.6, 75.2, 111.2, 124.9, 129.2, 130.5, 131.6, 135.0, 140.0, 141.8, 149.8, 163.1, 166.9, 169.0 ppm; δP (240 MHz, CDCl3): 15.6 ppm. HRMS (EI) mass calculated for C22H26N5O9P (M)+ 535.1468; Found: 535.1475.
4.5. General experimental procedure for deprotection reactions
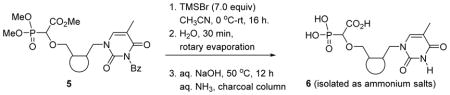
The deprotection of carboxyphosphono nucleoside derivatives 5 was done using TMSBr. The phosphonucleosides were treated with 7 equivalents of TMSBr at 0 °C, slowly allowed to return to room temperature and stirred overnight. Water was added to hydrolyze the resulting silyl ethers and the reaction mixture was concentrated. The residue was then dissolved in 1M NaOH (10 equivalents) and stirred at 50 °C overnight. The reaction mixture was concentrated and acidified to pH 1–2.5 and washed 3 times with DCM to remove the benzoic acid. A small pad of activated charcoal (G-60) was made in a sintered funnel and then washed with 20% ammonia and water. Next the acidified aqueous layer was packed on the charcoal column, which was then washed twice with water to remove the inorganic impurities and then the phosphonucleosides (6a–6f & 6i–6n) were eluted by using a 1:1 mixture of ethanol and a 20% ammonia solution. The fractions were spotted on a TLC plate and the UV active fractions were combined and concentrated by lyophilisation. Phosphonucleosides (6g & 6h) were eluted as ammonium salt by eluting with 20% ammonia solution.
The following compounds where prepared following the protocol for deprotection reactions:
2-cis-2-((2,4-Dioxo-5-methyl-pyrimidin-1-yl)methyl)cyclopentyl)methoxy)-2-phosphonoacetic acid 6a
White solid (55%, 79 mg, diastereomeric mixture); mp above 300 °C; δH (400 MHz, D2O): 1.32–1.80 (m, 6H), 1.85 (s, 3H), 2.37 (brs, 2H), 3.44–3.75 (m, 3H), 3.89–3.92 (m, 1H), 4.18 (d, 1H, JPH= 18.2 Hz), 7.54 (s, 1H) ppm; δC (150 MHz, D2O): 12.3, 21.7, 21.8, 27.5, 27.5, 39.9, 40.6, 48.4, 71.7, 82.1–82.4 (d), 82.9–83.3 (d), 110.1, 142.4, 159.9, 167.7, 176.2 ppm; δP (160 MHz, CDCl3): 11.7, 11.8 ppm; HRMS (ESI) mass calculated for C14H21N2O8P (M-H) 375.0962; Found: 375.0962.
2-cis-2-((2,4-Dioxo-5-methyl-pyrimidin-1-yl)methyl)cyclopropyl)methoxy)-2-phosphonoacetic acid 6b
White solid (49%, 69 mg, diastereomeric mixture); mp 230–232 °C; δH (400 MHz, D2O): 0.44–0.46 (m, 1H), 0.88–0.91 (m, 1H), 1.36–1.37 (m, 2H), 1.92 (s, 3H), 3.52–3.72 (m, 3H), 3.97–4.14 (m, 2H), 7.64–7.67 (m, 1H) ppm; δC (75 MHz, D2O): 11.1, 13.8, 16.5, 17.5, 50.5, 73.7, 84.1, 112.7, 146.3, 154.3, 169.5, 174.5 ppm; δP (160 MHz, CDCl3): 11.6, 11.7 ppm. HRMS (ESI) mass calculated for C12H17N2O8P (M-H) 347.0649; Found: 347.0646.
2-cis-6-((2,4-Dioxo-5-methyl-pyrimidin-1-yl)methyl)cyclohex-3-enyl)methoxy)-2-phosphonoacetic acid 6c
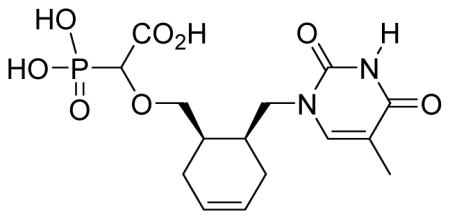
White solid (60%, 86 mg, diastereomeric mixture); mp 175–177 °C; δH (400 MHz, D2O): 1.87 (s, 3H), 1.93–2.27 (m, 6H), 3.42–3.46 (m, 1H), 3.59–3.67 (m, 2H), 3.74–3.91 (m, 3H), 5.70–5.79 (m, 2H), 7.67–7.68 (m, 1H) ppm; δC (75 MHz, D2O): 11.1, 25.5, 26.3, 29.6, 33.6, 34.3, 48.8, 80.0, 109.9, 125.1, 125.8, 143.9, 152.6, 166.8, 176.9 ppm; δP (160 MHz, CDCl3): 12.2 ppm; HRMS (ESI) mass calculated for C15H21N2O8P (M-H) 387.0962; Found: 387.0961.
2-(2-((2,4-Dioxo-5-methyl-pyrimidin-1-yl)methyl)benzyloxy)-2-phosphonoacetic acid 6d
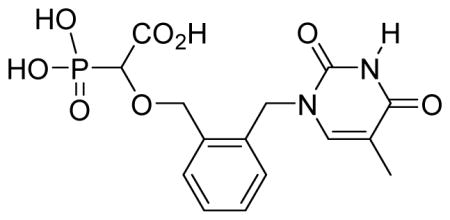
White solid (52%, 75 mg); mp 156–158 °C; δH (300 MHz, D2O): 1.86 (s, 3H), 3.95 (d, 1H, JPH= 17.4 Hz), 4.53–4.57 (d, 1H, J= 12 Hz), 4.65–4.66 (m, 1H), 5.15 (s, 2H), 7.07–7.11 (m, 1H), 7.33–7.39 (m, 2H), 7.44–7.48 (m, 1H), 7.55 (s, 1H) ppm; δC (75 MHz, D2O): 11.1, 48.4, 70.9, 78.9, 80.7, 111.3, 126.1, 128.1, 128.8, 130.1, 134.6, 143.3, 152.6, 166.5, 176.2 ppm; δP (240 MHz, CDCl3): 11.7 ppm; HRMS (ESI) mass calculated for C15H17N2O8P (M-H) 383.0649; Found: 383.0654.
2-cis-6-((2,4-Dioxo-5-methyl-pyrimidin-1-yl)methyl)cyclohexyl)methoxy)-2-phosphonoacetic acid 6e
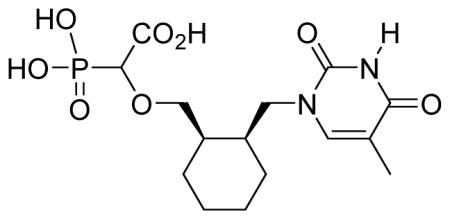
White solid (59%, 86 mg); mp 165–167 °C; δH (300 MHz, D2O): 1.33–1.36 (m, 4H), 1.47–1.61 (m, 4H), 1.86 (s, 3H), 2.06–2.13 (m, 2H), 3.38–3.64 (m, 3H), 3.87–3.96 (m, 2H), 7.58–7.60 (m, 1H) ppm; δC (150 MHz, D2O): 10.9, 22.7, 24.7, 25.0, 36.2, 36.5, 47.6, 71.9, 80.0, 81.0, 109.7, 143.1, 151.9, 166.7, 176.5 ppm; δP (160 MHz, CDCl3): 12.9, 13.0 ppm ; HRMS (ESI) mass calculated for C15H23N2O8P (M-H) 389.1119; Found: 389.1119.
2-(2-(4-((5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)-2-phosphonoacetic acid 6f
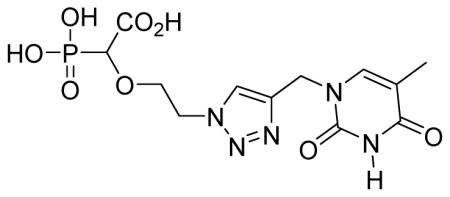
White solid (50%, 73 mg); mp 189–191 °C; δH (600 MHz, 323K, D2O): 1.97 (s, 3H), 3.96–3.98 (m, 1H), 4.06 (d, 1H, JPH= 12.0 Hz), 4.11–4.12 (m, 1H), 4.74 (s, 2H), 5.15 (s, 2H), 7.64 (s, 1H), 8.33 (s, 1H) ppm; δC (75 MHz, D2O): 11.1, 42.7, 50.5, 70.3, 76.7, 78.3, 110.9, 125.8, 142.6, 144.9, 152.0, 166.8, 172.2 ppm; δP (160 MHz, D2O): 10.7 ppm; HRMS (ESI) mass calculated for C12H16N5O8P (M-H): 388.0663; Found: 388.0666.
cis-2-(4-(2,4-Dioxo-5-methyl-pyrimidin-1-yl)but-2-enyloxy)-2-phosphonoacetic acid 6g
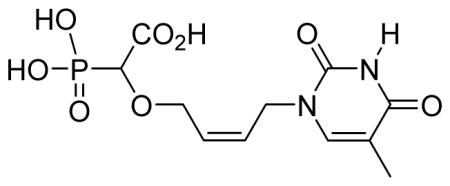
White solid (61%, 84 mg); mp 218–220 °C; δH (300 MHz, D2O): 1.86 (s, 3H), 3.98 (d, 1H, JPH= 18.6 Hz), 4.12–4.29 (m, 2H), 4.44 (d, 2H, J= 6.0 Hz), 5.59–5.67 (m, 1H), 5.84–5.92 (m, 1H), 7.49 (s, 1H) ppm; δC (75 MHz, D2O): 11.2, 45.4, 66.3, 66.5, 79.4, 81.1, 110.9, 126.6, 130.5, 142.8, 152.3, 167.1, 176.9 ppm; δP (160 MHz, D2O): 11.6 ppm; HRMS (ESI) mass calculated for C11H15N2O8P (M-H): 333.0493; Found: 333.0495.
2-(4-(2,4-Dioxo-5-methyl-pyrimidin-1-yl)butyloxy)-2-phosphonoacetic acid 6h
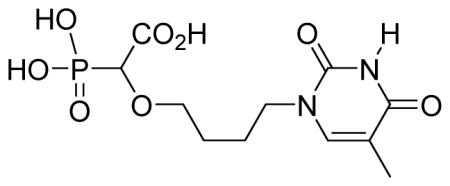
White solid (63%, 112 mg); mp 182–184 °C; δH (400 MHz, D2O): 1.68–1.82 (m, 4H), 1.93 (s, 3H), 3.47–3.56 (m, 1H), 3.67–3.69 (m, 1H), 3.82–3.96 (m, 2H), 3.97–4.02 (m, 1H), 7.58 (s, 1H) ppm; δC (100 MHz, D2O): 11.2, 18.3, 24.3, 25.3, 48.3, 73.9, 110.3, 143.3, 152.1, 166.1, 171.4 ppm; δP (160 MHz, CDCl3): 12.4, 12.5 ppm; HRMS (ESI) mass calculated for C11H17N2O8P (M-H) 335.0649; Found: 335.0652.
2-Cis-(4-(5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)but-2-enyloxy)-2-phosphonoacetic acid 6i
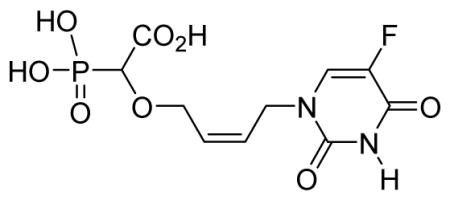
White solid (67%, 70 mg); mp 185–187 °C; δH (300 MHz, D2O): 3.95 (d, 1H, JPH= 17.7Hz), 4.09–4.26 (m, 2H), 4.42 (d, 2H, J = 6.9 Hz), 5.59–5.67 (m, 1H), 5.84–5.92 (m, 1H), 7.83–7.85 (m, 1H) ppm; δC (75 MHz, D2O): 45.8, 65.9, 79.3, 81.4, 126.1, 130.1, 131.1, 142.5, 150.0, 161.4, 177.6 ppm; δP (240 MHz, CDCl3): 11.7 ppm; HRMS (ESI) mass calculated for C10H12FN2O8P (M-H) 337.0242; Found: 337.0247.
2-(4-(5-Fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)butoxy)-2-phosphonoacetic acid 6j
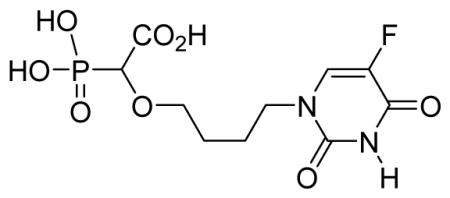
White solid (72%, 80 mg); mp 146–148 °C; δH (300 MHz, D2O): 1.55–1.64 (m, 2H), 1.69–1.79 (m, 2H), 3.38–3.45 (m, 1H), 3.55–3.62 (m, 1H), 3.76 (t, 2H, J = 7.2 Hz), 3.90 (d, 1H, JPH= 18.0 Hz), 7.88 (d, 1H, J= 6.0Hz) ppm; δC (75 MHz, D2O): 24.8, 25.5, 48.9, 71.2, 71.4, 80.0, 80.9, 131.4, 139.3, 151.1, 160.2, 176.6 ppm; δP (240 MHz, CDCl3): 12.3 ppm; HRMS (ESI) mass calculated for C10H12FN2O8P (M-H) 337.0242; Found: 337.0237.
2-Cis-(4-(6-amino-9H-purin-9-yl)but-2-enyloxy)-2-phosphonoacetic acid 6k
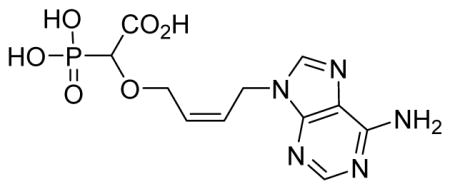
White solid (73%, 45 mg); mp 204–206 °C; δH (300 MHz, D2O): 4.01 (d, 1H, JPH= 17.4Hz), 4.17–4.32 (m, 2H), 4.86–4.92 (m, 2H), 5.75–5.83 (m, 1H), 5.87–5.95 (m, 1H), 8.13 (s, 1H), 8.17 (s, 1H) ppm; δC (150 MHz, D2O): 40.8, 66.1, 79.3, 80.3, 118.1, 126.5, 130.2, 142.0, 148.7, 152.4, 155.2, 176.4 ppm; δP (240 MHz, CDCl3): 11.9 ppm; HRMS (ESI) mass calculated for C11H14N5O6P (M-H) 342.0608; Found: 342.0619.
2-Cis-(4-(2-amino-6-oxo-1H-purin-9(6H)-yl)but-2-enyloxy)-2-phosphonoacetic acid 6l
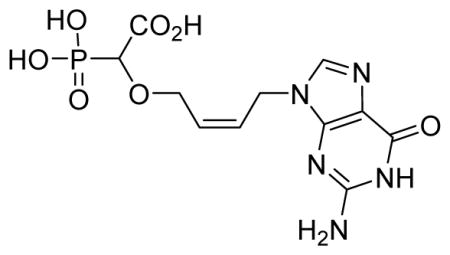
White solid (67%, 120 mg); mp 170–172 °C; δH (300 MHz, D2O): 4.07 (d, 1H, JPH= 18.0Hz), 4.19–4.36 (m, 2H), 4.72–4.74 (m, 2H), 5.69–5.77 (m, 1H), 5.83–5.91 (m, 1H), 7.80 (s, 1H) ppm; δC (75 MHz, D2O): 40.4, 66.6, 77.7, 80.4, 115.4, 126.8, 130.2, 139.5,151.0, 153.6, 158.4, 176.9 ppm; δP (240 MHz, CDCl3): 12.2 ppm; HRMS (ESI) mass calculated for C11H14N5O7P (M-H) 358.0557; Found: 358.0554.
2-Cis-(4-(4-amino-2-oxopyrimidin-1(2H)-yl)but-2-enyloxy)-2-phosphonoacetic acid 6m
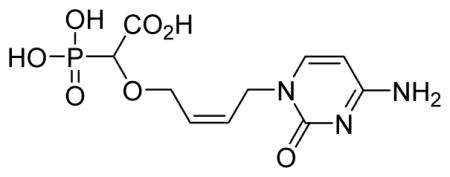
White solid (75%, 30 mg); mp 234–236 °C; δH (300 MHz, D2O): 3.91 (d, 1H, JPH= 18.0Hz), 4.03–4.19 (m, 2H), 4.37 (d, 2H, J = 6.3 Hz), 5.53–5.61 (m, 1H), 5.70–5.80 (m, 1H), 5.88 (d, 1H, J = 7.2 Hz), 7.54 (d, 1H, J = 7.5 Hz) ppm; δC (75 MHz, D2O): 46.4, 66.2, 78.3, 80.7, 95.5, 127.1, 129.8, 146.3, 157.7, 166.4, 176.3 ppm; δP (240 MHz, CDCl3): 12.1 ppm; HRMS (ESI) mass calculated for C10H14N3O7P (M-H) 318.0491; Found: 318.0494.
2-Trans-2-((5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)cyclopentyl)methoxy)-2-phosphonoacetic acid 6o
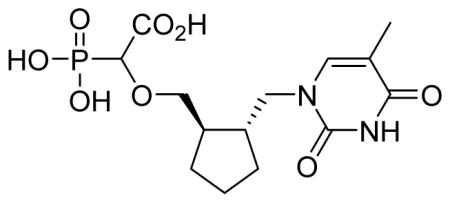
White solid (65%, 93 mg, diastereomeric mixture); mp above 300 °C; δH (300 MHz, D2O): 1.21–1.80 (m, 6H), 1.85 (s, 3H), 1.92–2.07 (m, 2H), 3.28–3.48 (m, 2H), 3.68–3.90 (m, 3H), 7.56 (s, 1H) ppm; δC (150 MHz, D2O): 11.2, 22.1, 23.3, 28.6, 34.4, 42.3, 42.6, 52.3, 75.6, 80.8, 81.1, 110.3, 143.7, 152.7, 166.9, 176.4 ppm; δP (160 MHz, CDCl3): 12.9, 13.0 ppm; HRMS (ESI) mass calculated for C14H21N2O8P (M-H) 375.0962; Found: 375.0966.
2-Cis-((4-hydroxybut-2-en-1-yl)oxy)-2-phosphonoacetic acid 6n
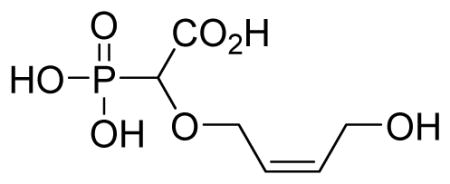
Isolated as a sodium salt. White solid (79%, 133 mg); mp above 300 °C; δH (300 MHz, D2O): 3.83 (d, 1H, JPH= 18.0Hz), 3.97–4.13 (m, 4H), 5.68–5.81 (m, 2H) ppm; δC (150 MHz, D2O): 57.1, 66.1, 66.2, 81.0, 81.9, 128.7, 131.6, 178.9 ppm; δP (160 MHz, CDCl3): 11.7, 13.5 ppm; HRMS (ESI) mass calculated for C6H11O7P (M-H) 225.0169; Found: 225.0168.
4.6. Reverse transcriptase assay with homopolymeric template/primers
HIV-1 RT assays were carried out in the presence of artificial homopolymeric template/primers. Poly(A), dT12-18, dC12-18, poly(I), dA12-18, poly(U),and poly(C) were from Pharmacia (Uppsala, Sweden). To prepare the template/primers for the RT experiments, 0.15 mM poly(U), poly(A), poly(I), and poly(C) were mixed with an equal volume of 0.0375 mM oligo(dA), oligo(dT), oligo(dC), and oligo(dG), respectively. The reaction mixture (50 μl) contained 50 mM Tris.HC1 pH 7.8, 5 mM dithiothreitol, 300 mM glutathione, 500 μM EDTA, 150 mM KCl, 5mM MgCl2, 1.25 μg of bovine serum albumin, an appropriate concentration of the tritium-labeled substrate [CH3-3H]dTTP, [5-3H]dCTP, [2,8-3H]dATP, or [8-3H]dGTP (2 μCi/assay), a fixed concentration of the template/primer poly(A).oligo(dT) (0.015 mM), poly(I).oligo(dC) (0.015 mM), poly(U).oligo(dA) (0.015 mM), or poly(C).oligo(dG) (0.015 mM), 0.06% Triton X-100, 10 μl of test compound solution (containing various concentrations of the compounds), and 1 μl of the HIV-1 RT preparation. The reaction mixtures were incubated at 37°C for 30 minutes, at which time 100 μl of calf thymus DNA (150 μg/ml), 2 ml of Na4P207 (0.1 M in 1 M HCl), and 2 ml of trichloroacetic acid (10% v/v) were added. The solutions were kept on ice for 30 minutes, after which the acid-insoluble material was washed and analyzed for radioactivity. For the experiments in which the 50% inhibitory concentration (IC50) of the test compounds was determined, fixed concentrations of 1.25 μM [3H]dTTP, 1.75μM [3H]dATP, 2.5 μM [3H]dCTP, or 2.0 μM [3H]dGTP were used.
4.7. Enzyme assay with HCMV DNA polymerase
The pGEM3Z-CMV UL54 plasmid for expression of the catalytic subunit (UL54 protein) of HCMV DNA polymerase was a generous gift from T. Cihlar (Gilead Sciences, Foster City, CA).27 Protein expression was performed with the TnT® SP6 Quick Coupled Transcription/Translation System (Promega).28 The plasmid was added (at 10 ng per μl volume) to the TnT® mix containing 0.5 mM MgCl2 and 10 mM potassium acetate, and the mixture was incubated for 3 hours at 30°C. To perform the HCMV DNA polymerase assay, 4 μl of the TnT® reaction product was added to a 46 μl mixture to obtain 25 mM Tris.HCl pH 8.0, 100 mM (NH4)2SO4, 0.5 mM dithiothreitol, 10 mM MgCl2, 0.2 mg/ml bovine serum albumin, 5 % glycerol, 150 ng per μl activated calf thymus DNA (from Amersham Biosciences, Piscataway, N.J.), 0.5 μM of the rate-limiting tritium-labeled dNTP, 100 μM of each of the three unlabeled dNTPs, and serial dilutions of the test compounds. After 60 minutes incubation at 37°C, nucleic acids were precipitated by addition of 1 ml of ice-cold 5% TCA and 20 mM Na4P2O7, then spotted onto glass microfiber filters (type G/C; GE Health Care UK Limited, Buckinghamshire, UK) and further washed with 5% TCA and ethanol to remove free radiolabeled dNTP. Radioactivity was determined in a Packard (Perkin Elmer, Zaventem, Belgium) Tri-Carb 2300 TR liquid scintillation counter. All radiolabeled materials were obtained from Moravek (Brea, CA).
4.8. Enzyme assay with herpes simplex virus type 1 (HSV-1) DNA polymerase and human DNA polymerases α and β
The reaction mixture (40 μl) for the HSV-1 DNA polymerase and human DNA polymerase α assays contained 4 μl of Premix (200 mM Tris.HCl pH 7.5; 2 mM DTT; 30 mM MgCl2), 4 μl of BSA (5 mg/ml), 1.6 μl of activated calf thymus DNA (1.0 mg/ml), 0.8 μl of dCTP (5 mM), 0.8 μl of dATP (5 mM), 0.8 μl of dGTP (5 mM), 2 μl of radiolabeled [3H]dTTP (1 mCi/ml) (3.2 μM), 18 μl of H2O, and 4 μl of test compound at different serial concentrations (i.e., 200, 40, 8, 1.6, 0.32 μM). The reaction was started by the addition of 4 μl of recombinant HSV-1 DNA polymerase (kindly provided by M.W. Wathen, at that time at Pfizer, Kalamazoo, MI) or human DNA polymerase α or β (Chimerix, Milwaukee, WI) (in 20 mM Tris.HCl pH 8.0; 1 mM DTT; 0.1 mM EDTA; 0.2 M NaCl; 40% glycerol), and the reaction mixture was incubated for 60 minutes at 37°C. Then, 1 ml of ice-cold 5% TCA in 0.02 M Na4P2O7.10 H2O was added to terminate the polymerization reaction. The consecutive steps (i.e., capture of the acid-insoluble precipitate onto filters, filter washing, and scintillation counting) were done as described above.
4.9. Enzyme assay with varicella-zoster virus (VZV) DNA polymerase
The VZV DNA polymerase assay was as follows: the 50 μl-reaction mixture contained 6.4 mM HEPES, 12 mM KCl, 25 mM NaCl, 5 mM MgCl2, 4.6 μg BSA, 2 mM CHAPS, 5 mM DTT, 5% glycerol, 1 μCi [3H]dTTP, 100 μM poly dA.oligo dT, and 5 μl of serial dilutions of the test compounds. The reaction was started by the addition (5 μl) of recombinant VZV DNA polymerase (kindly provided by M.W. Wathen) in 5 mM HEPES and incubated for 60 min at 37°C. The termination of the enzyme reaction and all consecutive steps to quantify the [3H]dTTP incorporation into the template/primer were done as described above.
4.10. Plasmid constructs and protein purification
RB69 gp43 WT and the RB69/HCMV chimera ABC5 were cloned into pPR-IBA1 and expressed in E.coli BL21(DE3) as described previously.23 RB69 ABC5 contains mutations V478W F479V, N480A, I557M, N558A, R559L, L561V, L562T, and to yield an enzyme derived from WT RB69 gp43 and the nucleotide binding site of HCMV UL54. HCMV UL54 was cloned into pFASTBac HTa and contains a C-terminal Strep tag allowing expression in the SF9-Baculovirus expression system.29 All enzymes used for biochemical studies also contain mutations to eliminate the 3′-5′ exonuclease activity eliminating the confounding effects of editing activities. These mutations are: D222A and D327A in RB69 gp43, and D542A in HCMV UL54.
4.11. Primer extension assay
The following sequences were used in a primer extension assay to monitor DNA synthesis and its inhibition. T50A6 (5′-CCA ATA TTC ACC ATC AAG GCT TGA TGA AAC TTC ACT CCA CTA TAC CAC TC-3′) was used as a template. The underlined residues are the portion of the template that anneals to the fluorescently labeled primer Cy5-P1 (5′-GAG TGG TAT AGT GGA GTG AA-3′). A 10 step, three-fold dilution series of the compound to be tested was prepare to obtain concentration range between 200 μM-10 nM. The reaction was carried out as follows: compound to be tested was pre-incubated 10 minutes at 37°C in buffer containing 25 mM Tris-HCL (pH8.0), 90 mM NaCl, 3 nM EDTA, 10 mM MgCl2, 1 mM DDT, 0.02% bovine serum albumin, 5% glycerol v/v with 75nM of Cy5-P1/T50A6 primer/template hybrid and 75 nM of enzyme. The enzymatic reaction was initiated by the addition of dNTP to a final concentration of 5 μM for RB69 WT and RB69 ABC5 and 250 μM for HCMV UL54. The final concentration of dNTP was increased in HCMV UL54 increase full length extension. The reaction was allowed to proceed for 5 minutes for RB69 WT and RB69 ABC5 and for 60 minutes for HCMV UL54. Reactions were stopped with 90% formamide containing traces of bromophenol blue and samples were resolved on a 12% denaturing polyacrylamide gel and visualized by fluorescence imaging. For dose response experiment, percentage inhibition was calculated as the fraction of full-length product divided by total primer. These values were then plotted against inhibitor concentration using GraphPad Prisim software. IC50 is defined as the concentration of compound required to inhibit full length extension by 50%.
4.12. Anti-HSV activity assays
The compounds were evaluated against herpes simplex virus type 1 (HSV-1) strain KOS, and herpes simplex virus type 2 (HSV-2) strain G. The antiviral assays were based on inhibition of virus-induced cytopathicity in human embryonic lung (HEL) fibroblasts. Confluent cell cultures in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) in the presence of varying concentrations of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds.
Supplementary Material
Acknowledgments
We thank Lizette van Berckelaer, Ria Van Berwaer and Kristien Minner for excellent technical assistance, and Christiane Callebaut and Dr. Annelies Stevaert for dedicated editorial assistance. The research was supported by Grants PF 10/018 and GOA 10/14 from the KU Leuven (University of Leuven) (to J.B.), from the National Institutes of Health Grant P50 GM103368 and R37 AI027690 MERIT Award (to E.A.), the Science Foundation Ireland (05/PICA/B802) (to A.R.M.), and the Canadian Institutes of Health Research (CIHR) (to M.G.). M.G. is the recipient of a career award from the Fonds de la recherche en santé du Québec (FRSQ).
Abbreviations
- α-CNP
α-carboxy nucleoside phosphonate
- RT
reverse transcriptase
- NRTI
nucleoside RT inhibitor
- NNRTI
non-nucleoside RT inhibitor
- NtRTI
nucleotide RT inhibitor
- NcRTI
nucleotide competing RT inhibitor
- BVDU
(E)-5-(2-bromovinyl)-2′-deoxyuridine
- BVDU-TP
BVDU-triphosphate
- PFA
phosphonoformic acid
- dNTP
2′-deoxynucleotide-5′-triphosphate
- dTTP
2′-deoxythymidine-5′-triphosphate
- dATP
2′-deoxyadenosine-5′-triphosphate
- dCTP
2′-deoxycytidine-5′-triphosphate
- dGTP
2′-deoxyguanosine-5′-triphosphate
- HCMV
human cytomegalovirus
- HSV
herpes simplex virus
- HIV
human immunodeficiency virus
- VZV
varicella-zoster virus
- WT
wild-type
- 3TC
lamivudine
- FTC
emtricitabine
- TFV-DP
tenofovir-diphosphate
- DIAD
diisopropyl azodicarboxylate
Footnotes
AUTHOR CONTRIBUTIONS
The manuscript was written through contribution of all authors. All authors have given approval to the final version of the manuscript.
Supporting information, including 1H, 13C and 31P NMR spectra and HPLC chromatograms of the test compounds.
References
- 1.Jochmans D. Novel HIV-1 reverse transcriptase inhibitors. Virus Res. 2008;134(1–2):171–185. doi: 10.1016/j.virusres.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Jochmans D, Deval J, Kesteleyn B, Van Marck H, Bettens E, De Baere I, Dehertogh P, Ivens T, Van Ginderen M, Van Schoubroeck B, Ehteshami M, Wigerinck P, Götte M, Hertogs K. Indolopyridones inhibit human immunodeficiency virus reverse transcriptase with a novel mechanism of action. J Virol. 2006;80(24):12283–12292. doi: 10.1128/JVI.00889-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Z, Walker M, Xu W, Shim JH, Girardet J-L, Hamatake RK, Hong Z. Novel nonnucleoside inhibitors that select nucleoside inhibitor resistance mutations in human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother. 2006;50(8):2772–2781. doi: 10.1128/AAC.00127-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajotte D, Tremblay S, Pelletier A, Salois P, Bourgon L, Coulombe R, Mason S, Lamorte L, Sturino CF, Bethell R. Identification and characterization of a novel HIV-1 nucleotide-competing reverse transcriptase inhibitor series. Antimicrob Agents Chemother. 2013;57(6):2712–2718. doi: 10.1128/AAC.00113-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moelling K, Broecker F, Kerrigan JE. RNase H: specificity, mechanism of action, and antiviral target. Methods Mol Biol. 2014;1087:71–84. doi: 10.1007/978-1-62703-670-2_7. [DOI] [PubMed] [Google Scholar]
- 6.Beilhartz GL, Ngure M, Johns BA, DeAnda F, Gerondelis P, Götte M. Inhibition of the ribonuclease H activity of HIV-1 reverse transcriptase by GSK5750 correlates with slow enzyme-inhibitor dissociation. J Biol Chem. 2014;289(23):16270–16277. doi: 10.1074/jbc.M114.569707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vernekar SK, Liu Z, Nagy E, Miller L, Kirby KA, Wilson DJ, Kankanala J, Sarafianos SG, Parniak MA, Wang Z. Design, synthesis, biochemical, and antiviral evaluations of C6 benzyl and C6 biarylmethyl substituted 2-hydroxylisoquinoline-1,3-diones: dual inhibition against HIV reverse transcriptase-associated RNase H and polymerase with antiviral activities. J Med Chem. 2015;58(2):651–664. doi: 10.1021/jm501132s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.James SH, Prichard MN. Current and future therapies for herpes simplex virus infections: mechanism of action and drug resistance. Curr Opin Virol. 2014;8:54–61. doi: 10.1016/j.coviro.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Freeman S, Gardiner JM. Acyclic nucleosides as antiviral compounds. Mol Biotechnol. 1996;5(2):125–137. doi: 10.1007/BF02789061. [DOI] [PubMed] [Google Scholar]
- 10.De Clercq E. (E)-5-(2-Bromovinyl)-2′-deoxyuridine (BVDU) Med Res Rev. 2005;25(1):1–20. doi: 10.1002/med.20011. [DOI] [PubMed] [Google Scholar]
- 11.Öberg B. Antiviral effects of phosphonoformate (PFA, foscarnet sodium) Pharmacol Ther. 1989;40(2):213–285. doi: 10.1016/0163-7258(89)90097-1. [DOI] [PubMed] [Google Scholar]
- 12.Lisco A, Vanpouille C, Tchesnokov EP, Grivel JC, Biancotto A, Brichacek B, Elliott J, Fromentin E, Shattock R, Anton P, Gorelick R, Balzarini J, McGuigan C, Derudas M, Götte M, Schinazi RF, Margolis L. Acyclovir is activated into a HIV-1 reverse transcriptase inhibitor in herpesvirus-infected human tissues. Cell Host Microbe. 2008;4(3):260–270. doi: 10.1016/j.chom.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMahon MA, Siliciano JD, Lai J, Liu JO, Stivers JT, Siliciano RF, Kohli RM. The antiherpetic drug acyclic inhibits HIV replication and select the V75I reverse transcriptase multidrug resistance mutation. J Biol Chem. 2008;283(46):31289–31293. doi: 10.1074/jbc.C800188200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perti T, Saracino M, Baeten JM, Johnston C, Diem K, Ocbamichael N, Huang ML, Selke S, Magaret A, Corey L, Wald A. High-dose valacyclovir decreases plasma HIV-1 RNA more than standard-dose acyclovir in persons coinfected with HIV-1 and HSV-2: a randomized crossover trial. J Acquir Immune Defic Syndr. 2013;63(2):201–208. doi: 10.1097/QAI.0b013e3182928eea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vanpouille C, Lisco A, Introini A, Grivel JC, Munawwar A, Merbah M, Schinazi RF, Derudas M, McGuigan C, Balzarini J, Margolis L. Exploiting the anti-HIV-1 activity of acyclovir: suppression of primary and drug-resistant HIV isolates and potentiation of the activity of ribavirin. Antimicrob Agents Chemother. 2012;56(5):2604–2611. doi: 10.1128/AAC.05986-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keane SJ, Ford A, Mullins ND, Maguire NM, Legigan T, Balzarini J, Maguire AR. Design and synthesis of α-carboxy nucleoside phosphonate analogues and evaluation as HIV-1 reverse transcriptase-targeting agents. J Org Chem. 2015;80(5):2479–2493. doi: 10.1021/jo502549y. [DOI] [PubMed] [Google Scholar]
- 17.Balzarini J, Das K, Bernatchez JA, Martinez SE, Ngure M, Keane S, Ford A, Maguire N, Mullins N, John J, Kim Y, Dehaen W, Vande Voorde J, Liekens S, Naesens L, Götte M, Maguire A, Arnold E. Alpha-carboxy nucleoside phosphonates as universal nucleoside triphosphate mimics. Proc Natl Acad Sci USA. 2015;112(11):3475–3480. doi: 10.1073/pnas.1420233112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balzarini J. Metabolism and mechanism of antiretroviral action of purine and pyrimidine derivatives. Pharm World Sci. 1994;16(2):113–126. doi: 10.1007/BF01880662. [DOI] [PubMed] [Google Scholar]
- 19.Debarge S, Balzarini J, Maguire AR. Design and synthesis of α-carboxy phosphononucleosides. J Org Chem. 2011;76(1):105–126. doi: 10.1021/jo101738e. [DOI] [PubMed] [Google Scholar]
- 20.(a) Yin X-q, Li W-k, Schneller SW. An efficient Mitsunobu coupling to adenine-derived carbocyclic nucleosides. Tetrahedron Lett. 2006;47(52):9187–9189. [Google Scholar]; (b) Ludek OR, Meier C. Influence of the N3-protection group on N1- vs.O2-alkylation in the Mitsunobu reaction. Eur J Org Chem. 2006;2006(4):941–946. [Google Scholar]; (c) Fletcher S. Regioselective alkylation of the exocyclic nitrogen of adenine and adenosine by the Mitsunobu reaction. Tetrahedron Lett. 2010;51(22):2948–2950. [Google Scholar]
- 21.(a) Elayadi H, Smietana M, Pannecouque C, Leyssen P, Neyts J, Vasseur J-J, Lazrek HB. Straightforward synthesis of triazoloacyclonucleotide phosphonates as potential HCV inhibitors. Bioorg Med Chem Lett. 2010;20(24):7365–7368. doi: 10.1016/j.bmcl.2010.10.046. [DOI] [PubMed] [Google Scholar]; (b) Piotrowska DG, Balzarini J, Glowacka IE. Design, synthesis, antiviral and cytostatic evaluation of novel isoxazolidine nucleotide analogues with a 1,2,3-triazole linker. Eur J Med Chem. 2012;47(1):501–509. doi: 10.1016/j.ejmech.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett N, Götte M. Utility of the bacteriophage RB69 polymerase gp43 as a surrogate enzyme for herpesvirus orthologs. Viruses. 2013;5(1):54–86. doi: 10.3390/v5010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tchesnokov EP, Obikhod A, Schinazi RF, Gotte M. Engineering of a chimeric RB69 DNA polymerase sensitive to drugs targeting the cytomegalovirus enzyme. J Biol Chem. 2009;284(39):26439–26446. doi: 10.1074/jbc.M109.012500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahn KE, Tchesnokov EP, Götte M, Doublié S. Phosphonoformic acid inhibits viral replication by trapping the closed form of the DNA polymerase. J Biol Chem. 2011;286(28):25246–25255. doi: 10.1074/jbc.M111.248864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarafianos SG, Das K, Clark AD, Jr, Ding J, Boyer PL, Hughes SH, Arnold E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc Natl Acad Sci U S A. 1999;96(18):10027–10032. doi: 10.1073/pnas.96.18.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das K, Bandwar RP, White KL, Feng JY, Sarafianos SG, Tuske S, Tu X, Clark AD, Jr, Boyer PL, Hou X, Gaffney BL, Jones RA, Miller MD, Hughes SH, Arnold E. Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and tenofovir resistance. J Biol Chem. 2009;284(50):35092–35100. doi: 10.1074/jbc.M109.022525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cihlar T, Fuller MD, Cherrington JM. Expression of the catalytic subunit (UL54) and the accessory protein (UL44) of human cytomegalovirus DNA polymerase in a coupled in vitro transcription/translation system. Protein Expr Purif. 1997;11(2):209–218. doi: 10.1006/prep.1997.0781. [DOI] [PubMed] [Google Scholar]
- 28.De Bolle L, Manichanh C, Agut H, De Clercq E, Naesens L. Human herpesvirus 6 DNA polymerase: enzymatic parameters, sensitivity to ganciclovir and determination of the role of the A961V mutation in HHV-6 ganciclovir resistance. Antiviral Res. 2004;64(1):17–25. doi: 10.1016/j.antiviral.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Bernatchez JA, Paul R, Tchesnokov EP, Ngure M, Beilhartz GL, Berghuis AM, Lavoie R, Li L, Auger A, Melnyk RA, Grobler JA, Miller MD, Hazuda DJ, Hecht SM, Götte M. Derivatives of mesoxalic acid block translocation of HIV-1 reverse transcriptase. J Biol Chem. 2015;290(3):1474–1484. doi: 10.1074/jbc.M114.614305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.